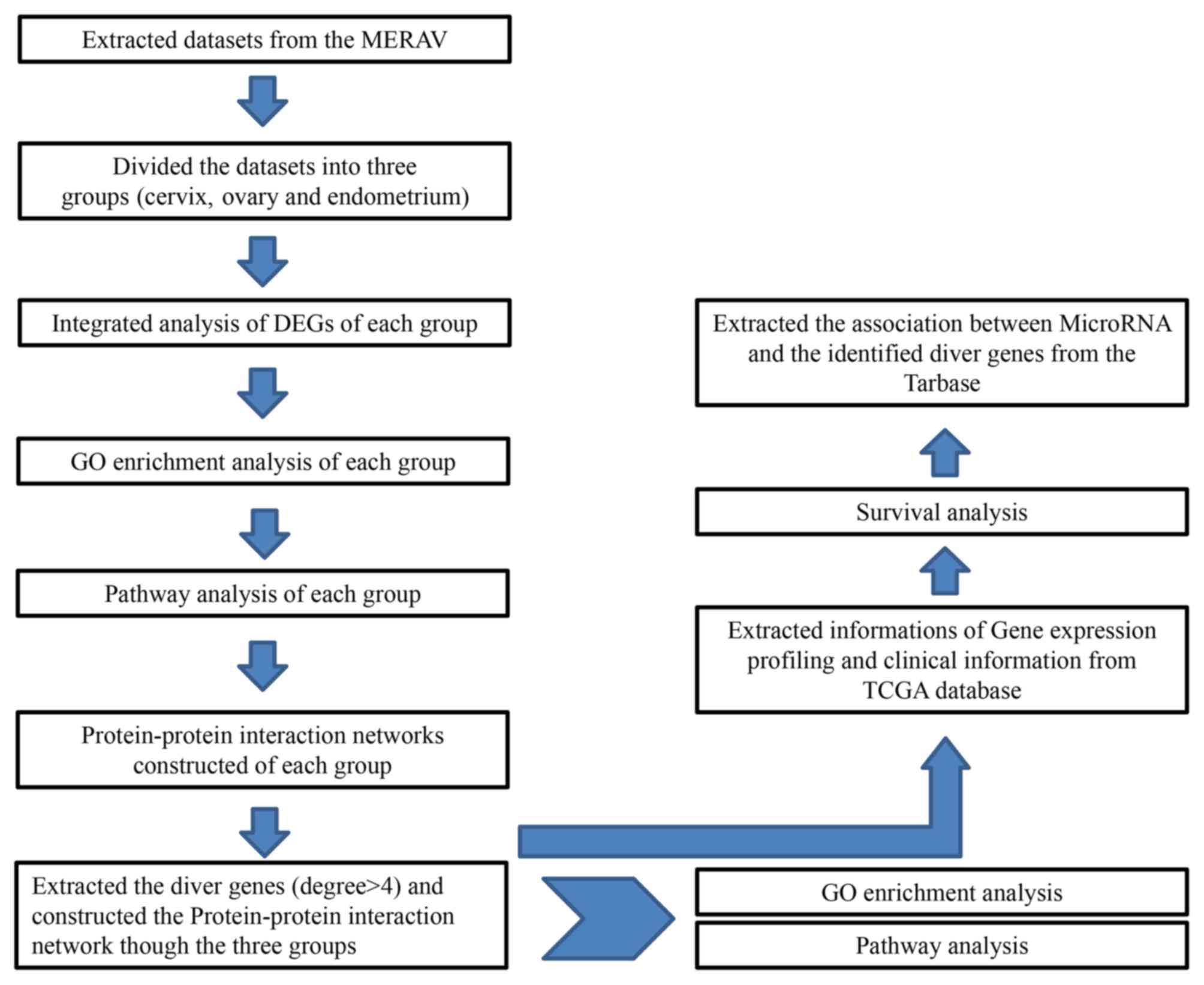
