Introduction
Atrial fibrillation (AF) is the most common
symptomatic cardiac arrhythmia in clinical practice, which exhibits
an increasing prevalence with age. As the currently available
therapeutic approaches for treating AF have a number of
limitations, including the adverse side effects that may occur with
antiarrhythmic drugs and arrhythmia recurrence following AF
ablation (1), AF contributes
substantially to worldwide morbidity and mortality (1–3). To
date, there are 4 principal pathophysiological mechanisms known to
contribute to AF including electrical and structural remodeling,
alterations in autonomic nervous system function, and abnormalities
in intracellular Ca2+ handling (4). Each of these may arise from
underlying cardiac disease and contribute to the subsequent
development of AF, which in turn may cause additional abnormalities
in the above aspects, further enhancing a patient's susceptibility
to AF induction and maintenance (4). Therefore, innovative studies of the
pathophysiology and underlying molecular mechanisms of AF are
required to develop novel therapeutic approaches for this
disease.
Ankyrins (Anks), a ubiquitously expressed family of
intracellular scaffolding proteins, are associated with a diverse
set of membrane, cytoskeletal and cytoplasmic proteins; they are
known to tether these proteins to specialized membrane signaling
domains. The Ank family is comprised of Ank-B, Ank-G and Ank-R,
which are encoded by the genes, Ankyrin-2 (ANK2),
ANK3 and ANK1, respectively (5). In particular, the targeting and
stability of sodium-calcium exchanger 1 (NCX1), inositol
triphosphate receptor and Na+K+-adenosine
triphosphatase (NKA) at the membrane of the transverse-tubule
(T-tubule)/sarcoplasmic reticulum microdomain in cardiomyocytes are
heavily reliant on Ank-B (6).
Ank-B also regulates the protein expression and membrane targeting
of the Kadenosine 5′-triphosphate (ATP) channel subunit
Kir6.2 to T-tubules, in addition to modulating KATP
channel ATP sensitivity (7). NCX1,
in turn, is an integral membrane protein that is expressed in
various tissues and is involved in Ca2+ homeostasis
(8). As aforementioned, a previous
study revealed that Ank-B binds to and targets NCX1, and
participates in the regulation of Ca2+ homeostasis
(9).
MicroRNAs (miR/miRNA) inhibit the expression of
specific mRNA targets through a base-pairing reaction between the
miRNA ‘seed region’ and sequences commonly located in the 3′
untranslated region (3′UTR) of the target mRNA (10). Recently, miR-1, together with
miR-133, were observed to regulate protein levels by repressing the
translation of genes involved in cardiac contractility, hypertrophy
and electrical conductance (11).
MiR-34a was reported to serve an important role in cardiac fibrosis
in patients and in mice, and it has been suggested that therapeutic
inhibition of members of the miR-34 family may attenuate
pathological cardiac remodeling and improve cardiac function in
patients, as this approach has been proven to be effective in mouse
models of cardiac disease (12,13).
Electrophysiological alterations and fibrosis are closely
associated with AF (5); however,
the potential functions of miR-1, miR-133 and miR-34a in AF have
been studied to a lesser extent.
In the present study, right atrial tissue samples
were collected from rheumatic heart disease patients with AF or
sinus rate (SR). As the aforementioned previous studies have
reported that miRNAs are associated with AF or with fundamental
pathophysiological processes of AF, the present study investigated
the expression differences between patient groups. By manipulating
the levels of miRNAs in cell culture through overexpression and
inhibition strategies, the role of miRNAs in the molecular
mechanisms underlying the early electrophysiological changes
observed in AF were further examined.
Materials and methods
Human tissue and animal samples
The present study, including all sample collection
procedures for human and animal studies, was approved by the Ethics
Committee of Xinqiao Hospital (Chongqing, China). Patients
participating in the present study were all recruited from the
Department of Cardiovascular Surgery of Xinqiao Hospital between
the July and October 2013. All patients provided written informed
consent. Right atrial tissue (100 mg/patient) was removed from
patients with rheumatic heart disease whilst undergoing valve
replacement (n=40; Table I); these
tissues were then either immediately snap-frozen in liquid nitrogen
or fixed in 4% paraformaldehyde at 4°C for 48 h. Right atrial
tissue was obtained from 20 patients with permanent AF (AF group;
n=20) and 20 patients with normal SR (SR group; n=20).
 | Table I.Clinical data of patients with atrial
fibrillation and sinus rate. |
Table I.
Clinical data of patients with atrial
fibrillation and sinus rate.
| Clinical data | AF group | SR group |
t/х2 | P-value |
|---|
| Age (years) | 52.9±5.8 | 52.4±6.2 | 0.237 | 0.814 |
| Sex (M/F) | 8/12 | 9/11 | 0.102 | 0.749 |
| Body weight
(kg) | 51.9±8.7 | 53.5±8.4 | 0.593 | 0.557 |
| LVEF (%) | 51.7±6.3 | 53.7±7.9 | 0.881 | 0.384 |
| LVFS (%) | 33.6±3.3 | 33.4±3.2 | 0.163 | 0.872 |
| Preoperative LA
(mm) | 49.6±5.0 | 48.5±6.6 | 0.617 | 0.541 |
| DVR/MVR (n) | 12/8 | 10/10 | 0.404 | 0.525 |
| Postoperative
hospital stay (days) | 10.7±2.0 | 11.1±2.0 | 0.720 | 0.476 |
The male neonatal Sprague Dawley (SD) rat pups (age,
<5 days; n=20) used in the present study were supplied by the
Laboratory Animal Center of the Third Military Medical University
(Chongqing, China). Neonatal rats were sacrificed as soon as they
were received.
Reagents
The reagents used for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) were
purchased from Takara Biotechnology Co., Ltd. (Dalian, China). The
agomir, antagomir and the corresponding negative controls (NC) for
miR-34a, as well as the recombinant plasmid vector containing the
predicted ANK-2-miR-34a sites and mutations of those sites
were obtained from Shanghai GenePharma Co., Ltd. (Shanghai, China).
