Introduction
Parkinson's disease (PD) is a common
neurodegenerative disorder characterized by the progressive
degeneration of dopaminergic neurons in the substantia nigra pars
compacta (SNpc) and abnormal aggregation of α-synuclein (α-syn)
(1). With increasing awareness of
the importance of genetic factors involved in the disease, several
genes that lead to hereditary PD were identified over a period of
ten years. Among them, α-syn was the first genetic factor found to
be linked to PD. At the time of writing, 6 pathogenic mutations of
α-syn were known to be involved in autosomal recessive
parkinsonism-A53T, E46K, A30P, H50Q, G51D and A53E (2–7).
A53T and A30P were the first two identified SNCA mutations and more
slightly common occurrence among the point mutations (8).
The aggregation of α-syn in the brain, from soluble
oligomers to insoluble inclusions, may be the initial
pathophysiological change in PD, and it also contributes to the
pathogenesis of familial or idiopathic PD (5). Moreover, there is accumulating
evidence that many cellular defects are implicated in the etiology
of synucleinopathies, including impairment of the
ubiquitin-proteasome system (UPS), oxidative stress, and
mitochondrial dysfunction (9). A
variety of conditions that disturb folding of proteins in the
endoplasmic-reticulum (ER) can trigger ER stress response (10). The accumulation of unfolded
proteins in the ER can trigger an evolutionarily conserved
response, termed the unfolded protein response (UPR). UPR is the
response which transiently clears unfolded proteins in order to
restores ER homeostasis and to promote cell survival (11). The typical UPR consists of three
pathways in eukaryotic cells, which are mediated by three ER
membrane-associated proteins: PKR-like eukaryotic initiation factor
2a kinase (PERK), inositol requiring enzyme 1 (IRE1), and
activating transcription factor-6 (ATF6) (12). Under stress-free conditions, these
sensors are combined with the ER chaperone Bip/GRP78 (glucose
regulated protein 78) and exist in their deactivated form (13). When misfolded proteins accumulate
in the ER lumen, UPR sensors detach from GRP78, causing PERK
oligomerization and autophosphorylation. Active PERK phosphorylates
eukaryotic translation initiation factor 2α (eIF2α), rendering it
inactive and blocking protein translation (14,15).
The phosphorylation of eIF2α inhibits the recycling of eIF2α to its
active GTP-bound form, which prevents the further influx of nascent
proteins into the already stressed ER lumen (16). If the various UPR-induced
mechanisms fail to alleviate ER stress, the PERK pathway activation
can induce expression of the proapoptotic transcription factor
C/EBP homologous protein (CHOP/GADD153), downstream of the
PERK-eIF2α-ATF4 pathway, which eventually cleaves caspase-3 to
mediate cell apoptosis (17).
The association between α-syn and ER stress, as well
as the role of ER and the Golgi apparatus (GA) in the
neurodegeneration observed in PD has attracted more attention in
recent years. Previous studies have demonstrated that the
accumulation of α-syn oligomers within the ER compartment causes
chronic ER stress which can induce cell death (11,18).
Moreover, studies in mutant mice showed that the overexpression of
α-syn leads to the fragmentation of GA in dopaminergic neurons in
the midbrain of mutant mice (19).
Furthermore, Cooper et al (20) have found evidence that α-syn
accumulation inhibits vesicular trafficking between the ER and GA
in vitro. However, all of these studies of the effects of
α-syn on the ER-Golgi compartment have focused exclusively on
neurons.
Recently, increasing evidence has suggested that
neurodegeneration associated with the expression of these muteins
is not restricted to dopaminergic neurons, indicating that
dysfunction of non-dopaminergic systems also contributes to the
pathogenesis of PD (21,22). In a 2011 systematic review of glial
involvement in PD, Halliday and Stevens drew the conclusion that
astrocytes play an important role in both the initiation and
progression of PD degeneration (23). Although astrocytes constitute the
largest population of non-excitable cells in the central nervous
system (CNS), they were initially considered to be passive
supporting cells. However, a growing list of studies indicates that
astrocytes are involved in a much wider range of brain functions,
including the active control of synaptogenesis (24) and plasticity (25,26),
the regulation of blood flow (27)
and restoration of neurons (28),
as well as the nourishing of nerves and the promotion of
myelination (29). Meanwhile,
increasing attention has been paid to the role of astrocytes in
neurodegenerative disorders, especially in PD. For example, direct
experiments confirmed that astrocytes take up altered α-syn that
has been released from axon terminals and astrocytes containing
α-syn aggregates, after which they produce proinflammatory
cytokines and chemokines, which in turn elicit microglial
activation and finally contribute to the degeneration of neurons
(30).
In our study, we obtained highly purified primary
rat astrocytes by a modification of a previously described method,
followed by infection with appropriate packaged lentiviral vectors
to establish astrocyte lines overexpressing wild-type and mutant
α-syn (A30P and A53T). Furthermore, western bolt analysis,
immunofluorescence, flow cytometry and ELISA were used to study in
detail the links between α-syn and ER stress, Golgi fragmentation,
apoptosis, secretion of neurotrophic factors, and growth of
neurons. In general, our research might provide new perspectives
for understanding the roles of astrocytes in the pathogenesis of
PD.
Materials and methods
Materials
Newborn Sprague-Dawley (SD) rats (day 0–3) were
obtained from the Experimental Animal Center of Xiangya Medical
College, Central South University (Changsha, Hunan, China). Animal
experiments were conducted in accordance with the guidelines of the
Institutional Animal Care and Use Committee of Xiangya Medical
College, Central South University. The experiments were approved by
the Ethics Committee of State Key Laboratory of Medical Genetics
(Hunan, China). The mammalian expression plasmids
α-syn-A53T-HA-FUIGW-GFP, α-syn-A30P-HA-FUIGW-GFP,
α-syn-WT-HA-FUIGW-GFP and FUIGW-GFP (31) were constructed by our group (State
Key Laboratory of Medical Genetics). 293FT cells were from the cell
bank of the Shanghai Institutes for Biological Sciences, Chinese
Academy of Sciences (Shanghai, China). The antibodies against HA
(1:1,000; SAB1306169), glial fibrillary acidic protein (GFAP;
1:500; SAB4300647), binding immunoglobulin heavy chain protein
(BiP; 1:3,000; G8918), MAP2 (1:1,000; HPA012828), and β-actin
(1:2,000; A5316) were all from Sigma-Aldrich (Merck-Millipore,
Darmstadt, Germany). Anti-α-syn (1:1,000; cat. no. 2642), PEARK
(1:1,000; cat. no. 5683), p-PEARK (1:1,000; cat. no. 3179), eIF2α
(1:1,000; cat. no. 5324), p-eIF2α (1:1,000; cat. no. 3398),
caspase-3 (1:1,000; cat. no. 9664) and anti-CHOP antibodies
(1:1,000; cat. no. 5554) were purchased from Cell Signaling
Technologies, Inc. (Danvers, MA, USA). The GDNF enzyme-linked
immunosorbent assay (ELISA) kit was from Promega Corp. (Madison,
WI, USA) and the Annexin V-FITC/PI Apoptosis Detection kit was
purchased from Invitrogen (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). The small interfering RNA (siRNA) for the CHOP protein
and the non-targeting scramble siRNA were purchased from
(Sigma-Aldrich; Merck-Millipore). The first RNA sequence was:
Sense, 5′GGAAGAACUAGGAAACGGA; antisense, 5′UCCGUUUCCUAGUUCUUCC. The
second siRNA sequence was: Sense, 5′CUGGGAAACAGCGCAUGAA; antisense,
5′UUCAUGCGCUGUUUCCCAG. The Lipofectamine® RNAiMAX
Transfection Reagent was from Invitrogen (Thermo Fisher Scientific,
Inc.). The ViraPower Packaging Mix and Lipofectamine 2000 were from
Invitrogen (Thermo Fisher Scientific, Inc.). DMEM/F12 and 0.25%
trypsin (with EDTA) were from Gibco (Thermo Fisher Scientific,
Inc.). Neurobasal-A medium, B27 supplement, Hank's Balanced Salt
Solution, 0.125% trypsin (without EDTA) and fetal bovine serum
(FBS) were from Gibco (Thermo Fisher Scientific, Inc.).