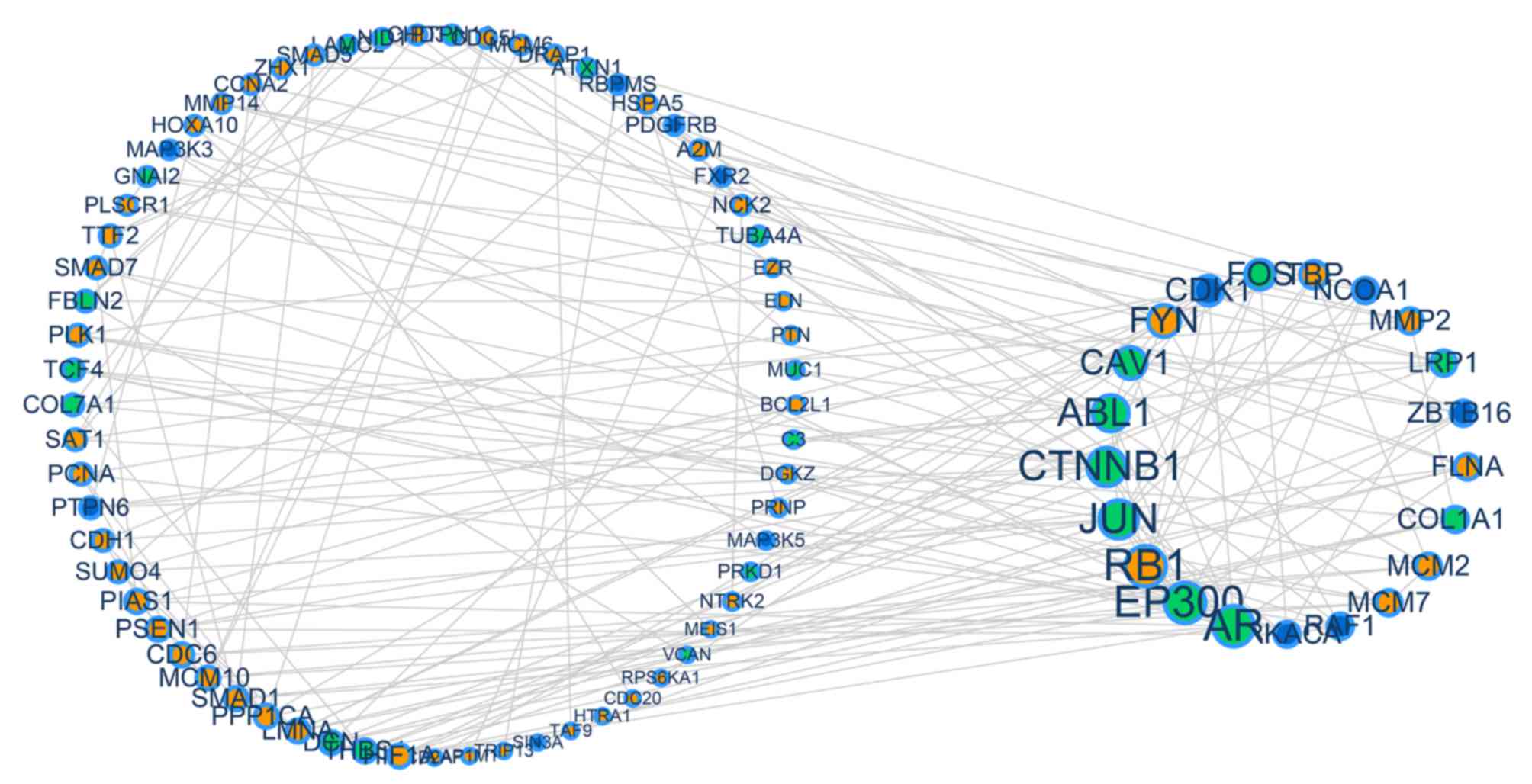
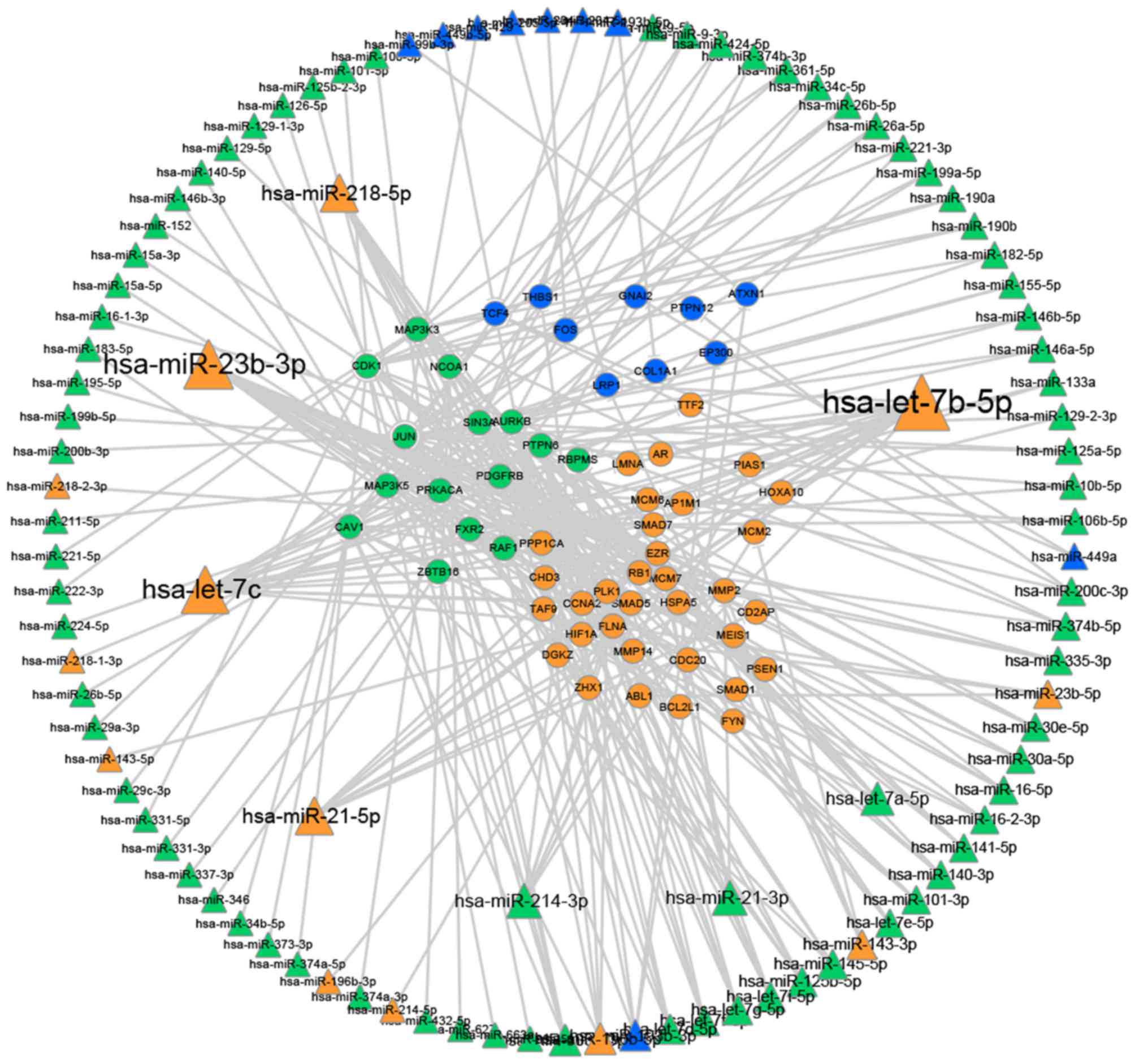