The pmirGLO Dual-Luciferase miRNA Target Expression Vector and Dual
luciferase assay system were purchased from Promega Corporation
(Madison, WI, USA). The recombinant plasmid containing the
ANK2 wild type (WT) and mutant (MUT) 3′UTR sequences were
purchased from Shanghai GenePharma Co., Ltd.
Cell culture
Atrial primary cardiomyocytes were isolated from the
auricular appendages and atrial tissues of male neonatal SD rats.
Briefly, the auricular appendages and atrial tissues were cut into
1 mm3 granules and digested with pancreatin and
collagenase II, filtered through a 200-mesh screen filter to remove
undigested cellular aggregates, non-cellular tissue components and
debris, and centrifuged at 700 × g for 5 min at 4°C. The cell
pellet was resuspended in Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.)
Cells (1.5×105 cm2) were subsequently placed
in a 25 cm culture flask and incubated in 95% air and 5%
CO2 at 37°C for 2 h to allow for the differential
attachment of non-myocardial cells. The non-adhesive cells
(cardiomyocytes) were transferred into a sterile tube, centrifuged
at 700 × g for 5 min at 4°C and subsequently plated into a novel
culture flask.
293T cells were purchased from American Type Culture
Collection (Manassas, VA, USA). Cells were cultured in 96-well
plates in DMEM with 10% FBS in 5% CO2 at 37°C at 24 h,
at which point transfection was performed. All cell culture
reagents were purchased from Gibco; Thermo Fisher Scientific, Inc.
(Waltham, MA, USA).
Bioinformatics analysis
Potential miR-34a targets were predicted using the
following online databases: TargetScan (version 7.1; www.targetscan.org/vert_71/), PicTar (2006
release; pictar.mdc-berlin.de), miRanda (2010
release; 34.236.212.39/microrna/home.do), RNA22-HAS (2006 release;
https://cm.jefferson.edu/rna22),
miRTarbase (version 6.0; http://mirtarbase.mbc.nctu.edu.tw/php/index.php) and
miRBD (2015 release; www.mirdb.org).
Transfection
Atrial cardiomyocytes were cultured to 70%
confluence in 6-well plates and transfected with a specific miR-34a
agomir (0.5 µM), antagomir (1 µM) or the respective matched miRNA
NC (Ago-N, 0.5 µM; Ant-N, 1 µM) using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) as the
transfection reagent. Briefly, transfection experiments were
carried out in 500 µl Opti-minimum essential medium (MEM; Gibco;
Thermo Fisher Scientific,) with the desired concentration of the
specific miR-34a interference reagent (agomir, Ago-N, antagomir and
Ant-N) Cells were removed following a 6 h incubation period with
the transfection reagent in serum free Opti-MEM medium at 37°C; the
medium was subsequently replaced with DMEM containing 10% FBS for
48 h at 37°C for the following experiments. The cells of
untransfected (Blank) group underwent the same procedure but the
interference reagent was replaced with serum free Opti-MEM.
293T cells were cultured to 50% confluence in
96-well plates and transfected with the WT or MUT vector,
containing the 3′UTR of ANK2 or the mutant 3′UTR sequence of
ANK2 (Shanghai GenePharma Co., Ltd.), respectively, along
with the miR-34a agomir, antagomir, Ago-N or Ant-N. Transfection
was carried out in 200 µl Opti-MEM with 2 µM of WT or MUT vector
and the specific miR-34a reagent. For all experiments, transfection
took place 24 h following the starvation of cells in serum-free
medium. Transfection experiments for each construct and/or miRNA
were performed in triplicate in each assay and three assays were
performed in total.
RT-qPCR
Total RNA was extracted using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) from cells transfected with each
specific miR-34a reagent or from untransfected controls. RT was
carried out using the RTPrimeScript™ RT reagent kit (Perfect Real
Time) as per the manufacturer's instructions (Takara Biotechnology
Co., Ltd.). The temperature protocol was as follows: 42°C for 30
min and 85°C for 5 sec. Amplification of cDNA products was
performed with the SYBR® Premix Ex Taq™ II (Perfect Real
Time) amplification kit (Takara Biotechnology Co., Ltd.) using U6
snRNA as the reference gene, and the Applied Biosystems 7500 Fast
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Initial denaturation occurred at 95°C for 30 sec, followed
by 40 cycles of 95°C for 5 sec and 60°C for 34 sec. Bulge-Loop™
Primers for miR-34a (hsa-miR-34a, cat. no. miRQ0000255-1-2l;
mmu-miR-34a, cat. no. miRQ0000542-1-2; sequences unavailable) and
U6 (cat. no. MQP-0202; sequence unavailable) detection were
purchased from Guangzhou RiboBio Co., Ltd, (Guangzhou, China). PCR
results were quantified using the 2−∆∆Cq method
(14).
Isoprenaline (ISO) mediated
Ca2+ signaling detection
Primary SD neonatal atrial cardiomyocytes were
treated with 10M ISO for different periods at 37°C (30 min, and 1,
1.5, 3, 6, 12, 24, 48 or 72 h) to mimic the early
electrophysiological change of atrial cardiomyocytes in AF
(15). Transfection of miR-34a
interference reagents was performed and miR-34a expression levels
from the various cultures were determined as aforementioned. The
calcium fluorescent dye Fluro-3 AM (10 µM; Thermo Fisher
Scientific, Inc.) was added into the serum-free DMEM and
subsequently added to atrial cardiomyocytes at 37°C for 30 min.
Cellular Ca2+ signaling of each group was detected at
37°C using a laser scanning confocal microscope in the line
scanning mode. Quantification of fluorescence intensity was
performed with the Leica Las AF lite software (version 2.6; Leica
Microsystems, Inc., Buffalo Grove, IL, USA).
