Introduction
Around 208,500 new cases of kidney cancer are
diagnosed annually worldwide (1).
It has been predicted that around 63,340 new cases of kidney cancer
will occur, and about 14,970 patients will die from kidney cancer
in the United States in 2018 (NIH). In China, the kidney cancer
incidence has been rising steadily in recent years (MOHC, Ministry
of Health of The People's Republic of China). Renal cell carcinoma
(RCC), the most common type of kidney cancer in adults, is
responsible for approximately 90–95% of cases (2,3). As
25–30% of RCC patients exhibit metastatic spread by the time they
are diagnosed, drug treatments targeting metastasis are needed
during early disease management. In this regard, Aurora kinase A
(AURKA) inhibitors have been shown to inhibit the growth and spread
of RCC (4–6).
AURKA, a serine-threonine-specific protein kinase,
is a member of the Aurora kinase (AURK) family, the members of
which serve as key regulators of mitosis; essential for accurate
and equal segregation of genomic material from parent to daughter
cells (7). Humans have three
classes of Aurora kinases, namely AURKA, AURKB and AURKC. AURKA is
a 403 amino-acid protein with a calculated molecular weight of 48
kDa (8). This protein has an
N-terminal regulatory domain and C-terminal catalytic domain. Two
amino acid sequences, the A-box in the regulatory domain and the
destruction-box (D-box) in the catalytic domain, are necessary for
recognition of AURKA by the APC/C complex, which mediates its
degradation at the end of mitosis or in G1 (9). Phosphorylation of a threonine residue
within the activation loop in the catalytic domain is critical for
Aurora kinase activity (10–12).
AURKA is mainly localized at spindle poles and the mitotic spindle
during mitosis, where it regulates centrosomes, spindles and
kinetochores. Recent studies have revealed that AURKA is frequently
overexpressed in several cancer cells, indicating its involvement
in tumor initiation and development (5,13–15).
Certain ongoing clinical trials and primary studies are assessing
the unique therapeutic potential of AURKA-targeted therapy for RCC.
However, RCC is relatively resistant to chemotherapy. Therefore,
identifying early-stage predictive biomarkers in RCC patients is
urgently needed.
In the present study, we showed that RCC cells with
loss of the VHL gene were more sensitive to AURKA inhibitor.
Moreover, we found that elevated pVHL levels improved AURKA
degradation, as a novel mechanism for the role of pVHL in
regulating AURKA.
Materials and methods
Cell culture and reagents
CAKI, ACHN, 786-O, 769-P and A498 cells were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA) and maintained in appropriate medium as suggested by the
ATCC. Cells were incubated in a humidified atmosphere of 95% air
plus 5% CO2 at 37°C. Alisertib and MG-132 were obtained
from Selleck Chemicals, (Houston, TX, USA). Cycloheximide (CHX) was
purchased from Solarbio Science and Technology (Beijing,
China).
Immunoblotting
After different types of treatment were performed,
cells were harvested and resuspended in RIPA lysis buffer (weak).
Equivalent amounts of proteins were analyzed by SDS-PAGE.
Appropriate antibodies against VHL (1:1,000), AURKA (1:1,000; Cell
Signaling Technology, Inc., Danvers, MA, USA), GAPDH (1:5,000;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) were used.
Proteins were visualized with peroxidase-coupled secondary antibody
from Sigma-Aldrich; Merck KGaA, (Darmstadt, Germany), using ECL
solution for detection.
Animals and anti-tumor activity assay
in vivo
BALB/Ca-nu/nu mice aged 4–5 weeks were obtained from
Zhejiang Academy of Medical Sciences. The animals were housed in
sterile cages under laminar airflow hoods in a specific
pathogen-free room with a 12-h light and 12-h dark schedule, and
fed autoclaved chow and water ad libitum. Animal experiments were
performed according to the institutional ethical guidelines of
animal care and were approved by Taizhou University (no.