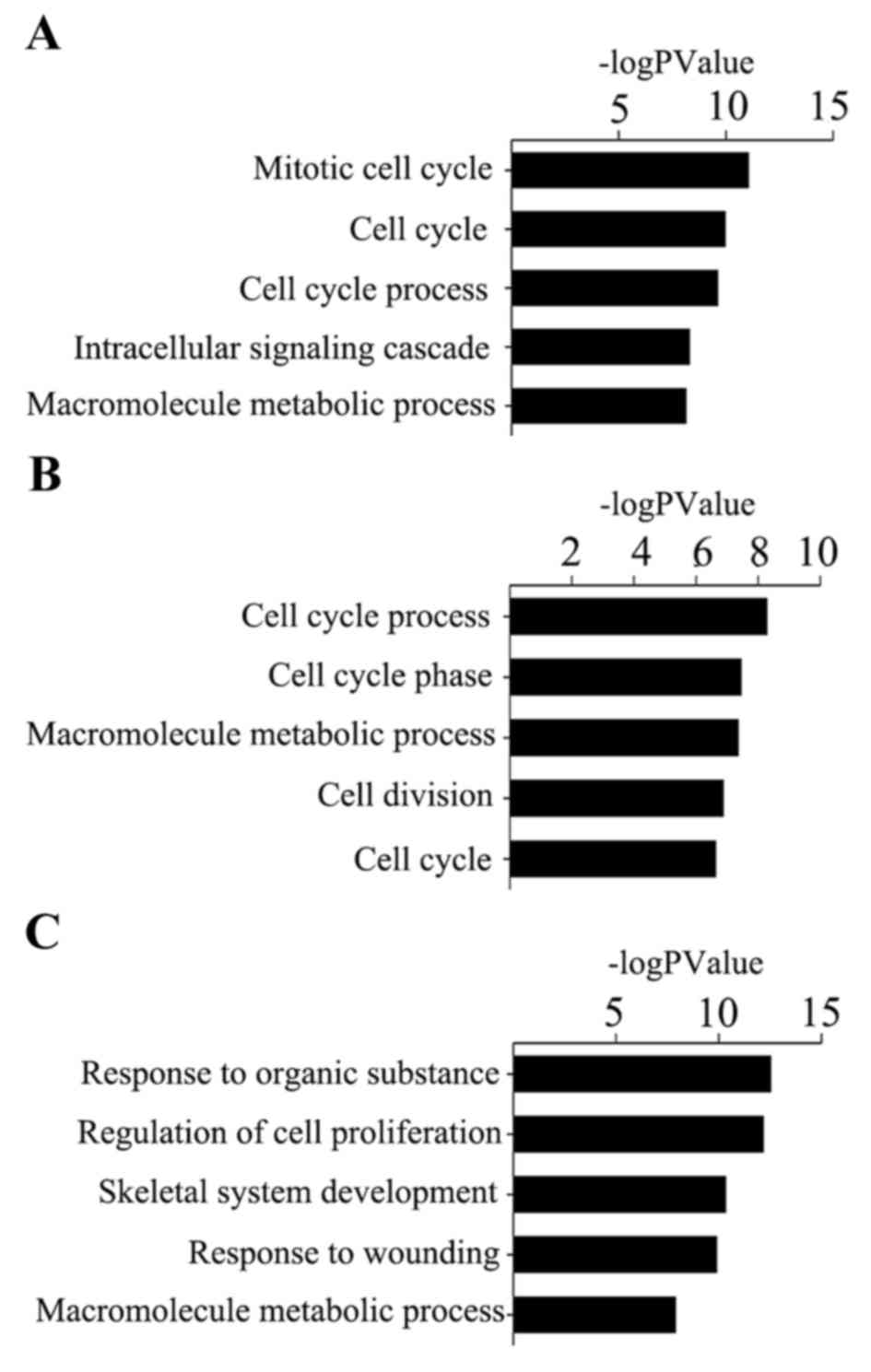