Isolation, purification and culture of
primary rat astrocytes and cortical neurons
Astrocytes were obtained as described previously
(32,33), with some modifications as follows:
Postnatal day 1–3, SD rats were decapitated and the cortices were
dissociated. The removed cortices were washed with precooled Hank's
balanced salt solution and minced, followed by incubating with
0.25% (wt/vol) trypsin/1 mM EDTA for 10 min at 37°C. After
centrifugation at 500 × g for 5 min, the pellet was resuspended in
DMEM and F12 (1:1) with 10% FBS, 2 mM L-glutamine, and 100 U/ml
penicillin-streptomycin (both Thermo Fisher Scientific, Inc.). The
cells were cultured for 1 h at 37°C in a humidified atmosphere
comprising 5% CO2 in an incubator (Thermo Fisher
Scientific, Inc.), and the flasks were shaken every 10 min to
remove fibroblasts and blood cells. After collecting the
supernatant, the cells were resuspended and seeded into culture
flasks. After 72 h, the medium was changed to fresh DMEM/F12 and
replaced every 3–4 days. The resulting mixed glial cells were
cultured for 7–10 days at 37°C, in an atmosphere comprising 5%
CO2. Astrocytes were purified from the mixed cultures by
mild trypsinization (0.05% trypsin, without EDTA) to remove
microglial cells and oligodendrocytes. The purified astrocytes were
cultured in DMEM/F12 with 10% FBS, 2 mM L-glutamine, and 100 U/ml
penicillin-streptomycin, and identified by phase-contrast
microscopy (Leica Microsystems Inc., Buffalo Grove, IL, USA) and
immunofluorescence.
Isolation and purification of cortical
neurons and co-culture with astrocytes
The method used was based on a previously described
protocol (34). Briefly, cortices
were isolated from neonatal SD rats within 24 h of birth. The
tissue was cut into small pieces and digested with 0.125% (wt/vol)
trypsin for 10 min at 37°C, followed by centrifugation at 500 × g
for 5 min and resuspension in DMEM/F12 medium supplemented with 10%
FBS, after which the resulting suspension was planted into the
bottom compartment of a porous Transwell cell culture chamber
treated with 1 mg/ml polylysine (Sigma-Aldrich; Merck-Millipore)
for co-culture. The medium was changed to Neurobasal/B27 medium
with 2 mM L-glutamine 4 h later. Astrocytes infected with
appropriate lentiviral vectors were placed on the top of the
culture chamber and co-cultured with neurons for 4 days at 37°C in
an atmosphere comprising 5% CO2.
Lentivirus vector construction and
infection of primary rat astrocytes
The protocol used is based on the method described
by Su et al (35). Briefly,
3 µg of the mammalian expression plasmids α-syn-A53T-HA-FUIGW-GFP,
α-syn-A30P-HA-FUIGW-GFP, α-syn-WT-HA-FUIGW-GFP and FUIGW-GFP were
individually used to transfected 293FT cells using Lipofectamine
2000 according to the manufacturer's instructions, according to the
steps in the lentivirus equipment package. Transfection efficiency
was assessed under a fluorescent microscope (Leica Microsystems
Inc., Buffalo Grove, IL, USA) and cell culture supernatant was
collected 72 h post-transfection. The lentiviral particles were
concentrated by centrifugation at 100,000 × g for 2 h and stored at
−70°C until use.
Astrocytes were seeded into the wells of 12-well
plates and grown to 70% confluence before infection. The following
day, an appropriate amount of lentiviral particle suspension was
diluted into medium containing 6 µg/ml of hexadimethrine bromide
(Polybrene; Sigma-Aldrich; Merck-Millipore), after which the old
medium was exchanged for the thus-prepared virion-containing
medium, and infection was conducted overnight. The
virion-containing medium was replaced with complete culture medium
the following day. At 7 days after infection, the expression of
α-syn was examined by western blot analysis and immunofluorescence
using an antibody against HA.
Knockdown of CHOP
Astrocytes were seeded into the wells of 10 cm
plates and grown to 70% confluence. Cells were cultured in 10 ml
medium composed of 8 ml of antibiotic-free growth medium and 2 ml
of Opti-MEM (Thermo Fisher Scientific, Inc.), 20 µl of
Lipofectamine RNAiMAX, and 200 pmol of CHOP siRNA. After 24 h of
transfection, cells were changed to normal growth medium and plated
into 12-well plates.
Immunofluorescence
Cells were washed with 1×PBS and fixed with 4% w/v
paraformaldehyde (Sigma-Aldrich; Merck-Millipore) in PBS for 15 min
at room temperature. After washing three times, the cells were
permeabilized by incubation in 1xPBS containing 1% Triton-X100 and
1% bovine serum albumin (BSA) for 1 h at room temperature, after
which they were incubated with the primary antibodies for 2 h at
room temperature. Appropriate secondary antibodies were used to
detect the corresponding proteins, after which the cells were
washed three times with 1xPBS. The cells were stored in the dark at
4°C until visualization under a confocal laser microscope (Zeiss,
Oberkochen, Germany).
Immunoassay for the detection of GDNF
Secretion
For the GDNF secretion assay, the supernatants of
primary rat astrocytes infected with lentiviral vectors
overexpressing wild-type α-syn, mutant α-syn or GFP-FUIGW (Control)
were collected at the 7th day after infection and stored at −80°C
until further use. Measurement of GDNF was performed using the GDNF
ELISA kit according to the manufacturer's instructions.
Western blot analysis
Cells were collected and lysed in RIPA buffer
containing protease inhibitors. 20 µg of protein were separated via
SDS-PAGE on a 12% polyacrylamide gel in running buffer. After
electrophoresis, the proteins were blotted onto nitrocellulose
membranes (Amersham, Bensheim, Germany), after which the membranes
were incubated in PBST containing 5% fat-free milk (Nestle,
Beijing, China) for 1 h at room temperature, followed by incubation
with different primary antibodies overnight at 4°C. The primary
antibodies were detected using a horse-radish peroxidase-linked
secondary antibody in conjunction with an enhanced
chemiluminescence (ECL) reagent (Pharmacia; GE Healthcare, Chicago,
IL, USA).
Annexin V and PI double staining
Different groups of cells were trypsinized and
gently washed once with medium, followed by washing with PBS before
re-suspension in 85 µl of binding buffer. Double staining was
performed with 10 µl of Annexin V-FITC and 5 µl of PI were added to
the re-suspended cells. After incubation at room temperature for 15
min in the dark, 400 µl of binding buffer was added to the cell
suspension. Measurement of the cell samples was performed on an
EPICS ALTRA flow cytometer (Beckman Coulter, Miami, US).
Statistical analysis
The data are presented as means ± standard
deviation. Analysis was conducted using ImageJ 1.51j8 (National
Institutes of Health, Bethesda, USA), GraphPad Prism 6 (GraphPad
Software, Inc., La Jolla, USA) and SPSS software (v16.0; IBM Corp,
Chicago, IL, USA). Comparisons between groups were determined by
one-way analysis of variance with Dunnett's t-test, P<0.05 was
considered to indicate a statistically significant difference. All
experiments were repeated three times.
Results
Highly purified astrocytes were
obtained and their derivative lines overexpressing wild-type or
mutant α-syn were established successfully
Astrocytes were isolated and purified by
differential adhesion and mild trypsinization as described in
Materials and Methods. When growing to 90% confluence, the
astrocytes displayed irregular shapes with long and rich processes
under the phase-contrast microscope (Fig. 1A). Additionally, immunofluorescence
with antibodies reactive against GFAP was used to confirm the
cells' identity (Fig. 1B). The
purity of the thus obtained astrocytes was almost 90%.