Luciferase activity assay
miRNA function following transfection of miRNA
modifier reagents was measured using a luciferase activity assay.
293T cells were transfected with pmirGLO™ luciferase miRNA
expression reporter vectors (Promega Corporation) carrying the
3′UTR of the potential miR-34a target gene ANK-2 or its
mutant variant, along with miR-34a modifier reagents as described
above using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.). Following transfection for 48 h,
luciferase activities were measured using a dual luciferase
reporter assay kit (Promega Corporation) on a luminometer (Lumat
LB9507; Berthold Technologies GmbH & Co. KG, Bad Wildbad,
Germany) according to the manufacturer's instructions.
Renilla luciferase activities were measured for
normalization.
Immunohistochemistry
Immunohistochemistry was performed on
paraffin-embedded explanted human atrial tissue fixed in 4%
paraformaldehyde at 4°C. Paraffin sections (6 µm) were immersed in
1,2-dimethylbenzene I for 10 min, 1,2-dimethylbenzene II for 10
min, 1,2-dimethylbenzene with ethyl alcohol (1:1), absolute ethyl
alcohol for 5 min, 95% ethyl alcohol for 5 min, 75% ethyl alcohol
for 5 min, 50% ethyl alcohol for 5 min in turn. Sections were
subsequently washed in PBS for 3 min three times. Following
microwave antigen retrieval at 120°C for 5 min and 85°C for 15 min
with the addition of citric acid buffer every 4 min to prevent
drying, 3% H2O2 was added for 15 min at room
temperature and sections were incubated overnight with the primary
antibody anti-Ank-B (1:50; cat. no. sc-12718; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) at 4°C. After washing the
sections with PBS-0.1% Triton X-100 for 5 min five times, sections
were incubated with horseradish peroxidase-labeled goat anti-mouse
secondary antibody (1:1,000, cat. no. A0216; Beyotime Institute of
Biotechnology, Shanghai, China) at 37°C for 50 min and subsequently
washed with PBS-0.1% Triton X-100 for 5 min five times.
Immunoreaction was revealed with 3,3′-diaminobenzidine and observed
using an inverted microscope. Results were analyzed with Image Pro
Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD,
USA).
Protein extraction and western blot
analysis
The expression of Ank-B in the atrial tissue of
patients and in the primary atrial cardiomyocytes of neonatal SD
rat pups transfected with miR-34a specific interference sequences
or in untransfected controls was determined using western blotting.
Total protein from tissue and cell cultures was extracted with SDS
Lysis Buffer (Beyotime Institute of Biotechnology) and protein
concentration was determined with the bicinchoninic acid protein
assay and proteins (20 µg/lane) were separated using 10% SDS-PAGE.
Gels were transferred using a wet transfer method to a
polyvinylidene fluoride membrane (Roche Diagnostics, Basel,
Switzerland) and blocked with 5% non-fat dried milk at room
temperature. Membranes were subsequently incubated overnight at 4°C
with the primary antibodies Ank-B (1:200; cat. no. sc-12718; Santa
Cruz Biotechnology, Inc.), NCX1 (1:100; cat. no. bs-1550R; Beijing
Biosynthesis Biotechnology Co., Ltd., Beijing, China) and β-actin
(1:1,000; cat. no. bs-0061R; Beijing Biosynthesis Biotechnology
Co., Ltd.). The membranes were washed in PBS with 0.1% Tween for 5
min five times, prior to incubation with horseradish
peroxidase-labeled goat anti-rabbit (1:1,000; cat. no. A0208;
Beyotime Institute of Biotechnology) and anti-mouse (1:1,000; cat.
no. A0216; Beyotime Institute of Biotechnology) for 1 h at 37°C.
Following washing the membrane in PBS with 0.1% Tween for 5 min
five times, immunoreactivity was revealed by chemiluminescence
using an Alpha Innotech FluorChem imaging system (Canberra Packard
Central Europe GmbH, Schwadorf, Austria) with Fluorchem SP software
(version 2.2; ProteinSimple, San Jose, CA, USA). The relative band
densities were analyzed with ImageJ 1.5.0 (National Institutes of
Health, Bethesda, MD, USA). β-actin was used as the internal
reference. The SR group was used as the control for protein level
analysis in atrial tissue, and the untransfected or untransfected
without ISO groups served as the controls for in vitro
analyses. All samples were run at least 3 times.
Statistical analysis
Student's t-test, chi-square test or one-way
analysis of variance followed by Tukey's post-hoc test was used to
analyze the statistical significance of data with SPSS 19.0 (IBM
Corp., Armonk, NY, USA). Data are presented as the mean ± standard
error mean of at least 3 independent experiments. P<0.05 was
considered to indicate a statistically significant difference.