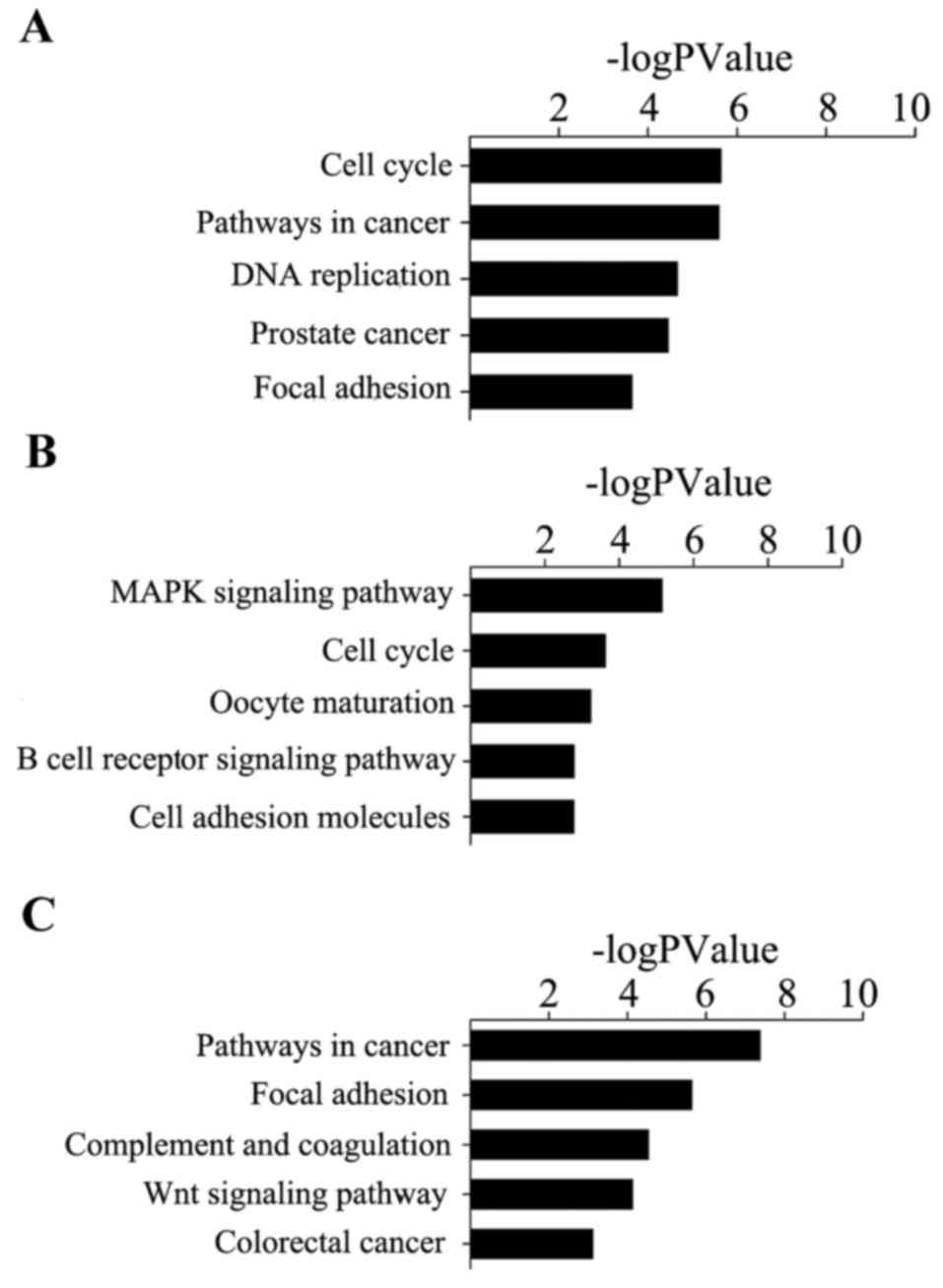
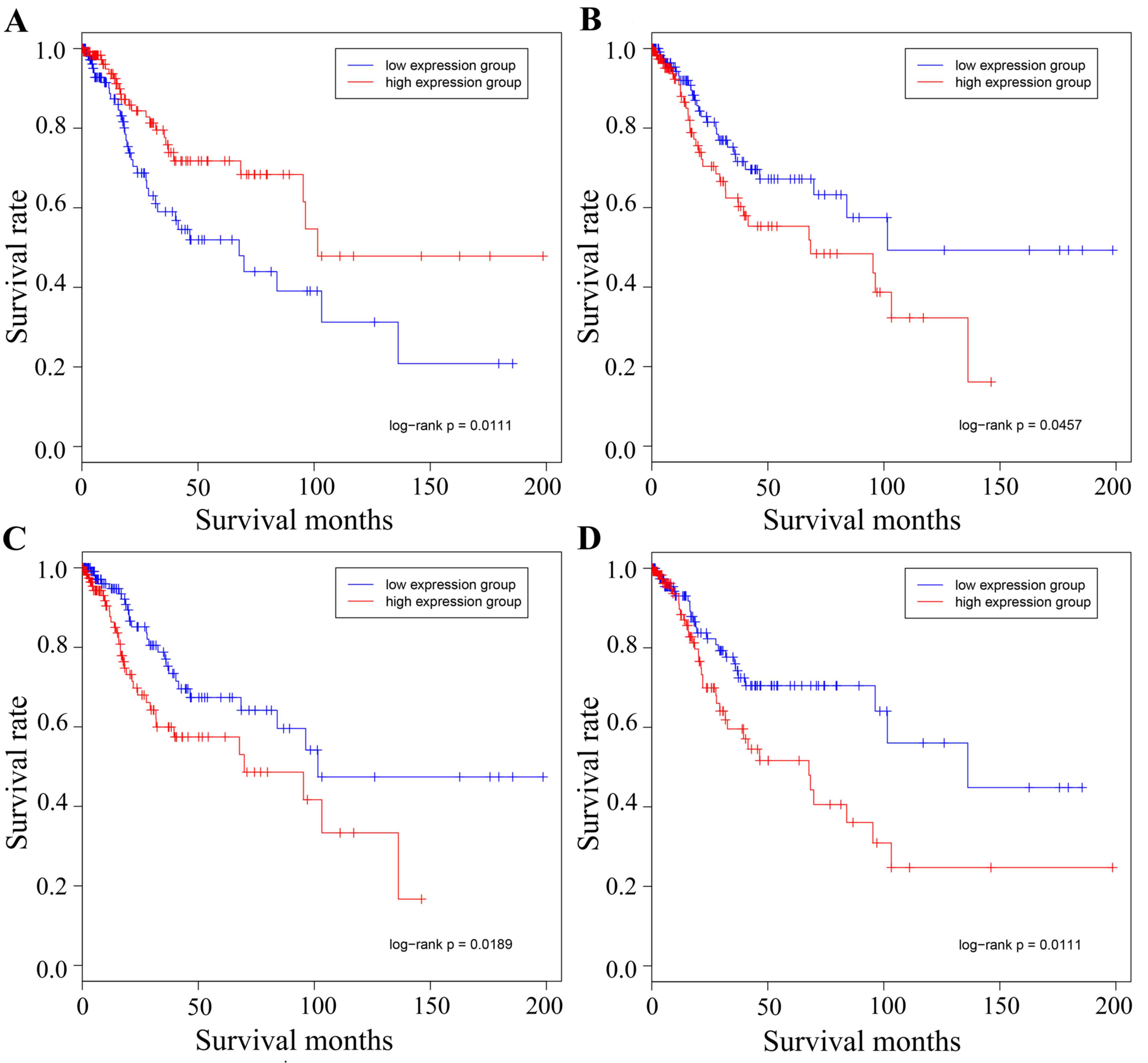
|
1
|
Li XY and Wang X: The role of human
cervical cancer oncogene in cancer progression. Int J Clin Exp Med.