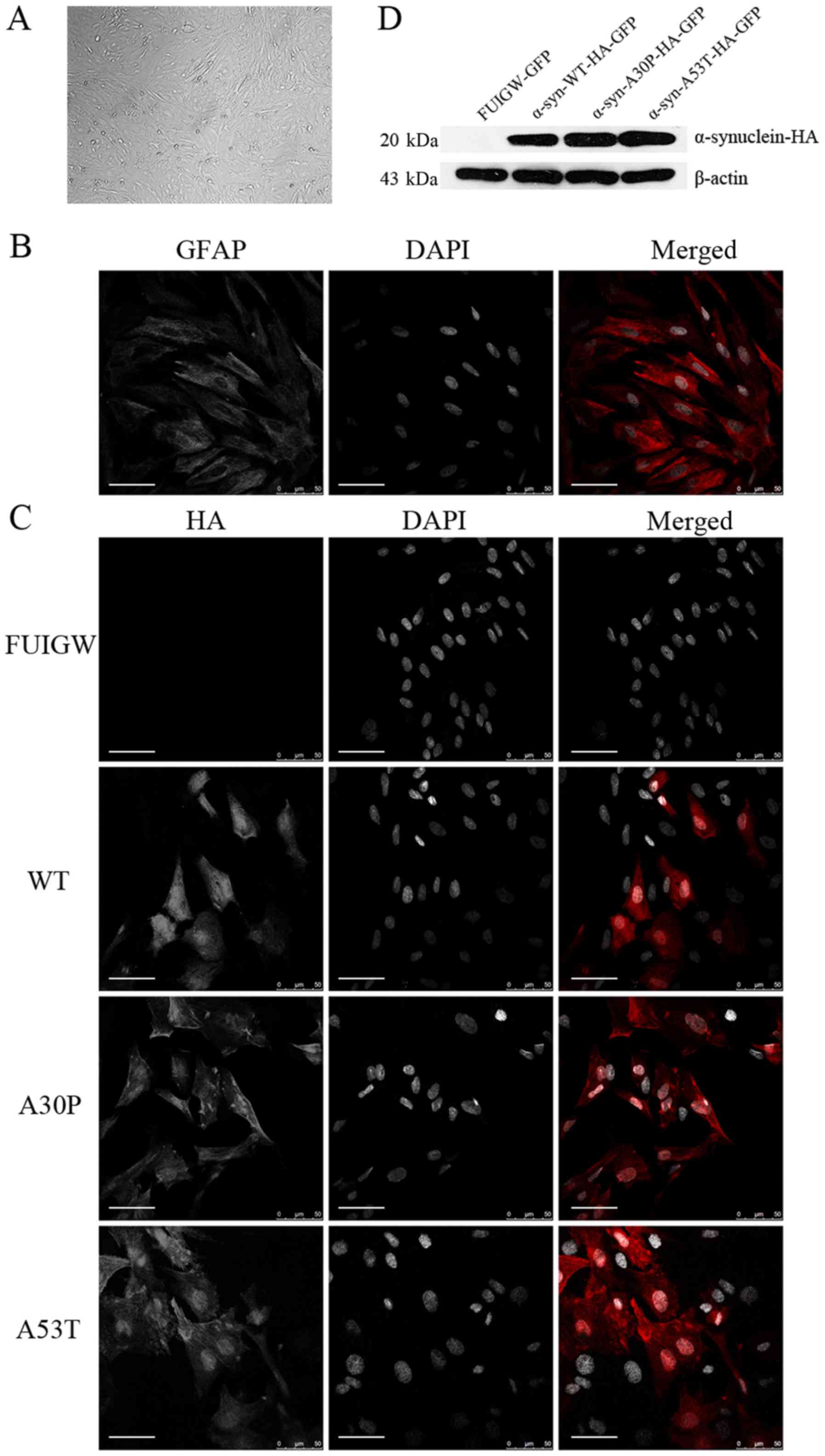 | Figure 1.Identification of cultured astrocytes
and the determination of the expression of the α-syn-HA fusion
protein following lentiviral infection. (A) Under a phase-contrast
microscope, the cultured primary astrocytes displayed an irregular
shape with long and rich processes (magnification, ×100). (B)
Astrocytes were immunostained with an anti-GFAP antibody (red) and
the nuclei were stained with DAPI (gray). (C) Immunofluorescence
localization analysis of the α-syn-HA fusion protein. Astrocytes
were infected with lentiviral vectors expressing
α-syn-WT-HA-FUIGW-GFP, α-syn-A53T-HA-FUIGW-GFP,
α-syn-A30P-HA-FUIGW-GFP and FUIGW-GFP, respectively. Using
immunofluorescent microscopy, the fusion proteins
α-syn-WT-HA-FUIGW-GFP, α-syn-A53T-HA-FUIGW-GFP and
α-syn-A30P-HA-FUIGW-GFP stained positively for HA (red), while
there was no red fluorescence in the astrocytes infected with the
control vector FUIGW-GFP. Nuclei are shown in gray (DAPI). DAPI
staining has been altered to gray to more clearly show the
morphological structure. Scale bars, 50 µm. (D) Western blot
analysis of the expression of the fusion protein α-syn-HA in the
three cell lines overexpressing WT and two mutant α-syn proteins,
and the negative control expressing FUIGW-GFP. The results were
consistent with the immunofluorescence observations. α-syn,
α-synuclein; WT, wild-type; HA, influenza A hemagglutinin; GFAP,
glial fibrillary acidic protein. |
To establish astrocyte lines overexpressing
wild-type or mutant α-syn, we infected the primary astrocytes with
lentiviral vectors. The results of immunofluorescence using anti-HA
antibodies to label the α-syn-HA fusion protein and DAPI to stain
the nuclei showed that the fusion protein was diffusely distributed
within the astrocytes infected with the α-syn-HA-FUIGW-GFP
lentivirus vector, while the astrocytes infected with the FUIGW-GFP
control vector showed negative immunostaining (Fig. 1C). Further western blot analysis
was in agreement with the immunofluorescence results (Fig. 1D). The data thus indicate that
astrocyte lines overexpressing wild-type or mutant α-syn were
established successfully.
ER stress in astrocytes is triggered
by the overexpression of wild-type or mutant α-syn
It has been suggested that ER stress caused by the
accumulation of α-syn in dopaminergic neurons is pivotal to the
pathogenesis of PD (36). To
confirm whether similar changes happened in astrocytes, primary rat
astrocytes were infected with lentiviral vectors expressing
wild-type or mutant α-syn, respectively. In addition, total
proteins from primary rat astrocytes treated with tunicamycin for 6
h were also analyzed as a positive control. Cells were cultured for
7 days after infection and we examined the expression of BiP,
phosphorylated or total forms of PERK and eIF2α. Bip is an
ER-resident chaperone, which is regarded as a marker of ER stress.
The activation of PERK-eIF2α axis was demonstrated by p-PERK and
p-eIF2α. Cells expressing mutant proteins showed increase in BiP,
p-PERK and p-eIF2α than the empty vector control (Fig. 2A-D). These results thus show that
overexpression of mutant α-syn can induce the PERK-eIF2α axis of ER
stress in astrocytes, whereas wild-type might induce ER stress
through other axises.
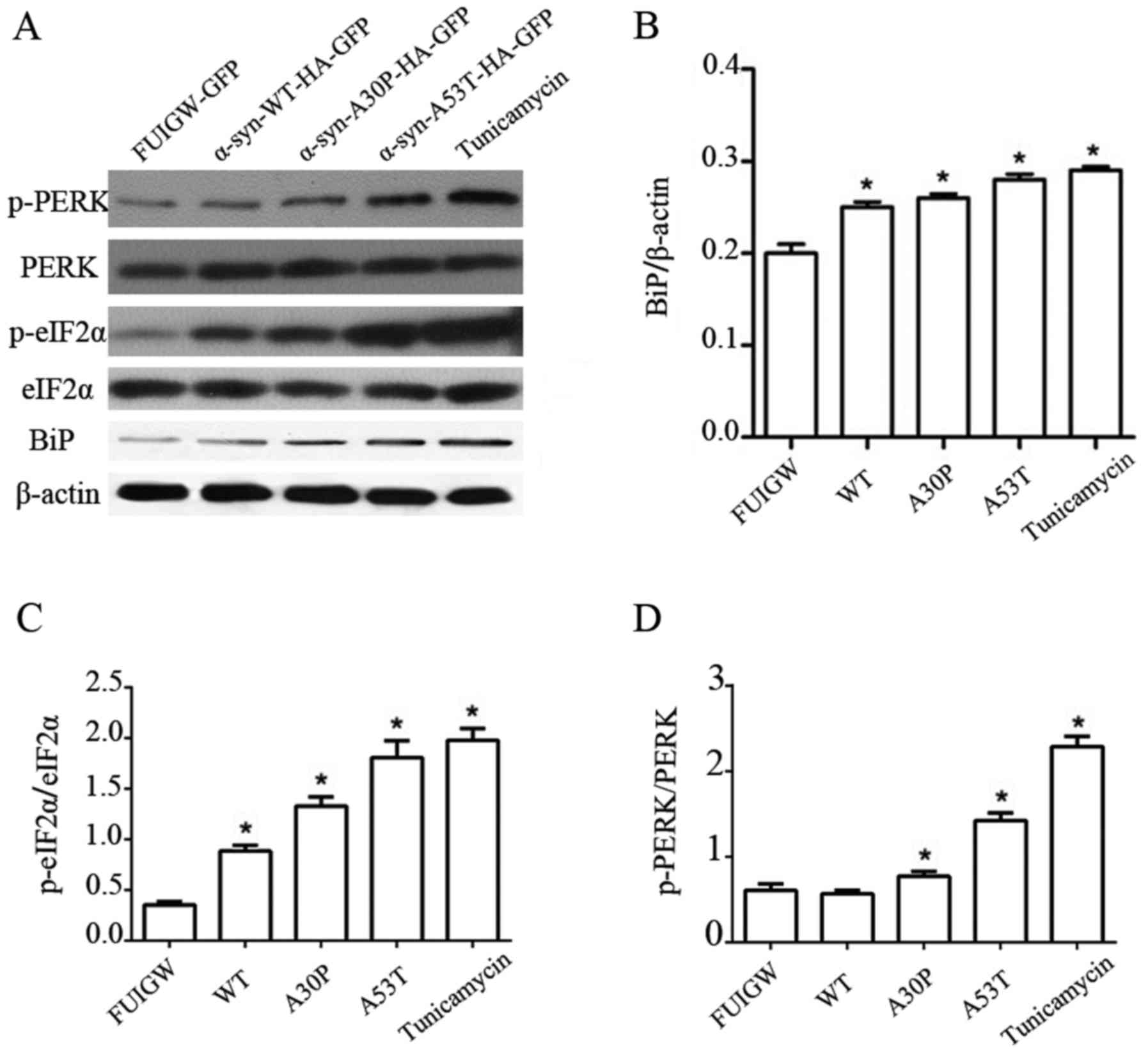 | Figure 2.Induction of UPR and PERK pathway in
astrocytes overexpressing α-syn. (A) Western blot analysis of
astrocytes 7 days following infection with lentiviral vectors
expressing α-syn-WT-HA-FUIGW-GFP, α-syn-A53T-HA-FUIGW-GFP,
α-syn-A30P-HA-FUIGW-GFP and the negative control FUIGW-GFP. Protein
levels of Bip, total PERK, phosphorylated PERK, total eIF2α and
phosphorylated eIF2α were determined by western blot analyses.