Results
Ank-B expression was inversely
proportional to miR-34a expression in the AF and SR patient
groups
The present study examined the protein expression
levels of Ank-B, an intracellular scaffolding protein that is
important for ion homeostasis and is associated with AF (16), in cardiac tissue from patients with
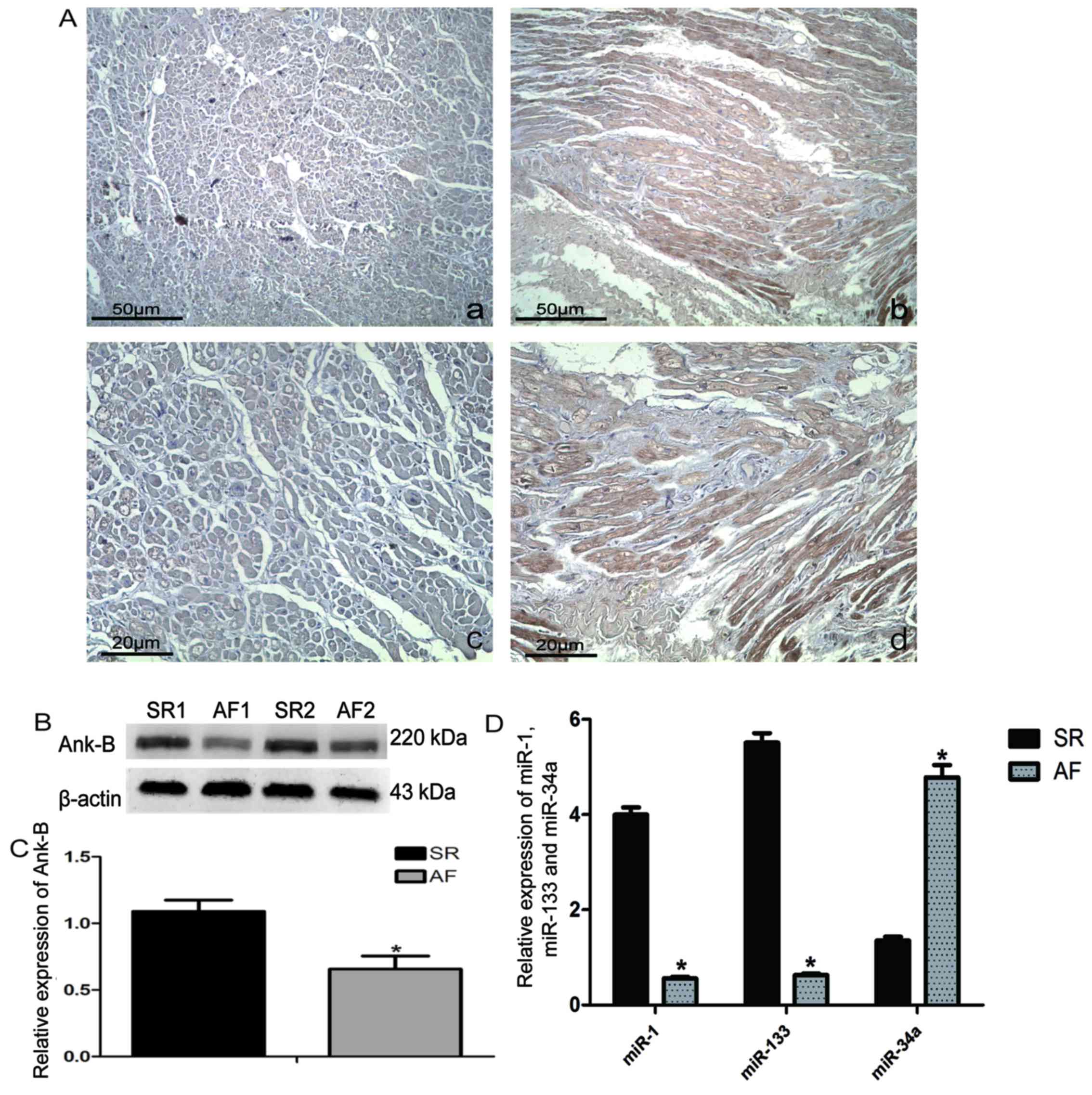
AF or SR. As presented in Fig. 1A,
Ank-B expression in the atrial tissue from the AF group (Fig. 1A-a and c) was lower than that
observed in the SR group (Fig. 1A-b
and d), as determined by the decreased amount of positive brown
signaling in the AF images. The results of western blot analysis of
atrial tissue protein were consistent with those observed from
immunohistochemistry staining, as measured by chemiluminescent
quantitation (Fig. 1B and C).
Notably, as shown in Fig. 1D, the
relative expression of miR-34a, as measured by RT-qPCR, was higher
in the atrial tissue of the AF group when compared with the SR
group; however, the relative expression of miR-1 and miR-133 was
lower in the AF group than in the SR group. MiR-1 and miR-133 serve
important roles in cardiac electrophysiological remodeling, and are
known to be closely associated with congenital and acquired heart
disease (11,17). Recently, miR-34a was noted for its
significant effect on the development of fibrosis (18) and, in particular, of cardiac
fibrosis (12,13). Therefore, the present study
performed bioinformatics analysis using online databases, which
revealed that 4 of the 6 databases predicted that the 3′UTR of
ANK2 (Ank-B) contained potential miR-34a target binding
sites; while one website predicted that the 3′UTR of ANK2
(Ank-B) also contained potential miR-133 target sites. These
results, combined with the accepted mechanism of miRNA function,
indicated that miR-34a may have some co-regulatory connection with
Ank-B.