8:8363–8368. 2015.PubMed/NCBI
|
|
2
|
Nisker JA: Screening for endometrial
cancer. Can Fam Physician. 29:961–965. 1983.PubMed/NCBI
|
|
3
|
Setiawan VW, Yang HP, Pike MC, McCann SE,
Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, et al:
Type I and II endometrial cancers: Have they different risk
factors? J Clin Oncol. 31:2607–2618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wright JD: Take 'em or leave 'em:
Management of the ovaries in young women with endometrial cancer.
Gynecol Oncol. 131:287–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lukk M, Kapushesky M, Nikkilä J, Parkinson
H, Goncalves A, Hube W, Ukkonen E and Brazma A: A global map of
human gene expression. Nat Biotechnol. 28:322–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cancer Genome Atlas Research Network, .
McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ,
Mastrogianakis GM, Olson JJ, Mikkelsen T, Lehman N, et al:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Research Network:
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guichard C, Amaddeo G, Imbeaud S, Ladeiro
Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M,
Degos F, et al: Integrated analysis of somatic mutations and focal
copy-number changes identifies key genes and pathways in
hepatocellular carcinoma. Nat Genet. 44:694–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu P, Morrison C, Wang L, Xiong D, Vedell
P, Cui P, Hua X, Ding F, Lu Y, James M, et al: Identification of
somatic mutations in non-small cell lung carcinomas using
whole-exome sequencing. Carcinogenesis. 33:1270–1276. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nik-Zainal S, Alexandrov LB, Wedge DC, Van
Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J,
Stebbings LA, et al: Mutational processes molding the genomes of 21
breast cancers. Cell. 149:979–993. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baudot A, Real FX, Izarzugaza JM and
Valencia A: From cancer genomes to cancer models: Bridging the
gaps. EMBO Rep. 10:359–366. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kreeger PK and Lauffenburger DA: Cancer
systems biology: A network modeling perspective. Carcinogenesis.
31:2–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee S, Stewart S, Nagtegaal I, Luo J, Wu
Y, Colditz G, Medina D and Allred DC: Differentially expressed
genes regulating the progression of ductal carcinoma in situ to
invasive breast cancer. Cancer Res. 72:4574–4586. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wray CJ, Ko TC and Tan FK: Secondary use
of existing public microarray data to predict outcome for
hepatocellular carcinoma. J Surg Res. 188:137–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barrett T, Troup DB, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, et al: NCBI GEO: Archive for functional genomics data
sets-10 years on. Nucleic Acids Res. 39:(Database Issue).
D1005–D1010. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chung VY, Tan TZ, Tan M, Wong MK, Kuay KT,
Yang Z, Ye J, Muller J, Koh CM, Guccione E, et al:
GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian
cancer through transcriptional regulation and histone modification.
Sci Rep. 6:199432016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Halabi NM, Martinez A, Al-Farsi H, Mery E,
Puydenus L, Pujol P, Khalak HG, McLurcan C, Ferron G, Querleu D, et
al: Preferential allele expression analysis identifies shared
germline and somatic driver genes in advanced ovarian cancer. PLoS
Genet. 12:e10058922016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Espinosa AM, Alfaro A, Roman-Basaure E,
Guardado-Estrada M, Palma Í, Serralde C, Medina I, Juárez E,
Bermúdez M, Márquez E, et al: Mitosis is a source of potential
markers for screening and survival and therapeutic targets in
cervical cancer. PLoS One. 8:e559752013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shaul YD, Yuan B, Thiru P, Nutter-Upham A,
McCallum S, Lanzkron C, Bell GW and Sabatini DM: MERAV: A tool for
comparing gene expression across human tissues and cell types.
Nucleic Acids Res. 44:D560–D566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
R Development Core Team, . R: A language
and environment for statistical computingR Foundation for
Statistical Computing. Vienna, Austria: 2017
|
|
21
|
Prasad Keshava TS, Goel R, Kandasamy K,
Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R,
Shafreen B, Venugopal A, et al: Human protein reference
database-2009 update. Nucleic Acids Res. 37:(Database Issue).