β-actin blotting was used as a loading control. (B) Integrated
density values for BiP relative to β-actin. (C) Integrated density
values for phosphorylated eIF2α relative to total eIF2α. (D)
Integrated density values for phosphorylated PERK relative to total
PERK. The bars represent the mean ± standard deviation. *P<0.05
vs. FUIGW (negative control). α-syn, α-synuclein; WT, wild-type;
HA, influenza A hemagglutinin; GFAP, glial fibrillary acidic
protein; GFP, green fluorescence protein; UPR, unfolded protein
response; PERK, protein kinase RNA-like endoplasmic reticulum
kinase; Bip, binding immunoglobulin heavy chain protein; p-,
phosphorylated; eIF2α, eukaryotic translation initiation factor
2α. |
Overexpression of wild-type or mutant
α-syn damages the GA of astrocytes
To examine whether the GA was also affected by the
overexpression of wild-type or mutant α-syn, immunofluorescence was
used. We infected primary rat astrocytes as described before, while
cells expressing GFP-FUIGW were used as a control. We analyzed the
morphology of the GA in the primary rat astrocytes at the 7th day
post-transfection using the cis-Golgi matrix protein marker GM130
(37,38), which can be used to visualize the
morphology of GA, as well as its location. At least 200 cells were
analyzed in each group and the percentage of fragmented GA was
calculated as described previously (19). Under the confocal laser microscope,
the GA of astrocytes overexpressing wild-type or mutant α-syn was
diffusely distributed in the cytoplasm, reticulate structures were
apparently disturbed, which was accompanied by apparent breakdown.
By contrast, the GA of the cells expressing GFP-FUIGW maintained a
normal juxtanuclear anastomosing linear profile (Fig. 3A). Compared with the control group,
in which Golgi fragmentation was observed in 16.9±5.8% of cells,
the percentage of fragmented GA in the cells overexpressing
wild-type or mutant α-syn (A30P and A53T) was much higher, at
73.7±10.1, 75.5±11.1 and 82.3±9.9%, respectively. Moreover, the
results were statistically significant (Fig. 3B). Furthermore, immunofluorescence
showed that the α-syn-HA fusion protein did not co-localize with
the GA, which was consistent with previous studies (39). The results therefore indicate that
the overexpression of wild-type or mutant α-syn causes
fragmentation of the GA in astrocytes.
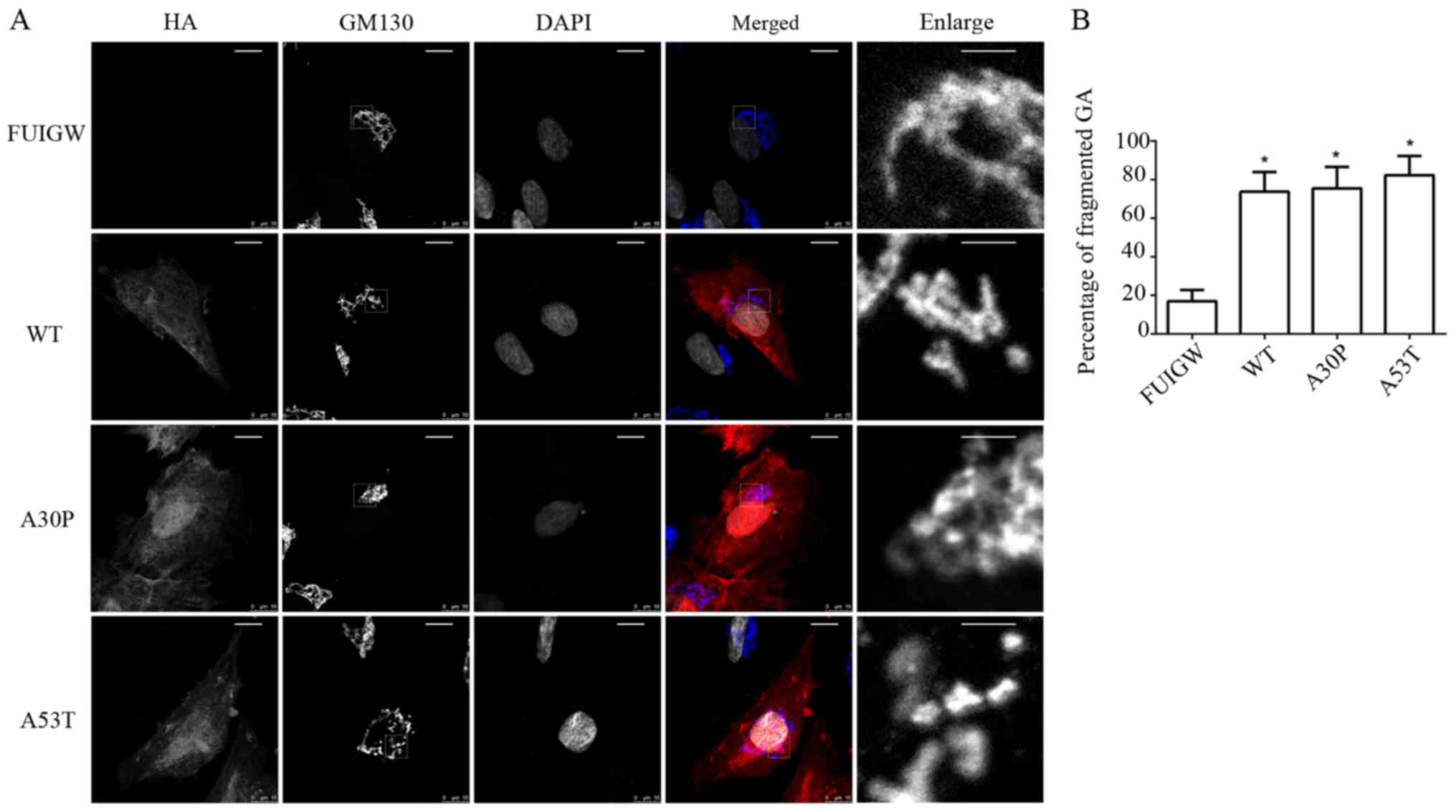 | Figure 3.Golgi fragmentation in astrocytes
overexpressing WT and mutant α-syn. (A) Morphology of GA was
observed 7 days following infection. α-syn-HA and GA were stained
using antibodies reactive against HA (red) and GM130 (blue, in
merged image), and nuclei were stained with DAPI (altered to gray).
A breakdown of the GA was visible in the astrocytes overexpressing
WT or mutant α-syn (A30P and A53T, respectively), while they were
intact in the control group (scale bars, 10 µm). The single-channel
staining images for GM130 and DAPI have been altered to gray to
more clearly show the morphological structure. The images labelled
‘enlarge’ are the enlarged images of the section surrounded by a
dotted line in the GM130 images (scale bars, 2 µm). (B)
Quantitative analysis of GA fragmentation among the different
groups. The bars represent mean values ± standard deviation.