miR-34a overexpression enhances
Ca2+ signaling in atrial cardiomyocytes treated with
ISO
Atrial electrical remodeling is an important
component of the primary pathophysiological process in the
development of AF, and intracellular calcium signaling is the
principle constituent of early electrical remodeling (4). To imitate the early
electrophysiological change of atrial cardiomyocytes in AF, the
present study treated primary SD neonatal atrial cardiomyocytes
with 10M ISO (15). ISO, a
non-selective β-adrenergic agonist, has been shown to increase the
intracellular Ca2+ transient and contraction amplitudes
in cardiomyocytes via the β-adrenergic receptor-adenylyl
cyclase-cyclic adenosine 5′-phosphate-protein kinase A signaling
pathway (19). qPCR analysis
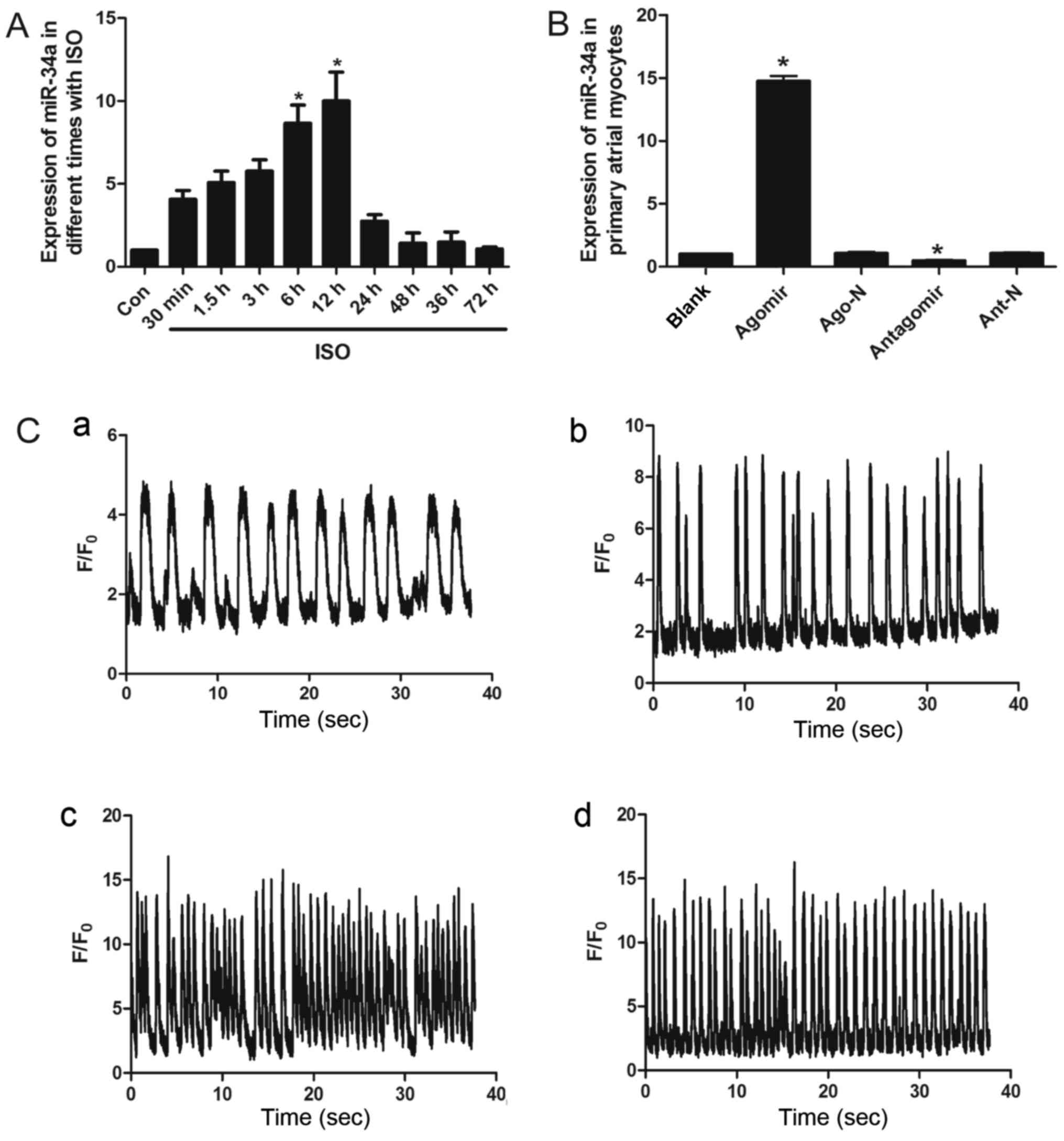
demonstrated that the relative expression of endogenous miR-34a
increased with ISO treatment duration, prior to peaking at 12 h and
then declining (Fig. 2A). In order
to observe and analyze the effects of miR-34a on intracellular
Ca2+ signaling, the present study induced the
overexpression or silencing of miR-34a in primary SD neonatal
atrial cardiomyocytes via transfection with an miR-34a specific
agomir or antagomir (chemically modified double stranded miRNA
mimic, or single stranded complementary, interfering,
oligonucleotides), respectively (Fig.
2B). Following 12 h of ISO treatment, the intracellular
Ca2+ signal of treated cells was stronger than that of
the untreated group. Simultaneous overexpression of miR-34a further
enhanced intracellular Ca2+ signaling. The strength and
frequency of the signal were increased in treated cells
(Agomir+ISO; Fig. 2C-d), compared
with the blank (Fig. 2C-a), Ago
(Fig. 2C-b) and ISO groups
(Fig. 2C-c). By contrast,
inhibition of miR-34a did not lead to a significant difference
between antagomir transfected and untransfected control groups.
Thus, inhibition of miR-34a did not appear to be sufficient to
counterbalance the change in intracellular Ca2+
signaling caused by ISO; however, miR-34a overexpression did
enhance it. It was therefore hypothesized that the increased
expression of miR-34a may elicit, or cause the further
deterioration of, early electrophysiological changes in the atrial
cardiomyocytes.
ANK2 is a target of miR-34a
ANK2, the gene encoding Ank-B, was the
predicted potential target gene of miR-34a, as predicted by 4 of
the 6 bioinformatics databases (Fig.
3A). Ank-B participates in cellular Ca2+ signaling
via direct physical connections with a number of ion channels and
exchangers (6); it is unknown
whether miR-34a affects intracellular Ca2+ signaling via
a direct physical connection with ANK2 as well, albeit
through a completely different mechanism. To further elucidate the
underlying mechanism of the apparent miR-34a Ank-B co-regulation,
and to confirm our previous hypothesis, the present study cloned
the WT 3′UTR of ANK2 into the pmirGLO-vector, a reporter
construct for the measurement of miRNA activity, and co-transfected
the miR-34a agomir or antagomir with the vector into 293T cells
(Fig. 3B). The results revealed
that the normalized luciferase activity of the agomir-transfected
group was significantly reduced, while the normalized luciferase
activity of the antagomir-transfected group increased, when
compared with their respective negative (scrambled) and
untransfected controls (Fig. 3C).