TZYXY2016-302). The largest diameter of tumors developed in all
mice examined is 1.745 cm. shRNA-transfected cells were
transplanted subcutaneously into the flanks of nude mice. When the
tumor volume reached about 100 mm3, the mice were
randomly assigned into control and treatment groups, 8 mice each
group. Control groups were given vehicle, and treatment groups
received alisertib at the indicated doses per os for five days per
week for 3 weeks. Tumor volumes were measured twice per week. After
treatment, animals were killed by cervical dissociation, and solid
tumors were removed and weighed. The inhibition rate was calculated
as [(average tumor weight of vehicle control group/average tumor
weight of test group)/average tumor weight of normal saline group]
X100%.
Knockdown of VHL by shRNA
The lentivirus vector pLKO.1 was used. The inserted
shRNA-targeting vhl sequence was as follows:
5′-CCGGTATCACACTGCCAGTGTATACCTCGAGGTATACACTGGCAGTGTGATATTTTTG-3′.
Lentiviral packaging was conducted in 293T cells following standard
procedures. The shRNA was transfected into cells using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the instructions of the
manufacturer.
Statistical analysis
The data were analyzed using SPSS v13.0 (SPSS, Inc.,
Chicago, IL, USA). The results were compared using one-way analysis
of variance followed by Dunnett's post hoc test for multiple
comparisons. All results are expressed as the mean ± standard
deviation from three replicates. P<0.05 was considered to
indicate a statistically significant difference.
Results
VHL gene expression profile and
anti-proliferation effect of AURKA inhibitor in RCC cells
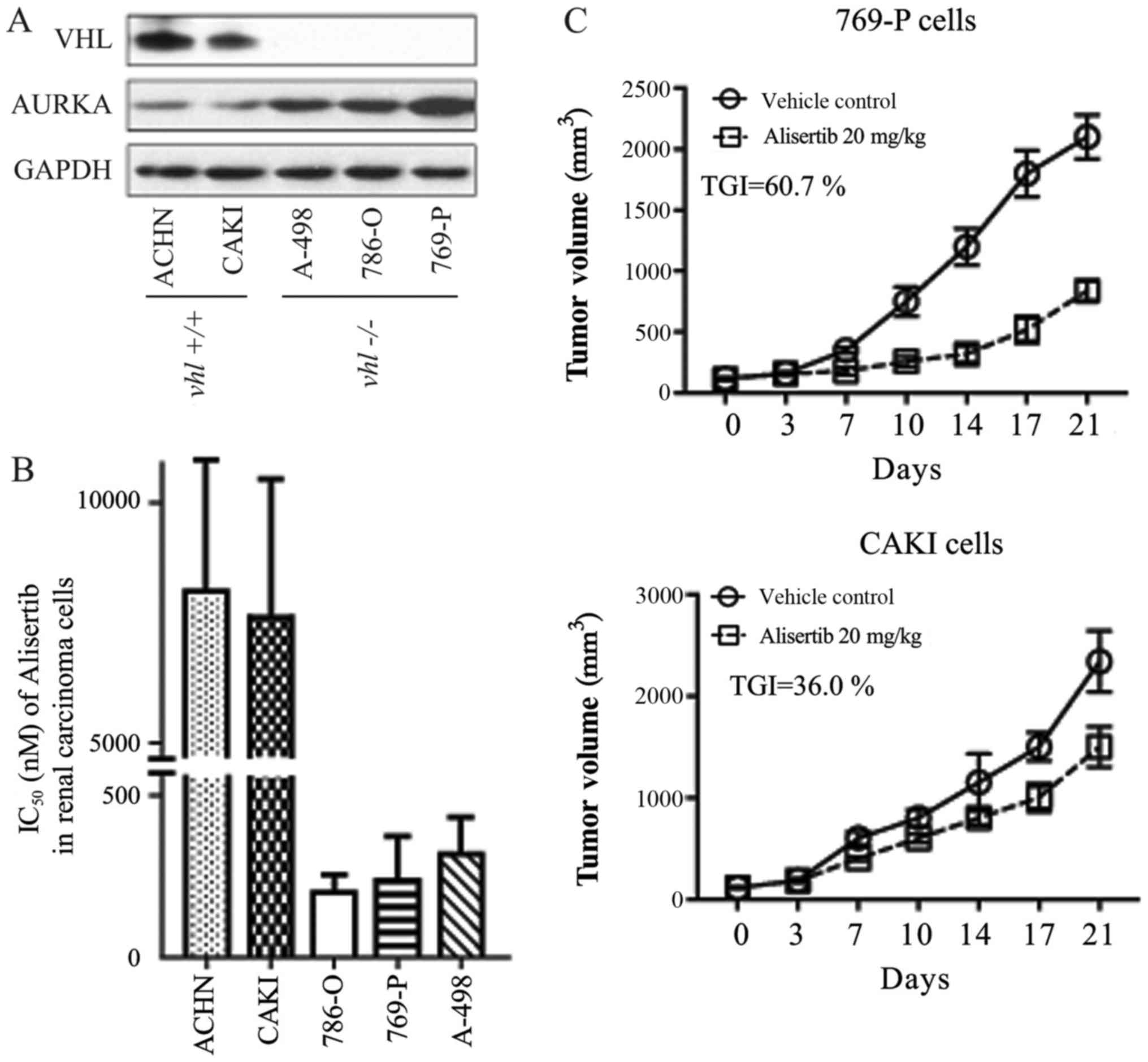
Immunoblot detection revealed that two RCC cell
lines (ACHN and CAKI) expressed the wild type VHL gene, while the
other three (A498, 769-P and 786-O) were VHL-deficient cells
(Fig. 1A), consistent with reports
by the National Cancer Institute using cDNA micro-arrays
(genome-www.edu/nci60/).
Inhibition of cellular proliferation was evaluated
using a CCK-8 kit (Beyotime Institute of Biotechnology, Haimen,
China). In the present study, the AURKA-specific chemical inhibitor
alisertib more readily inhibited the growth of ACHN and CAKI cells
when compared with its effect in the other three RCC cell lines
(Fig. 1B). IC50 values for
alisertib in each cell line were as follows: ACHN, ~8 µmol/l; CAKI,