*P<0.05 vs. FUIGW (negative control). α-syn, α-synuclein; WT,
wild-type; HA, influenza A hemagglutinin; GFP, green fluorescence
protein; GA, Golgi apparatus. |
Overexpression of wild-type or mutant
α-syn can activate CHOP and induce apoptosis in astrocytes
Although the exact molecular mechanism remains
speculative, several lines of evidence indicate that the
accumulation of misfolded α-syn within ER induces ER stress and,
ultimately, neuronal apoptosis, which contributes to
neurodegeneration in PD (11). The
results of this study demonstrated that ER stress and Golgi injury
were present in astrocytes overexpressing α-syn. To confirm whether
apoptosis was induced by the overexpression of α-syn, 7 days after
infecting the primary rat astrocytes with lentiviral vectors
overexpressing wild-type α-syn, mutant proteins and GFP-FUIGW,
respectively, we performed Annexin V and PI double staining
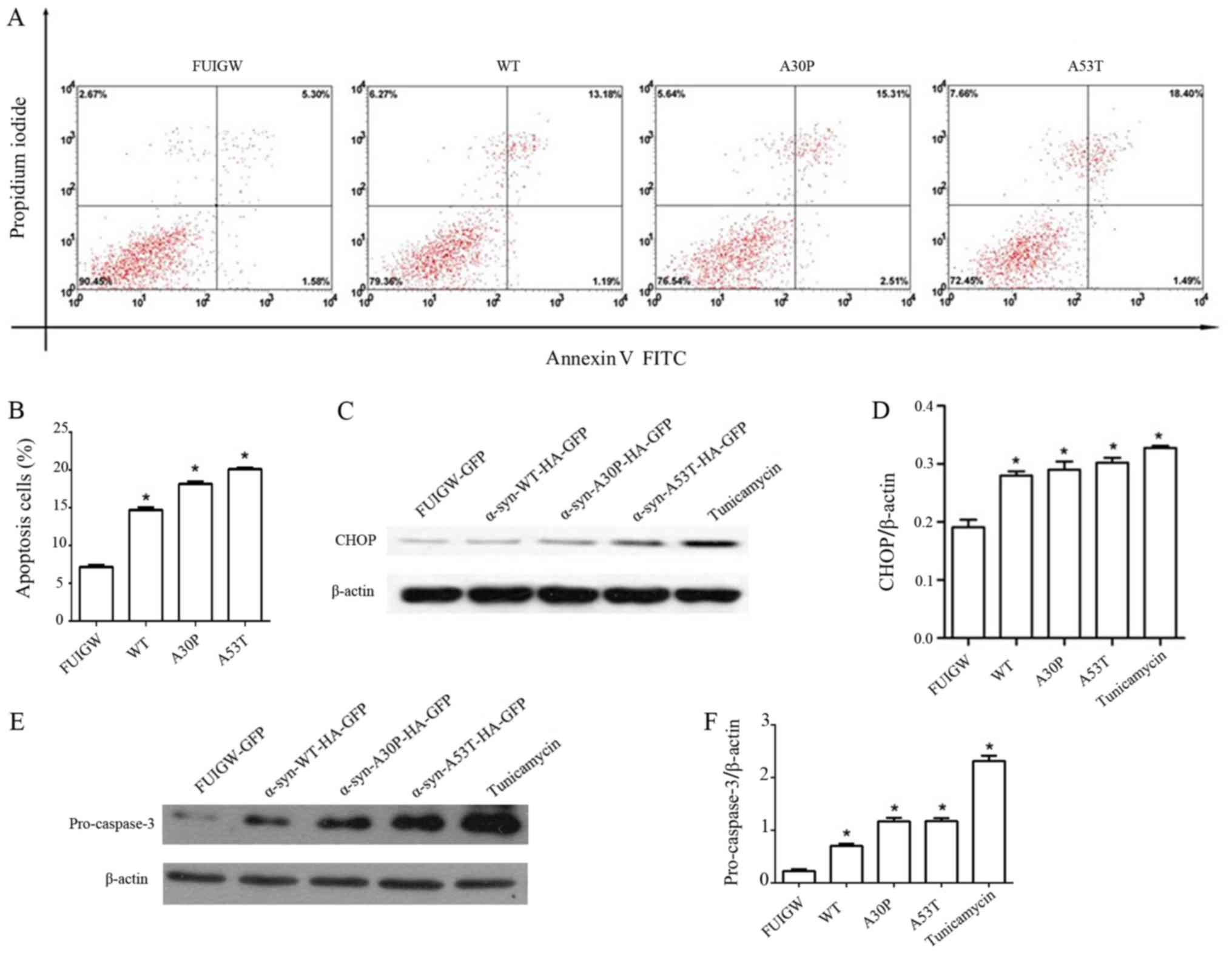
followed by flow cytometry. The results revealed four groups of
astrocytes in varying stages of apoptosis (Fig. 4A). Quantitative analyses revealed
that the overexpression of wild-type or mutant α-syn caused a
2-fold increase of the apoptosis rate relative to the control group
(Fig. 4B). Different lines of
evidence have shown that CHOP mediates apoptosis during ER stress
(17,40). Given this, we further examined the
levels of CHOP and cleaved caspase-3 by western blot. Comparing
with astrocytes expressing GFP-FUIGW, the ones overexpressing
either wild-type or mutant α-syn had higher CHOP and cleaved
caspase-3 levels (Fig. 4C-F).
These results suggest that ER stress induced by the overexpression
of α-syn in astrocytes might further lead to apoptosis through a
CHOP-mediated pathway.
Inhibiting of CHOP partially decreases
the apoptosis induced by overexpression of A53T
CHOP plays a convergent role in the UPR and it has
been identified as one of the most important chaperonins mediated
ER stress-induced apoptosis (41).
We took a direct approach to knockdown CHOP gene to examine its
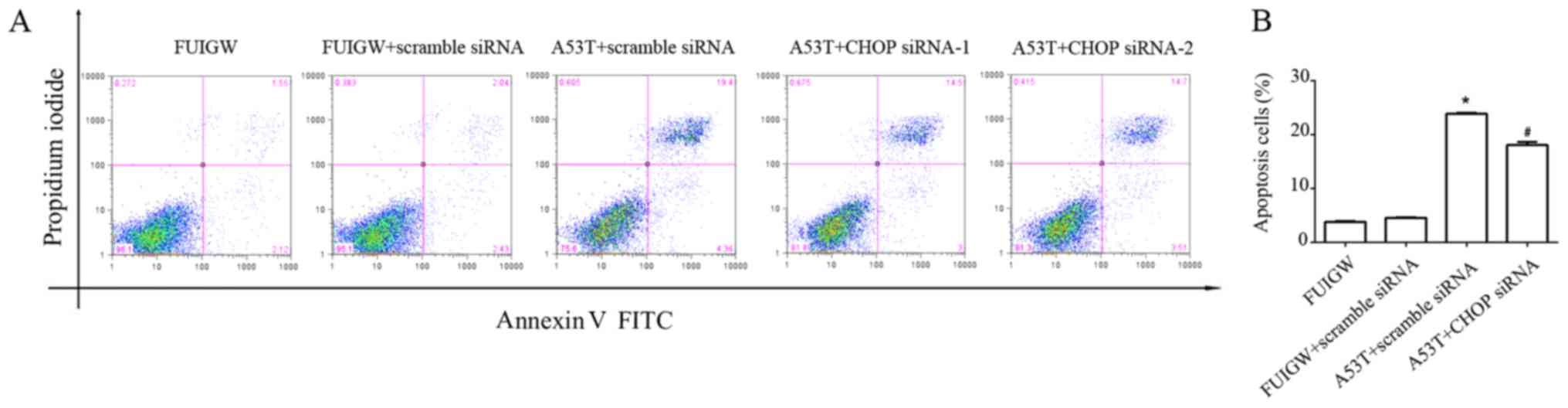
effect on ER stress-induced apoptosis. As shown in Fig. 5, overexpression of A53T increased
apoptosis in astrocytes compared with the control cells. The
apoptotic effect of overexpressed A53T was partially decreased by
knocking down CHOP with siRNA compared with A53T-mutant type
transduced with scrambled siRNA. Combine with the previous results,
parallel to the CHOP-mediated pathway, overexpression of A53T in
astrocytes may lead to apoptosis through other pathways.