In addition, a vector construct containing a version of the
ANK2 3′UTR sequence was generated, in which the predicted
miR-34a binding site had been mutated (MUT type) and co-transfected
along with the miR-34a agomir and antagomir into 293T cells. In
these experiments, no significant difference in the normalized
luciferase activities between the groups was observed (P>0.05;
Fig. 3C). Furthermore,
transfection of primary SD neonatal atrial cardiomyocytes with the
miR-34a agomir (mimicking overexpression) significantly reduced the
protein levels of Ank-B; however, transfection with the miR-34a
antagomir increased the levels of Ank-B (P<0.05; Fig. 3D). Together, these results
suggested that miR-34a may inhibit the expression of Ank-B by
directly binding to the 3′UTR of ANK2, and that the effect
of miR-34a expression on intracellular Ca2+ signaling
may also be explained by this mechanism.
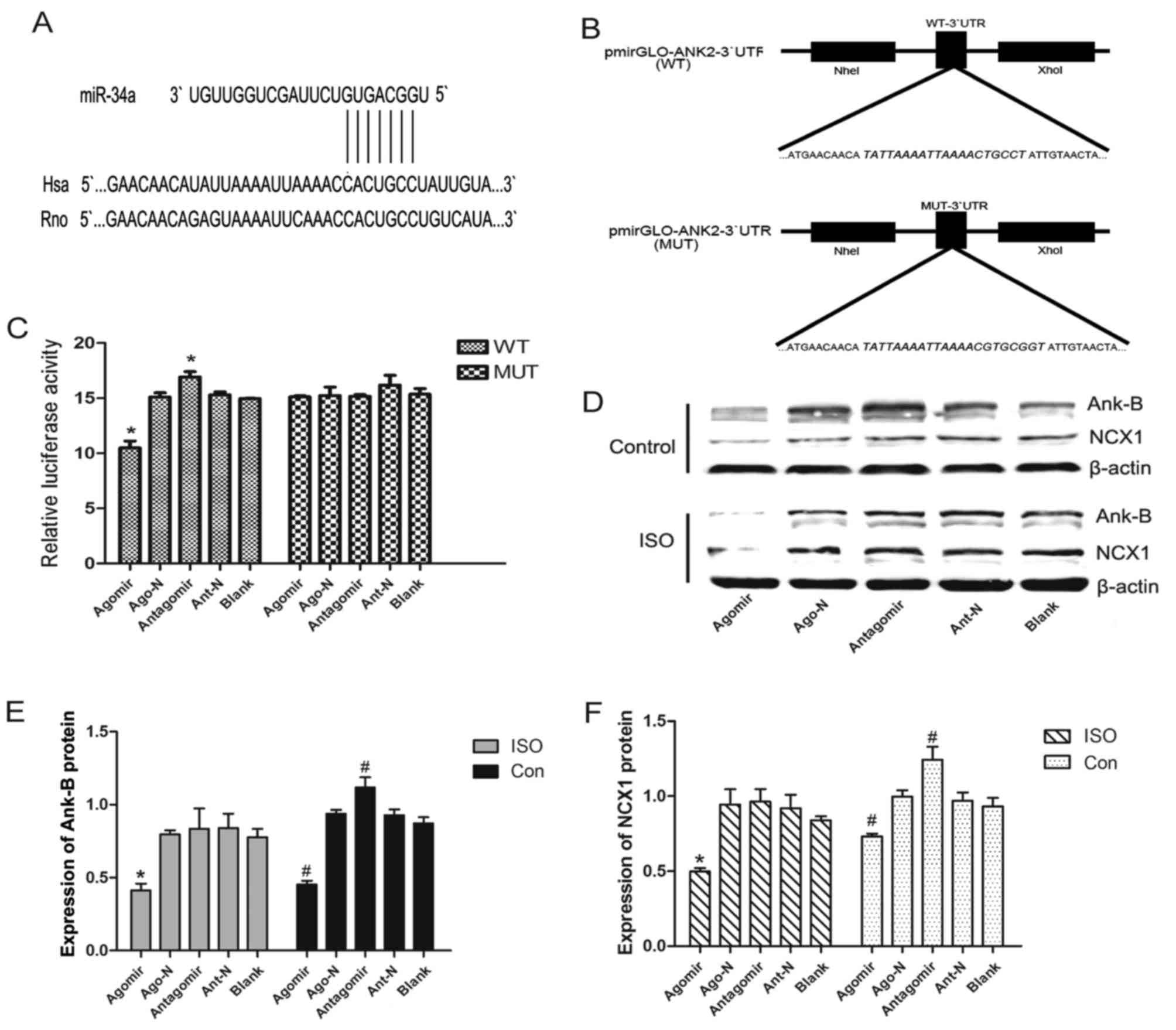 | Figure 3.ANK2 is a target of miR-34a.
(A) Sequence alignment of miR-34a and 3′UTR of ANK2 using
the TargetScan algorithm. (B) Plasmid construction: 3′UTR of
ANK2 or a mutated sequence was cloned into pmirGLO-vector.
(C) Luciferase reporter assay with co-transfection of WT or MUT
3′UTR, and agomir, Ago-N, antagomir, Ant-N or blank control in 293T
cells. or MUT group, WT, wild-type; MUT, mutant (D) Western
blotting results revealed the relative expression of (E) Ank-B and
(F) NCX1. Values are presented as the mean ± standard error.
*P<0.05 vs. the corresponding blank group. Con, control;
ANK2, Ankyrin 2; Ank-B, Ankyrin B; miR, microRNA; UTR,
untranslated region; NCX1, sodium-calcium exchanger 1; ISO,
Isoprenaline; Ago-N, agomir-negative control; Ant-N,
antagomir-negative control. |
Effects of miR-34a modulation on Ank-B
and NCX1 expression in primary rat neonatal atrial cardiomyocytes
treated with ISO
One of the membrane proteins that serves as a
binding partner with Ank-B is NCX1, a
Na+/Ca2+ exchanger involved in the regulation
of Ca2+ homeostasis (9). The present study demonstrated that
overexpression of miR-34a in primary rat neonatal atrial
cardiomyocytes led to lower levels of Ank-B and NCX1, while
inhibition of miR-34a led to higher levels (Fig. 3D-F), suggesting that miR-34a may
impact cellular Ca2+ signaling directly by regulating
the expression of Ank-B and indirectly by affecting NCX1.