~7.5 µmol/l; 786-O, ~0.20 µmol/l; 769-P, ~0.25 µmol/l; A-498, ~0.33
µmol/l. We found a clear positive relationship between VHL gene
expression and alisertib sensitivity (IC50) in the RCC cells. In
other words, RCC cells expressing wild type VHL were more sensitive
to alisertib (Fig. 1A and B). This
Pearson's correlation was further confirmed in a xenograft animal
model. As shown in Fig. 1C,
alisertib displayed differential anti-tumor activity in CAKI and
769-P xenografts, with inhibitory rates of 36.0 and 60.7%,
respectively.
Alteration in alisertib
anti-proliferation activity by VHL
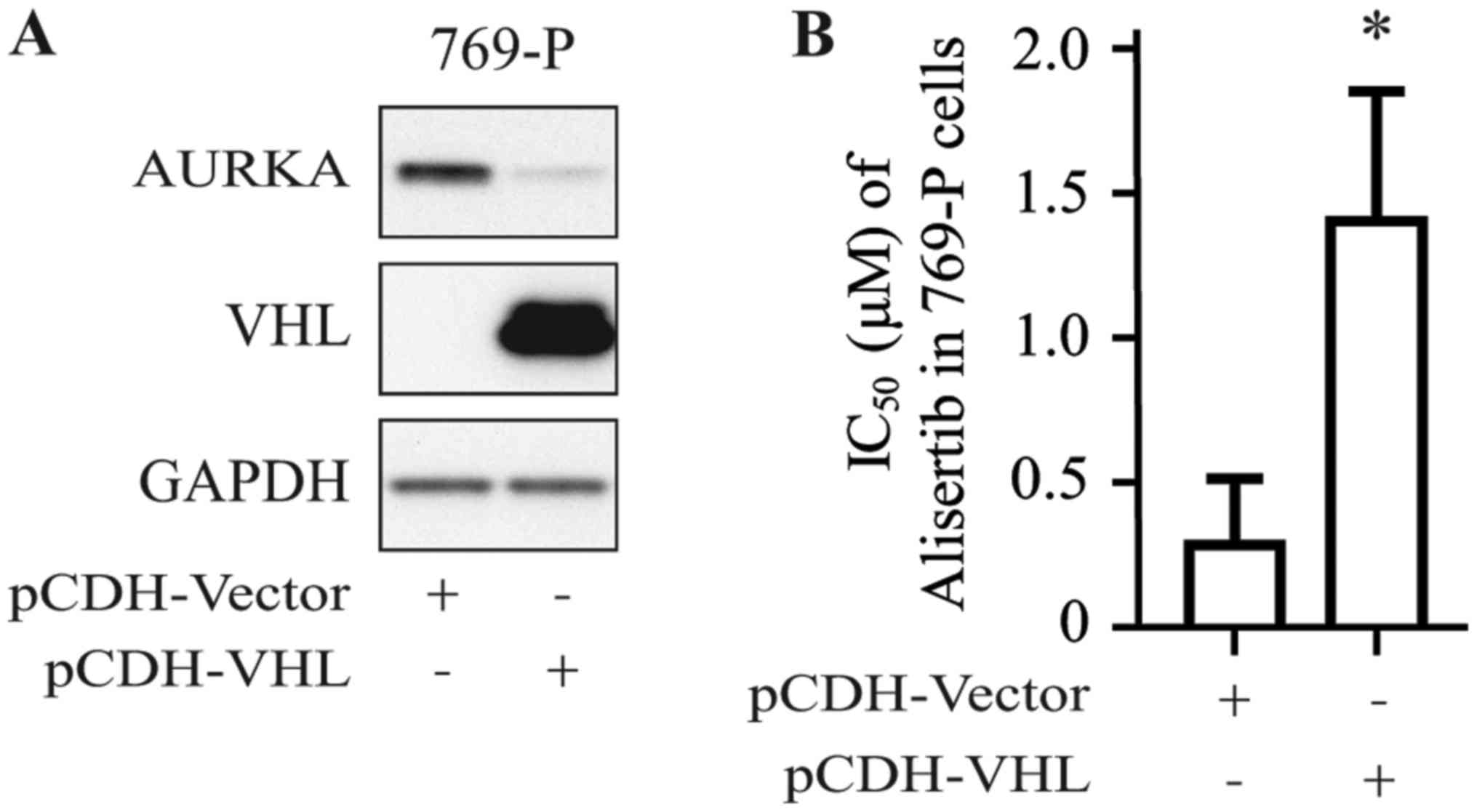
To confirm the positive relationship between the VHL
gene expression profile and alisertib sensitivity in RCC cell
lines, 769-P cells were stably transfected with pCDH-VHL plasmid;
cells transfected with empty pCDH-vector served as the control. As
shown in Fig. 2A, VHL protein
could be detected in 769-P cells transfected with pCDH-VHL.
Furthermore, 769-P cells transfected with pCDH-VHL were shown to be
more resistant to alisertib than those transfected with empty
vector (Fig. 2B), with the IC50
increasing from 0.25 to 1.25 µmol/l.