Overexpression of wild-type or mutant
α-syn in astrocytes inhibits neurite outgrowth likely by reducing
the secretion of GDNF
Our work demonstrated that the overexpression of
wild-type or mutant α-syn in astrocytes causes ER-Golgi dysfunction
and even induces apoptosis. In recent years, increasing evidence
appears to suggest that astrocytes can also regulate neuronal
activity and synaptic transmission and plasticity, and are thus far
from being mere passive supportive cells (42). We therefore presumed that astrocyte
dysfunction may affect the outgrowth and function of neighboring
neurons. To investigate the relationship of dysfunctional
astrocytes and neurite outgrowth, we co-cultured cortical neurons
with the primary rat astrocytes overexpressing wild-type or mutant
α-syn in Transwell cell culture inserts containing a permeable
collagen-coated PTFE membrane. After 4 days, neurites were clearly
identified under the fluorescence microscope upon immunostaining
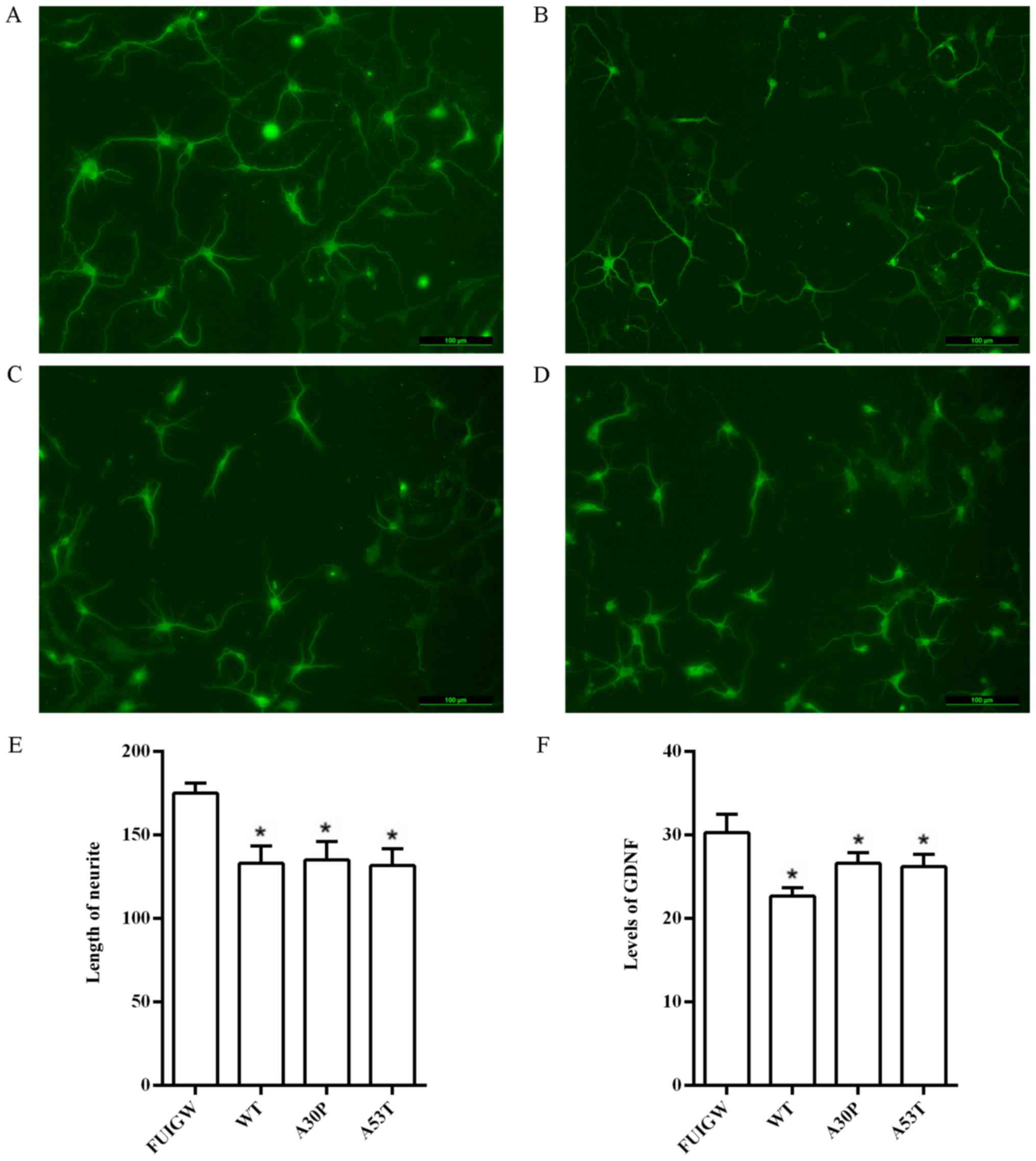
with an antibody against MAP2 (Fig.
6A-D). The lengths of the longest neurites of 100 neurons were
measured using the Image J software, and statistical analysis
suggested that the neurons in the control group had significantly
longer neurites than the neurons co-cultured with astrocytes
overexpressing α-syn (Fig. 6E).
The results thus demonstrated that the overexpression of α-syn in
astrocytes can attenuate neurite outgrowth, at least in
vitro.
Astrocytes have the ability to synthesize and
secrete a variety of neurotrophic factors, which are thought to
play a prominent role in regulating the growth of neurons (43). The question is therefore prescient
whether the inhibition of neurite outgrowth involves a decrease of
the secretion of neurotrophic factors from astrocytes induced by
the overexpression of wild-type or mutant α-syn. To investigate
this question, we further measured the effects of α-syn on the
secretory function of astrocytes. GDNF, a neurotrophic factor which
is intimately related to PD (44),
was chosen as an efficient evaluation index. We therefore infected
primary rat astrocytes with lentiviral vectors overexpressing
wild-type and mutant α-syn, as well as GFP-FUIGW as a control, and
measured the levels of GDNF in the cell culture supernatants 7 days
after infection using a commercially available ELISA kit. We found
that the levels of GDNF were significantly lower in primary rat
astrocytes overexpressing wild-type or mutant α-syn than in the
control group (Fig. 6F). These
findings indicate that the overexpression of α-syn can decrease the
secretion of neurotrophic factors, as observed for GDNF. Taken
together, the data indicate that the inhibition of neurite
outgrowth may be linked to the diminished secretion of neurotrophic
factors by astrocytes that accumulate α-syn.
Discussion
Aberrant aggregation of α-syn in neurons, resulting
in the so-called Lewy bodies (LBs), is a defining feature of
idiopathic and most autosomal dominant forms of PD (45). Homeostasis can be reestablished
upon abnormal aggregation of α-syn via induction of the UPR and the
autophagy-lysosome pathway (ALP) (11). However, when α-syn inclusions
overwhelm the ability of the degradation pathway to successfully
process and remove excess α-syn, oxidative stress, mitochondrial
damage and ER stress are triggered (41). In spite of the apparent relevance
of these mechanisms in astrocytes, most studies have focused on
dopaminergic neurons, and we presently have only a limited
understanding of the relationship between α-syn and astrocytes. In
addition, astrocytes play a key role in neuron physiology,
including trophic support, gliotransmission and antioxidant
activity (46,47). Therefore, we studied the effects of
α-syn on physiological processes in primary rat astrocytes and
their relation to the growth of co-cultured neurons.
It should be noted that the ER stress response can
be triggered by a variety of conditions that disturb folding of
proteins in the ER (10). The thus
induced stress can be reversed by the UPR, which aims to clear
unfolded proteins and restore ER homeostasis (11). BiP assists the folding of proteins
and regulates the activity of transmembrane-signaling proteins
during ER stress (48). IRE1,
PERK, and ATF6 are the three transmembrane signaling proteins that
associate with BiP in their inactive state, in resting cells. In
conditions of ER stress, BiP is sequestered through binding to
unfolded or misfolded proteins, which lead to the releasing from
IRE1/PERK/ATF6 and activation of ER stress sensors. BiP/GRP78
expression is therefore widely used as a marker for ER stress
(49). The three UPR transmembrane
signaling proteins, as mentioned above, use unique mechanisms of
signal transduction to regulates the expression of various
transcriptional factors and signaling events to adapt to ER stress
(40). We only explored the effect
of overexpression of α-syn in astrocytes on the PERK branch of UPR
and the impact of CHOP on apoptosis. The ATF6 and the IRE1 branches
of UPR were not examined. We used western blot analysis to show
that there was a significant increase in the levels of BiP in
astrocytes overexpressing α-syn, which indicated that ER stress was
caused by the overexpression of α-syn in astrocytes.
If the UPR fails, impaired protein homeostasis can
lead to chronic ER stress which induces cell death (19). At least three apoptosis pathways
are known to be involved in the cell apoptotic. The first is
transcriptional activation of the gene for CHOP. The second is
activation of the cJUN NH2-terminal kinase (JNK) pathway. The third
is activation of ER-associated caspase-12 (17). CHOP appears to be a crucial
proapoptotic factor found at the convergence point in the
regulatory network used to initiate apoptosis caused by ER stress.