Following exposure to ISO, the expression of Ank-B
and NCX1 was lower than that in cells without ISO treatment;
however, the trends in the expression of these proteins following
the overexpression and inhibition of miR-34a in cells exposed to
ISO were consistent with those exhibited by cells without ISO
treatment (Fig. 3E and F).
Therefore, ISO exposure had no influence on the regulation of
miR-34a to the expression of Ank-B and NCX-1, while miR-34a with
ISO exposure had a synergistic effect on calcium signaling.
Discussion
Atrial electrical remodeling, which can cause
impairment in atrial conduction, is characterized by a reduction of
the effective refractory period and the action potential duration
(APD), the dispersion in refractoriness and a reduction in the
conduction velocity of impulse propagation (20). These alterations may often have
been elicited during the early process of remodeling, and are
maintained in the development of AF (20). The atrial enlargement and fibrosis
observed in AF is characterized by structural remodeling, which
causes further conduction disturbances. These electrophysiological
changes have, until recently, provided the basis for the majority
of the therapies for AF; however, AF itself promotes the remodeling
process, which in turn contributes to the therapeutic resistance
observed in patients with long-standing arrhythmia (21).
The fundamental molecular basis of electrical
remodeling is an alteration in the function and/or expression of
ion channels and exchangers, including the NCX1, NKA, and L-type
Ca2+-channels and ryanodine receptors (4). As aforementioned, Ank-B binds to a
number of different types of ion channels and exchangers in
cardiomyocytes, and serves an important role in maintaining
cellular homeostasis for various ions, contributing to proper cell
functioning (6). ANK2 gene
variants are associated with sinus node dysfunction in humans,
which are often accompanied by AF (22). A previous study revealed that mice
heterozygous for an ANK2 deletion (Ank-B+/−)
exhibited atrial electrophysiological dysfunction and increased
susceptibility to AF, and that Ank-B+/− atrial myocytes
displayed shortened action potentials (23). In the present study, the expression
of Ank-B was decreased in cardiac dysfunction patients with AF when
compared with those with SR, and that the level of Ank-B was
decreased in atrial myocytes treated with ISO, which mimics the
early electrophysiological changes seen in AF. Furthermore, it was
observed that the cells with decreased levels of Ank-B expression
displayed abnormal Ca2+ signaling as well. It was
therefore hypothesized that a hereditary ANK2 gene mutation
may cause AF, and an acquired aberrant function and/or expression
of Ank-B may also be associated with AF. However, the underlying
mechanism that may be accountable for the acquired aberrant
expression of Ank-B in these experiments was not immediately
evident.
The role of miRNAs in the development of the heart
and of heart diseases has also become more apparent. In the present
study, some of the miRNAs associated with electrophysiological
processes, such as miR-1 and miR-133, and with the development of
fibrosis, such as miR-34a, were investigated. The results
demonstrated that the levels of miR-1 and miR-133 were decreased in
the atrial tissue from patients with AF when compared with those in
tissues from patients with SR, while miR-34a exhibited the opposite
trend. Bioinformatics analysis predicted that the 3′UTR of
ANK2 may contain homology to the ‘seed region’ of miR-34a
and miR-133, with a higher likelihood of being assigned to miR-34a
as it was flagged by several different analysis programs. However,
the changes in miR-1 and miR-133 expression presented a similar
trend to that of Ank-B in the atrial tissue of AF patients.
Although miR-1 and miR-133 are associated with various ion channels
(10,24), of which, some directly bind to
Ank-B (7,25), as the majority of miRNAs tend to
downregulate the expression of their target genes, it was suggested
that miR-1 and miR-133 may participate in the development of AF via
different signaling pathways, as opposed to through the direct
regulation of Ank-B expression.
MiR-34a was revealed to be induced in the aging
heart and was associated with heart fibrosis; the inhibition of
members of the miR-34 family could also attenuate pathological
cardiac remodeling and improve cardiac function (12). The present study identified direct
potential binding sites for miR-34a in the 3′UTR of ANK2 and
demonstrated that the expression of Ank-B could be regulated by the
modulation of miR-34a. It was also demonstrated that the levels of
NCX1, an ion exchanger, were indirectly affected by miR-34a
modulation, potentially through Ank-B as an intermediary, which may
account for the smaller degree of alterations observed in the
expression of NCX1 than of Ank-B. In addition, it was further
deduced that the changes in intracellular Ca2+ signaling
associated with modulation of miR-34a expression may be as a result
of Ank-B functioning. Lower levels of Ank-B and NCX1 influenced the
outward currents in the plateau phase of atrial myocytes, leading
to the reduction in APD and Ca2+ signal enhancement.
Upon treatment with ISO, the overexpression of miR-34a enhanced the
Ca2+ signal in terms of strength and frequency, while
inhibition of miR-34a was unable to counteract the effect of ISO,
which was attributed to the complexities of the response to ISO
treatment. The aberrant cellular Ca2+ signaling observed
in the present study following miR-34a modulation was similar to
the early electrophysiological changes seen in AF. Therefore, it
was hypothesized that miR-34a may be involved in the development of
AF by regulating the expression of Ank-B, and in turn, disrupting
normal cell signaling.
In conclusion, the results of the present study
demonstrated that miR-34a is upregulated in AF, and that it may
serve an important role in the early electrophysiological changes
and development of AF via the regulation of the expression of
Ank-B. Notably, miR-34a was also a miRNA identified as having a
role in fibrosis, and the present study revealed that it is also
associated with electrophysiological changes. Taken together, these
results provide a significant and valuable basis for the further
study of the underlying mechanism of AF, and also a promising
target for the development of clinical diagnosis strategies and
therapies to treat patients with AF.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from The
National Natural Science Foundation of China (grant no.