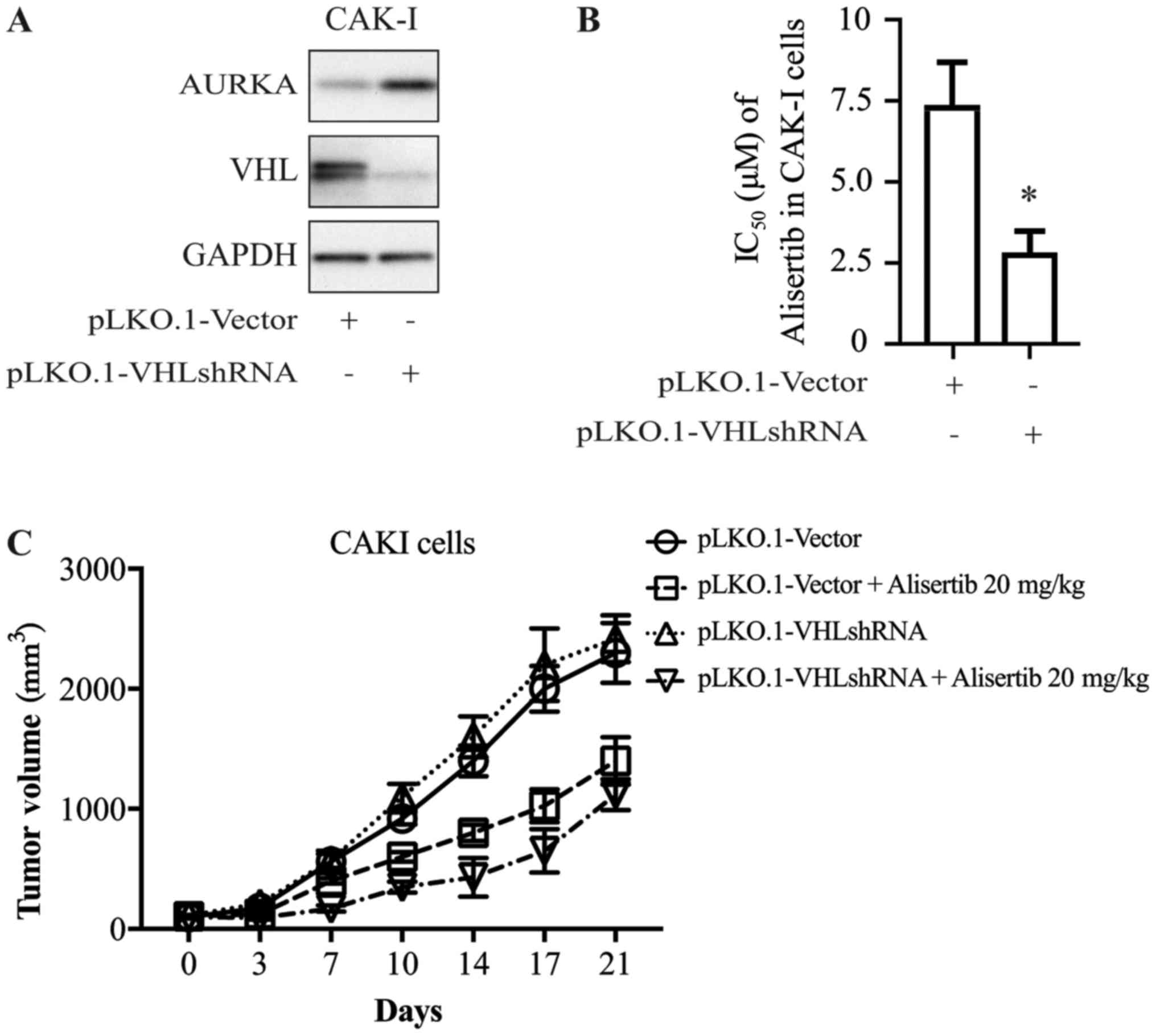
To further explore whether there is a causal
relationship between VHL expression and alisertib sensitivity, CAKI
cells were transfected with shRNA directed against VHL; cells
transfected with empty vector as the control. Cells were harvested
72 h after transfection and extracts were prepared and analyzed by
western blotting. Expression of the VHL gene decreased
significantly after shRNA transfection (Fig. 3A). At 24 h after transfection with
VHL shRNA or control vector, CAKI cells were treated with alisertib
for 72 h. The cytotoxicity of alisertib was significantly higher
for cells in the presence of VHL shRNA than for the control group
(Fig. 3B), which indicated that
decreasing the expression of VHL altered the anti-proliferation
activity of alisertib.
This association was further confirmed in the animal
model. As shown in Fig. 3C,
alisertib displayed differential anti-tumor activity in the
xenograft model of CAKI cells transfected with VHL-shRNA or control
vector, with inhibitory rates of ~54 and ~32%, respectively
(Fig. 3C).