Further investigations in our test suggested that ER stress
contributed to the activation of CHOP and induced apoptosis.
CHOP−/− astrocytes overexpressing A53T were
partially resistant to ER stress-mediated apoptosis, suggesting the
presence of other pathways mediating apoptosis in astrocytes
overexpressed wide-type and mutant α-syn besides the CHOP signaling
pathway. Further studies are warranted to investigate the role of
other pathways in α-syn induced apoptosis of astrocytes. Previous
work revealed the that pro-apoptotic transcriptional factor CHOP
activates the expression of apoptosis-related proteins such as
GADD34, TRAIL receptor-2, and endoplasmic reticulum
oxidoreductase-1 (Ero1α) (11).
Another possible mechanism by which CHOP induces apoptosis is via
direct inhibition of Bcl-2 transcription and induction of the
expression of the pro-apoptotic BH3-only protein (Bim) (11).
Immunostaining with an antibody against GM130
revealed Golgi fragmentation in astrocytes overexpressing α-syn
under laser confocal microscopy, which demonstrated that the
overexpression of α-syn in astrocytes induced Golgi damage.
Mukherjee et al (50)
stated that the fragmentation of GA is an early event during
apoptosis that occurs independently of major changes of the
cytoskeleton. Human and animal studies have shown clear evidence of
morphological changes of GA in neurons affected by a variety of
neurodegenerative diseases, including Alzheimer's disease (51) and amyotrophic lateral sclerosis
(52,53). Fragmentation of GA may be explained
by the fact that aggregated α-syn blocks ER-Golgi traffic, which
leads to further cell defects (20).
In order to understand changes of neurite outgrowth
in cocultures with astrocytes infected with lentiviral vectors
overexpressing α-syn-A53T-HA-FUIGW-GFP, α-syn-A30P-HA-FUIGW-GFP,
and α-syn-WT-HA-FUIGW-GFP, respectively, we established a primary
neuronal-astroglial co-culture system in which primary neurons were
seeded into the lower compartment of Transwell cell culture
inserts, and primary rat astrocytes were grown in the upper
compartment. Both primary neurons and primary rat astrocytes could
not touch each other directly, but a permeable polycarbonate
template membrane with 0.4 µm diameter between them allowed
substrate exchange. As a member of the microtubules-associated
protein family, MAP2 is involved in the formation of microtubules
in neurons, and is consequently used to label neurites (54). Using immunofluorescence, we proved
that the overexpression of either wild-type or mutant α-syn in
astrocytes inhibited neurite outgrowth to a similar degree. We
further showed that astrocytes overexpressing wild-type or mutant
α-syn had reduced levels of GDNF. It is known that neurotrophic
factors play an important role in the growth of neurites,
differentiation of neurons, synaptogenesis, synaptic plasticity and
maturation of electrophysiological characteristics (55). In fact, several neurotrophic
factors are closely associated with PD, and this includes GDNF.
GDNF specifically promotes the survival and morphological
differentiation of dopaminergic neurons and increases their
high-affinity dopamine uptake (44), which is more effective than other
neurotrophic factors (56). A
large number of tests have demonstrated that the suppression of
GDNF could inhibit the neurites produced by neurons in vitro
(56–59). According to our results, we
hypothesized that diminished GDNF levels might account for the
observed inhibition of neurite outgrowth, but the exact mechanism
underlying it will require further experimental investigation. In
addition, soluble proteins other than GDNF that are secreted by
astrocytes also contribute to the growth of neurons (60). It is believed that functions of
astrocytes depend on the amounts of proteins synthesized and
secreted by the normal ER-Golgi compartment (47). Further evidence will be provided to
demonstrate whether inhibition of neurite outgrowth by
overexpression of α-syn in astrocytes can be rescued by supplement
GDNF.
In conclusion, our results demonstrate that the
mutant α-syn (A53T and A30P) in astrocytes triggered ER stress via
PERK/eIF2α signaling pathway. Astrocytes apoptosis were induced
through a CHOP-mediated pathway. In addition, Golgi fragmentation
was found in the process. Overexpressing wild-type or mutant α-syn
in astrocytes significantly decreased the levels of GDNF, and
partly inhibited neurite outgrowth. Further study of the effects of
α-syn on astrocytes might help us understand the exact role of
astrocytes in the pathogenesis of PD, possibly revealing novel
therapeutic targets in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from The
Major State Basic Research and Development Program of China (973
Program; grant no. 2011CB510001), and grants from The National
Natural Science Foundation of China (grant nos. 81000542, 81200870,
30900469, 81430023, 81130021, 81371405, 31500832 and 81361120404),
as well as The Science and Technology Program of Hunan province
(grant no. 2014TT2014), and SRFDP (grant no. 20120162120079).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
All authors had full access to the data, and take
responsibility for the integrity of the data and the accuracy of
the data analysis. CW, JTan and LT contributed to the study
conception and design. ML and LW performed the majority of the
experiments, and LQ participated in the isolation and purification
of primary rat astrocytes and cortical neurons. ML, LW, JTan and CW
acquired the data. JTan, HZ, XS and JTang analyzed and interpreted
the data. ML, LW and LQ drafted the manuscript. ML, JTan, CW and LQ
critically revised the manuscript for important intellectual
content. HZ, XS and JTang performed statistical analysis. CW, JTan
and LT supervised the study and gave final approval of the version
to be published. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The experiments were approved by the Ethics
Committee of State Key Laboratory of Medical Genetics.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Greenamyre JT: Glutamate-dopamine
interactions in the basal ganglia: Relationship to Parkinson's
disease. J Neural Transm Gen Sect. 91:255–269. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kruger R, Kuhn W, Müller T, Woitalla D,
Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L and Riess O:
Ala30Pro mutation in the gene encoding alpha-synuclein in
Parkinson's disease. Nat Genet. 18:106–108. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Appel-Cresswell S, Vilarino-Guell C,
Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu
C, Trinh J, et al: Alpha-synuclein p.H50Q, a novel pathogenic
mutation for Parkinson's disease. Mov Disord. 28:811–813. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lesage S, Anheim M, Letournel F, Bousset
L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, et al:
G51D α-synuclein mutation causes a novel parkinsonian-pyramidal
syndrome. Ann Neurol. 73:459–471. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Polymeropoulos MH, Lavedan C, Leroy E, Ide
SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et
al: Mutation in the alpha-gene identified in families with
Parkinson's disease. Science. 276:2045–2047. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zarranz JJ, Alegre J, Gómez-Esteban JC,
Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O,
Atarés B, et al: The new mutation, E46K, of alpha-synuclein causes
Parkinson and Lewy body dementia. Ann Neurol. 55:164–473. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pasanen P, Myllykangas L, Siitonen M,
Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M and Paetau
A: Novel α-synuclein mutation A53E associated with atypical
multiple system atrophy and Parkinson's disease-type pathology.
Neurobiol Aging. 35(2180): e1–5. 2014.PubMed/NCBI
|
|
8
|
Deng H and Yuan L: Genetic variants and
animal models in SNCA and Parkinson disease. Ageing Res Rev.
15:161–76. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martin LJ, Pan Y, Price AC, Sterling W,
Copeland NG, Jenkins NA, Price DL and Lee MK: Parkinson's disease
alpha-synuclein transgenic mice develop neuronal mitochondrial
degeneration and cell death. J Neurosci. 26:41–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rao RV and Bredesen DE: Misfolded
proteins, endoplasmic reticulum stress and neurodegeneration. Curr
Opin Cell Biol. 16:653–662. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Colla E, Coune P, Liu Y, Pletnikova O,
Troncoso JC, Iwatsubo T, Schneider BL and Lee MK: Endoplasmic
reticulum stress is important for the manifestations of
α-synucleinopathy in vivo. J Neurosci. 32:3306–3320. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lai E, Teodoro T and Volchuk A:
Endoplasmic reticulum stress: Signaling the unfolded protein
response. Physiology (Bethesda). 22:193–201. 2007.PubMed/NCBI
|
|
14
|
Shi Y, Vattem KM, Sood R, An J, Liang J,
Stramm L and Wek RC: Identification and characterization of
pancreatic eukaryotic initiation factor 2 alpha-subunit kinase,
PEK, involved in translational control. Mol Cell Biol.