D767–D772. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang Q, Wang Y, Hao Y, Juan L, Teng M,
Zhang X, Li M, Wang G and Liu Y: miR2Disease: A manually curated
database for microRNA deregulation in human disease. Nucleic Acids
Res. 37:(Database Issue). D98–D104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vlachos IS, Paraskevopoulou MD, Karagkouni
D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL,
Maniou S, Karathanou K, Kalfakakou D, et al: DIANA-TarBase v7.0:
Indexing more than half a million experimentally supported
miRNA:mRNA interactions. Nucleic Acids Res. 43:(Database Issue).
D153–D159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaimal V, Bardes EE, Tabar SC, Jegga AG
and Aronow BJ: ToppCluster: A multiple gene list feature analyzer
for comparative enrichment clustering and network-based dissection
of biological systems. Nucleic Acids Res. 38:W96–W102. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Altermann E and Klaenhammer TR:
PathwayVoyager: Pathway mapping using the Kyoto encyclopedia of
genes and genomes (KEGG) database. BMC Genomics. 6:602005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stamoulis C and Betensky RA: A novel
signal processing approach for the detection of copy number
variations in the human genome. Bioinformatics. 27:2338–2345. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miller CA, Settle SH, Sulman EP, Aldape KD
and Milosavljevic A: Discovering functional modules by identifying
recurrent and mutually exclusive mutational patterns in tumors. BMC
Med Genomics. 4:342011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ciriello G, Cerami E, Sander C and Schultz
N: Mutual exclusivity analysis identifies oncogenic network
modules. Genome Res. 22:398–406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vandin F, Upfal E and Raphael BJ: De novo
discovery of mutated driver pathways in cancer. Genome Res.
22:375–385. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsaniras Champeris S, Kanellakis N,
Symeonidou IE, Nikolopoulou P, Lygerou Z and Taraviras S: Licensing
of DNA replication, cancer, pluripotency and differentiation: An
interlinked world? Semin Cell Dev Biol. 30:174–180. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pruitt SC, Bailey KJ and Freeland A:
Reduced Mcm2 expression results in severe stem/progenitor cell
deficiency and cancer. Stem Cells. 25:3121–3132. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Simon NE and Schwacha A: The Mcm2-7
replicative helicase: A promising chemotherapeutic target. Biomed
Res Int. 2014:5497192014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pittayapruek P, Meephansan J, Prapapan O,
Komine M and Ohtsuki M: Role of matrix metalloproteinases in
photoaging and photocarcinogenesis. Int J Mol Sci. 17:pii:
E8682016. View Article : Google Scholar
|
|
35
|
Chan TF, Poon A, Basu A, Addleman NR, Chen
J, Phong A, Byers PH, Klein TE and Kwok PY: Natural variation in
four human collagen genes across an ethnically diverse population.
Genomics. 91:307–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bosman FT and Stamenkovic I: Functional
structure and composition of the extracellular matrix. J Pathol.
200:423–428. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Uitto VJ and Larjava H: Extracellular
matrix molecules and their receptors: An overview with special
emphasis on periodontal tissues. Crit Rev Oral Biol Med. 2:323–354.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Deyl Z, Miksik I and Eckhardt A:
Preparative procedures and purity assessment of collagen proteins.
J Chromatogr B Analyt Technol Biomed Life Sci. 790:245–275. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dai J, Wang T, Wang W, Zhang S, Liao Y and
Chen J: Role of MAPK7 in cell proliferation and metastasis in
ovarian cancer. Int J Clin Exp Pathol. 8:10444–10451.
2015.PubMed/NCBI
|
|
40
|
Lee YS and Dutta A: The tumor suppressor
microRNA let-7 represses the HMGA2 oncogene. Genes Dev.
21:1025–1030. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly
MB, Wang Y, Qian Z, Jin J, Zhang Y, et al: MicroRNA expression
signatures accurately discriminate acute lymphoblastic leukemia
from acute myeloid leukemia. Proc Natl Acad Sci USA.
104:19971–19976. 2007. View Article : Google Scholar : PubMed/NCBI
|