VHL down-regulates AURKA in RCC
cells
According to our results, we may conclude that VHL
loss makes human RCC cells more sensitive to AURKA inhibitor. This
increased sensitivity might be caused by VHL down-regulating AURKA
via the HIF pathway. Consistent with previous reports (16,17),
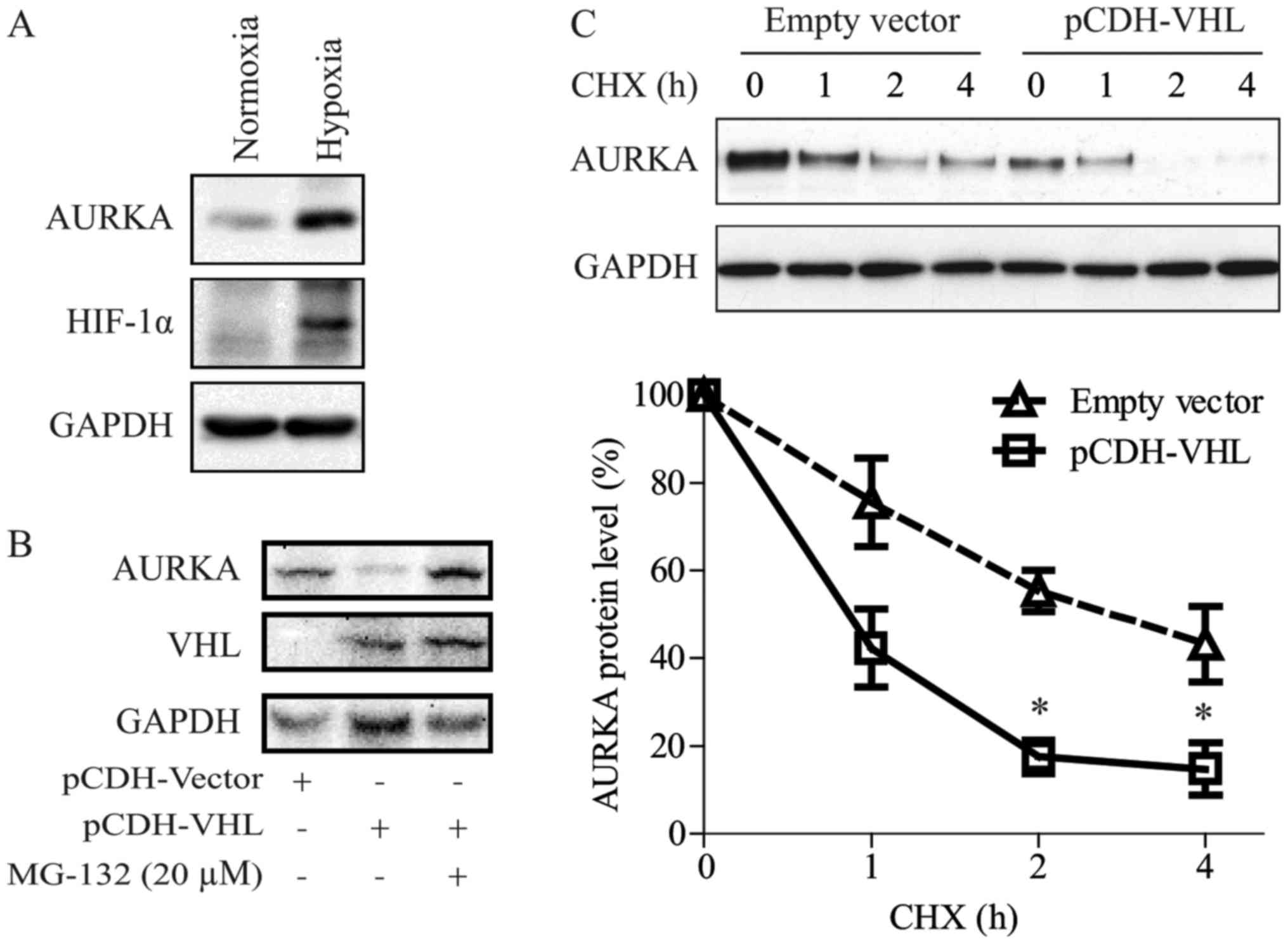
we showed that VHL down-regulated AURKA expression (Figs. 1A, 2A and 3A).
As reported by Xu et al (16), VHL inactivation induced AURKA
expression in clear cell RCC (CCRCC) cells via the HIF pathway. In
the present study, we also observed that hypoxic conditions could
induce AURKA protein expression in RCC (Fig. 4A).