18:7499–7509. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harding HP, Zhang Y and Ron D: Protein
translation and folding are coupled by an
endoplasmic-reticulum-resident kinase. Nature. 397:271–274. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Harding HP, Zhang Y, Bertolotti A, Zeng H
and Ron D: Perk is essential for translational regulation and cell
survival during the unfolded protein response. Mol Cell. 5:897–904.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kalia LV, Kalia SK, McLean PJ, Lozano AM
and Lang AE: α-Synuclein oligomers and clinical implications for
Parkinson disease. Ann Neurol. 73:155–169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gonatas NK, Stieber A and Gonatas JO:
Fragmentation of the Golgi apparatus in neurodegenerative diseases
and cell death. J Neurol Sci. 246:21–30. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cooper AA, Gitler AD, Cashikar A, Haynes
CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al:
Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron
loss in Parkinson's models. Science. 313:324–328. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lim SY, Fox SH and Lang AE: Overview of
the extranigral aspects of Parkinson disease. Arch Neurol.
66:167–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karpinar DP, Balija MB, Kügler S, Opazo F,
Rezaei-Ghaleh N, Wender N, Kim HY, Taschenberger G, Falkenburger
BH, Heise H, et al: Pre-fibrillar alpha-synuclein variants with
impaired beta-structure increase neurotoxicity in Parkinson's
disease models. EMBO J. 28:3256–3268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Halliday GM and Stevens CH: Glia:
Initiators and progressors of pathology in Parkinson's disease. Mov
Disord. 26:6–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barres BA: The mystery and magic of glia:
A perspective on their roles in health and disease. Neuron.
60:430–440. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Faissner A, Pyka M, Geissler M, Sobik T,
Frischknecht R, Gundelfinger ED and Seidenbecher C: Contributions
of astrocytes to synapse formation and maturation-Potential
functions of the perisynaptic extracellular matrix. Brain Res Rev.
63:26–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Haydon PG and Nedergaard M: How do
astrocytes participate in neural plasticity? Cold Spring Harb
Perspect Biol. 7:a0204382014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koehler RC, Roman RJ and Harder DR:
Astrocytes and the regulation of cerebral blood flow. Trends
Neurosci. 32:160–169. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Silver J, Schwab ME and Popovich PG:
Central nervous system regenerative failure: Role of
oligodendrocytes, astrocytes, and microglia. Cold Spring Harb
Perspect Biol. 7:a0206022014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lundgaard I, Osório MJ, Kress BT,
Sanggaard S and Nedergaard M: White matter astrocytes in health and
disease. Neuroscience. 276:161–173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH,
Rho S, Hwang D, Masliah E and Lee SJ: Direct transfer of
alpha-synuclein from neuron to astroglia causes inflammatory
responses in synucleinopathies. J Biol Chem. 285:9262–9272. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
La Rocca R, Fulciniti M, Lakshmikanth T,
Mesuraca M, Ali TH, Mazzei V, Amodio N, Catalano L, Rotoli B,
Ouerfelli O, et al: Early hematopoietic zinc finger protein
prevents tumor cell recognition by natural killer cells. J Immunol.
182:4529–4537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang C, Hu ZL, Wu WN, Yu DF, Xiong QJ,
Song JR, Shu Q, Fu H, Wang F and Chen JG: Existence and distinction
of acid-evoked currents in rat astrocytes. Glia. 58:1415–1424.
2010.PubMed/NCBI
|
|
33
|
Shu Q, Hu ZL, Huang C, Yu XW, Fan H, Yang
JW, Fang P, Ni L, Chen JG and Wang F: Orexin-A promotes cell
migration in cultured rat astrocytes via Ca2+-dependent PKCα and
ERK1/2 signals. PLoS One. 9:e952592014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaech S and Banker G: Culturing
hippocampal neurons. Nat Protoc. 1:2406–2415. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Su YR, Wang J, Wu JJ, Chen Y and Jiang YP:
Overexpression of lentivirus-mediated glial cell line-derived
neurotrophic factor in bone marrow stromal cells and its
neuroprotection for the PC12 cells damaged by lactacystin. Neurosci
Bull. 23:67–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sugeno N, Takeda A, Hasegawa T, Kobayashi
M, Kikuchi A, Mori F, Wakabayashi K and Itoyama Y: Serine 129
phosphorylation of alpha-synuclein induces unfolded protein
response-mediated cell death. J Biol Chem. 283:23179–23188. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ramirez IB and Lowe M: Golgins and GRASPs:
Holding the Golgi together. Semin Cell Dev Biol. 20:770–779. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Seemann J, Pypaert M, Taguchi T, Malsam J
and Warren G: Partitioning of the matrix fraction of the Golgi
apparatus during mitosis in animal cells. Science. 295:848–851.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fujita Y, Ohama E, Takatama M, Al-Sarraj S
and Okamoto K: Fragmentation of Golgi apparatus of nigral neurons
with alpha-synuclein-positive inclusions in patients with
Parkinson's disease. Acta Neuropathol. 112:261–265. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: Cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pérez-Alvarez A and Araque A:
Astrocyte-neuron interaction at tripartite synapses. Curr Drug
Targets. 14:1220–1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oliveira SL, Pillat MM, Cheffer A, Lameu
C, Schwindt TT and Ulrich H: Functions of neurotrophins and growth
factors in neurogenesis and brain repair. Cytometry A. 83:76–89.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lin LF, Doherty DH, Lile JD, Bektesh S and
Collins F: GDNF: A glial cell line-derived neurotrophic factor for
midbrain dopaminergic neurons. Science. 260:1130–1132. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Simón-Sánchez J, Schulte C, Bras JM,
Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW,
Hernandez DG, et al: Genome-wide association study reveals genetic
risk underlying Parkinson's disease. Nat Genet. 41:1308–1312. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang DD and Bordey A: The astrocyte
odyssey. Prog Neurobiol. 86:342–367. 2008.PubMed/NCBI
|
|
47
|
Bélanger M and Magistretti PJ: The role of
astroglia in neuroprotection. Dialogues Clin Neurosci. 11:281–295.
2009.PubMed/NCBI
|
|
48
|
Tiffany-Castiglioni E and Qian Y: ER
chaperone-metal interactions: Links to protein folding disorders.
Neurotoxicology. 33:545–557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li J, Ni M, Lee B, Barron E, Hinton DR and
Lee AS: The unfolded protein response regulator GRP78/BiP is
required for endoplasmic reticulum integrity and stress-induced
autophagy in mammalian cells. Cell Death Differ. 15:1460–1471.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mukherjee S, Chiu R, Leung SM and Shields
D: Fragmentation of the Golgi apparatus: An early apoptotic event
independent of the cytoskeleton. Traffic. 8:369–378. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tillement JP and Papadopoulos V:
Subcellular injuries in Alzheimer's disease. CNS Neurol Disord Drug
Targets. 13:593–605. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sundaramoorthy V, Walker AK, Yerbury J,
Soo KY, Farg MA, Hoang V, Zeineddine R, Spencer D and Atkin JD:
Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology
characteristic of amyotrophic lateral sclerosis in neuronal cells.
Cell Mol Life Sci. 70:4181–4195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fujita Y, Okamoto K, Sakurai A, Kusaka H,
Aizawa H, Mihara B and Gonatas NK: The Golgi apparatus is
fragmented in spinal cord motor neurons of amyotrophic lateral
sclerosis with basophilic inclusions. Acta Neuropathol.
103:243–247. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dehmelt L and Halpain S: The MAP2/Tau
family of microtubule-associated proteins. Genome Biol. 6:2042005.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Airavaara M, Voutilainen MH, Wang Y and
Hoffer B: Neurorestoration. Parkinsonism Relat Disord. 18 Suppl
1:S143–S146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jiao L, Zhang Y, Hu C, Wang YG, Huang A
and He C: Rap1GAP interacts with RET and suppresses GDNF-induced
neurite outgrowth. Cell Res. 21:327–337. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Takaku S, Yanagisawa H, Watabe K, Horie H,
Kadoya T, Sakumi K, Nakabeppu Y, Poirier F and Sango K: GDNF
promotes neurite outgrowth and upregulates galectin-1 through the
RET/PI3K signaling in cultured adult rat dorsal root ganglion
neurons. Neurochem Int. 62:330–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gavazzi I, Kumar RD, McMahon SB and Cohen
J: Growth responses of different subpopulations of adult sensory
neurons to neurotrophic factors in vitro. Eur J Neurosci.
11:3405–3414. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zurn AD, Winkel L, Menoud A, Djabali K and
Aebischer P: Combined effects of GDNF, BDNF, and CNTF on motoneuron
differentiation in vitro. J Neurosci Res. 44:133–141. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sampaio TB, Savall AS, Gutierrez MEZ and
Pinton S: Neurotrophic factors in Alzheimer's and Parkinson's
diseases: implications for pathogenesis and therapy. Neural Regen
Res. 12:549–557. 2017. View Article : Google Scholar : PubMed/NCBI
|