30600252).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ performed the experiments and was a major
contributor in writing the manuscript. YZ and ZF analyzed the data.
WC and YX conceptualized the study design and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study, including all sample collection
procedures for human and animal studies, were approved by the
Ethics Committee of Xinqiao Hospital (Chongqing, China). All
participants provided written informed consent.
Consent for publication
All participants provided written informed
consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Heijman J, Guichard JB, Dobrev D and
Nattel S: Translational challenges in atrial fibrillation. Circ
Res. 122:752–773. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miyasaka Y, Barnes ME, Gersh BJ, Cha SS,
Bailey KR, Abhayaratna WP, Seward JB and Tsang TS: Secular trends
in incidence of atrial fibrillation in Olmsted County, Minnesota,
1980 to 2000 and implications on the projections for future
prevalence. Circulation. 114:119–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dobrev D and Nattel S: New antiarrhythmic
drugs for treatment of atrial fibrillation. Lancet. 375:1212–1223.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iwasaki YK, Nishida K, Kato T and Nattel
S: Atrial fibrillation pathophysiology: Implications for
management. Circulation. 124:2264–2274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kashef F, Li J, Wright P, Snyder J,
Suliman F, Kilic A, Higgins RS, Anderson ME, Binkley PF, Hund TJ
and Mohler PJ: Ankyrin-B protein in heart failure: Identification
of a new component of metazoan cardioprotection. J Biol Chem.
287:30268–30281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peter J, Mohler JQD and Bennett V:
Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger and
insP3Receptor in a cardiac T-Tubule/SR microdomain. PLoS Biol.
3:e4232005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Kline CF, Hund TJ, Anderson ME and
Mohler PJ: Ankyrin-B regulates Kir6.2 membrane expression and
function in heart. J Biol Chem. 285:28723–28730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schulze DH, Muqhal M, Lederer WJ and
Ruknudin AM: Sodium/calcium exchanger (NCX1) macromolecular
complex. J Biol Chem. 278:28849–28855. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohler TJ and Ha PJ: Ankyrin-based
targeting pathway regulates human sinoatrial node automaticity.
Channels (Austin). 2:404–406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Care A, Catalucci D, Felicetti F, Bonci D,
Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, et al:
MicroRNA-133 controls cardiac hypertrophy. Nat Med. 13:613–618.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boon RA, Iekushi K, Lechner S, Seeger T,
Fischer A, Heydt S, Kaluza D, Tréguer K, Carmona G, Bonauer A, et
al: MicroRNA-34a regulates cardiac ageing and function. Nature.
495:107–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bernardo BC, Gao XM, Winbanks CE, Boey EJ,
Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du XJ, et
al: Therapeutic inhibition of the miR-34 family attenuates
pathological cardiac remodeling and improves heart function. Proc
Natl Acad Sci USA. 109:17615–17620. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen CL, Lin JL, Lai LP, Pan CH, Huang SK
and Lin CS: Altered expression of FHL1, CARP, TSC-22 and P311
provide insights into complex transcriptional regulation in
pacing-induced atrial fibrillation. Biochim Biophys Acta.
1772:317–329. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mohler PJ, Splawski I, Napolitano C,
Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT and Bennett
V: A cardiac arrhythmia syndrome caused by loss of ankyrin-B
function. Proc Natl Acad Sci USA. 101:9137–9142. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Terentyev D, Belevych AE, Terentyeva R,
Martin MM, Malana GE, Kuhn DE, Abdellatif M, Feldman DS, Elton TS
and Györke S: miR-1 overexpression enhances Ca(2+) release and
promotes cardiac arrhythmogenesis by targeting PP2A regulatory
subunit B56alpha and causing CaMKII-dependent hyperphosphorylation
of RyR2. Circ Res. 104:514–521. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tao H, Shi KH, Yang JJ, Huang C, Liu LP
and Li J: Epigenetic regulation of cardiac fibrosis. Cell Signal.
25:1932–1938. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song LS, Wang SQ, Xiao RP, Spurgeon H,
Lakatta EG and Cheng H: Beta-adrenergic stimulation synchronizes
intracellular Ca(2+) release during excitation-contraction coupling
in cardiac myocytes. Circ Res. 88:794–801. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pang H, Ronderos R, Pérez-Riera AR,
Femenía F and Baranchuk A: Reverse atrial electrical remodeling A
systematic review. Cardiol J. 18:625–631. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Verheule S, Tuyls E, Gharaviri A, Hulsmans
S, van Hunnik A, Kuiper M, Serroyen J, Zeemering S, Kuijpers NH and
Schotten U: Loss of continuity in the thin epicardial layer because
of endomysial fibrosis increases the complexity of atrial
fibrillatory conduction. Circ Arrhythm Electrophysiol. 6:202–211.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Le Scouarnec S, Bhasin N, Vieyres C, Hund
TJ, Cunha SR, Koval O, Marionneau C, Chen B, Wu Y, Demolombe S, et
al: Dysfunction in ankyrin-B-dependent ion channel and transporter
targeting causes human sinus node disease. Proc Natl Acad Sci USA.
105:15617–15622. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cunha SR, Hund TJ, Hashemi S, Voigt N, Li
N, Wright P, Koval O, Li J, Gudmundsson H, Gumina RJ, et al:
Defects in ankyrin-based membrane protein targeting pathways
underlie atrial fibrillation. Circulation. 124:1212–1222. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ono K, Kuwabara Y and Han J: MicroRNAs and
cardiovascular diseases. FEBS J. 278:1619–1633. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hund TJ, Wright PJ, Dun W, Snyder JS,
Boyden PA and Mohler PJ: Regulation of the ankyrin-B-based
targeting pathway following myocardial infarction. Cardiovasc Res.
81:742–749. 2009. View Article : Google Scholar : PubMed/NCBI
|