We have shown in a previous study that pVHL regulate
AURKA directly via an HIF-independent pathway (17). As shown in Fig. 4B and C, in the present study we
confirmed that VHL can promote AURKA degradation. Moreover, the
proteasome inhibitor, MG-132, blocked pVHL-induced AURKA
degradation. Interestingly, the AURKA protein had a shorter
half-life in 769-P cells re-expressed with VHL compared with that
in control cells.
Discussion
In this study, we showed that 786-O, 769-P and A498
cells deficient in the VHL gene were sensitive to alisertib, an
AURKA-specific chemical inhibitor. Conversely, alisertib-resistant
CAKI and ACHN cells expressed the wild type VHL gene. Re-expression
or knockdown of VHL reversed the anti-proliferation activity of
alisertib in RCC cells. The inverse association between VHL gene
expression profile and alisertib sensitivity was confirmed in human
cancer xenografts models. Taken together, we suggest that VHL loss
could potentially serve as a biomarker for predicting the efficacy
of AURKA inhibitors.
It is well known that VHL protein serves as a
substrate recognition component of an E3-ubiquitin ligase complex
that targets hypoxia inducible factor (HIF) for ubiquitination and
degradation (18). In addition to
HIF-α, pVHL interacts with certain other proteins and has multiple
functions, including in microtubule dynamics, cell proliferation
and growth, neuronal apoptosis, extracellular matrix deposition,
DNA damage response/repair and cilia maintenance. It has been
extensively reported in human cancers that loss of VHL is a
predictive biomarker for the efficacy of neoadjuvant therapy
(19,20). In the present study, we proposed
the potential of VHL to be a predictive marker of response to AURKA
inhibitors and tested our hypothesis in vitro and in
vivo. We further speculated that pVHL regulated AURKA levels
via HIF-dependent and HIF-independent pathways. As it is
technically feasible and reproducible to measure VHL loss in
patients, which should be easier than detecting AURKA expression
levels, VHL meets the criteria to serve as a predictive marker of
efficacy of alisertib-based therapy.
Our data supported that VHL loss could be
potentially applied as a biomarker to predict the efficacy of AURKA
inhibitors. Additional pre-clinical and clinical trials are needed
to validate our findings. It is necessary to confirm our primary
data with results obtained from the patients' samples (e.g. PDTX
model) and correlate the results with the clinic pathological
data.
Acknowledgements
The authors would like to thank Mr. Jian-Xing Zhang
(Laboratory for Biological Medicine, School of Medicine, Taizhou
University, Zhejiang, China) for their technical assistance during
the animal work.
Funding
This work was supported by the Zhejiang Provincial
Natural Science Foundation (grant nos. LY15H310002, LY15H310001 and
LY16H310006), the National Natural Science Foundation of China
(grant no. 81201530), the Public Technology Research Projects of
the Science Technology Department of Zhejiang Province (grant nos.
2016C37111 and 2015C37093, and the Science Technology Department of
Taizhou City (grant no. 15yw08).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XFD, GC and YLW designed the research. XFD, JZ and
GC performed the research. XFD, GC and YLW analyzed the data, and
GC and YLW wrote the manuscript.
Ethics approval and consent to
participate
Animal experiments were performed according to the
institutional ethical guidelines of animal care and were approved
by Taizhou University (no. TZYXY2016-302).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kidney cancer update the year in review in
kidney cancer. Clin Adv Hematol Oncol. 13:327–329. 2015.PubMed/NCBI
|
|
2
|
Massari F, Di Nunno V, Ciccarese C, Graham
J, Porta C, Comito F, Cubelli M, Iacovelli R and Heng DYC: Adjuvant
therapy in renal cell carcinoma. Cancer Treat Rev. 60:152–157.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Posadas EM, Limvorasak S and Figlin RA:
Targeted therapies for renal cell carcinoma. Nat Rev Nephrol.
13:496–511. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cha TL, Chuang MJ, Wu ST, Sun GH, Chang
SY, Yu DS, Huang SM, Huan SK, Cheng TC, Chen TT, et al: Dual
degradation of aurora A and B kinases by the histone deacetylase
inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer
cells. Clin Cancer Res. 15:840–850. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kurahashi T, Miyake H, Hara I and Fujisawa
M: Significance of Aurora-A expression in renal cell carcinoma.
Urol Oncol. 25:128–133. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y, Zhou W, Wei L, Jin J, Tang K, Li C,
Teh BT and Chen X: The effect of Aurora kinases on cell
proliferation, cell cycle regulation and metastasis in renal cell
carcinoma. Int J Oncol. 41:2139–2149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karthigeyan D, Prasad SB, Shandilya J,
Agrawal S and Kundu TK: Biology of Aurora A kinase: Implications in
cancer manifestation and therapy. Med Res Rev. 31:757–793.
2011.PubMed/NCBI
|
|
8
|
Borisa AC and Bhatt HG: A comprehensive
review on Aurora kinase: Small molecule inhibitors and clinical
trial studies. Eur J Med Chem. 140:1–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lim SK and Gopalan G: Aurora-A kinase
interacting protein 1 (AURKAIP1) promotes Aurora-A degradation
through an alternative ubiquitin-independent pathway. Biochem J.
403:119–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eckerdt F, Pascreau G, Phistry M, Lewellyn
AL, DePaoli-Roach AA and Maller JL: Phosphorylation of TPX2 by Plx1
enhances activation of Aurora A. Cell Cycle. 8:2413–2419. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang Y, Li T, Ems-McClung SC, Walczak CE,
Prigent C, Zhu X, Zhang X and Zheng Y: Aurora A activation in
mitosis promoted by BuGZ. J Cell Biol. 217:107–116. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Korobeynikov V, Deneka AY and Golemis EA:
Mechanisms for nonmitotic activation of Aurora-A at cilia. Biochem
Soc Trans. 45:37–49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Compérat E, Bièche I, Dargère D,
Laurendeau I, Vieillefond A, Benoit G, Vidaud M, Camparo P, Capron
F, Verret C, et al: Gene expression study of Aurora-A reveals
implication during bladder carcinogenesis and increasing values in
invasive urothelial cancer. Urology. 72:873–877. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goktas S, Yildirim M, Suren D, Alikanoglu
AS, Dilli UD, Bulbuller N, Sezer C and Yildiz M: Prognostic role of
Aurora-A expression in metastatic colorectal cancer patients. J
BUON. 19:686–691. 2014.PubMed/NCBI
|
|
15
|
Twu NF, Yuan CC, Yen MS, Lai CR, Chao KC,
Wang PH, Wu HH and Chen YJ: Expression of Aurora kinase A and B in
normal and malignant cervical tissue: High Aurora A kinase
expression in squamous cervical cancer. Eur J Obstet Gynecol Reprod
Biol. 142:57–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu J, Li H, Wang B, Xu Y, Yang J, Zhang X,
Harten SK, Shukla D, Maxwell PH, Pei D and Esteban MA: VHL
inactivation induces HEF1 and Aurora kinase A. J Am Soc Nephrol.
21:2041–2046. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hasanov E, Chen G, Chowdhury P, Weldon J,
Ding Z, Jonasch E, Sen S, Walker CL and Dere R: Ubiquitination and
regulation of AURKA identifies a hypoxia-independent E3 ligase
activity of VHL. Oncogene. 36:3450–3463. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gossage L, Eisen T and Maher ER: VHL, the
story of a tumour suppressor gene. Nat Rev Cancer. 15:55–64. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim BJ, Kim JH, Kim HS and Zang DY:
Prognostic and predictive value of VHL gene alteration in renal
cell carcinoma: A meta-analysis and review. Oncotarget.
8:13979–13985. 2017.PubMed/NCBI
|
|
20
|
Ferchichi I, Kourda N, Sassi S, Romdhane
KB, Balatgi S, Cremet JY, Prigent C and Elgaaied AB: Aurora A
overexpression and pVHL reduced expression are correlated with a
bad kidney cancer prognosis. Dis Markers. 33:333–340. 2012.
View Article : Google Scholar : PubMed/NCBI
|