Introduction
Acute myeloid leukemia (AML) is a highly
heterogeneous hematopoietic malignancy with an incidence of
approximately 3/100,000 per year (1). Current treatment approaches have
resulted in a cure rate of 35% in younger AML patients and 15% in
older patients (2,3). Based on the NCCN Guidelines for AML,
prognosis prediction methods and therapy selected predominantly
based on validated cytogenetics which divide the AML patients into
favorable, intermediate and poor risk status (2,4).
However, some patients eventually relapse despite the lack of
adverse risk factors (5).
Therefore, optimized biomarkers are necessary in order to refine
the prognosis of AML patients.
Long non-coding RNAs (lncRNAs) are defined as
non-protein-coding RNA transcripts longer than 200 bp. Recently,
certain lncRNAs have been reported to exhibit a role in AML. For
example, lncRNA CRNDE has been demonstrated to be associated with
the classification and total survival of AML patients through
regulating proliferation, apoptosis and cell cycle of AML cells
(6). Furthermore, Garzon et
al (7) derived a lncRNA score
composed of 48 lncRNAs for prognostic prediction of cytogenetically
normal AML. Notably, the majority of the 48 lncRNAs associated with
survival do not associate with known prognostic gene mutations in
the study (7). LncRNA prognostic
marker independent of known specific subtype will improve survival
prediction of AML.
LncRNAs function through regulation of mRNA
expression. Previous studies reported that lncRNAs regulate
transcription via local (cis) and long distance (trans) mechanisms.
Cis-regulation is identified as when the transcription of an lncRNA
affects the expression levels of its neighbor genes.
Trans-regulation is when that lncRNA can interact with a
transcriptional factor (TF) thereby influencing the expression of
the TF of target genes. In the competing endogenous (ce) RNA
hypothesis, lncRNAs compete with the mRNAs containing the same
microRNA (miRNA) response elements for binding the miRNAs, thereby
influencing the expression of the miRNA target genes (8).
The present study aimed to build an lncRNA risk
score to refine AML patient prognostic classification by analyzing
AML patients from Therapeutically Available Research to Generate
Effective Treatments (TARGET) and The Cancer Genome Atlas (TCGA)
project. Furthermore, we predicted the potential target of the
prognostic lncRNAs in cis/trans/ceRNA regulation.
Materials and methods
Acquisition of TCGA and TARGET AML
data
The RNA-seq data and corresponding clinical
information of AML patients in TARGET and TCGA project were
downloaded from Genomic Data Commons Data Portal (portal.gdc.cancer.gov/). Data of acute promyelocytic
leukemia in TCGA were filtered out as the therapy and prognosis for
acute promyelocytic leukemia patients differs from other subtypes
of AML and there were no acute promyelocytic leukemia patients in
the TARGET project. Following further removal of data without
complete clinical information, a total of 340 TARGET AML samples
and 162 TCGA AML samples were investigated in the present study.
Based on the GENCODE project long non-coding RNA annotation file
(version 26, GRCh38), we obtained the Reads Per Kilobase per
Million (RPKM) mapped reads expression data of lncRNAs from level 3
RNA-seq data of TARGET and TCGA.
Identification of subtype-specific
lncRNAs
Differentially expressed subtype-specific lncRNAs
were calculated using DESeq (version 1.6.1) (9) by comparing the lncRNA expression of a
specific class of AML samples with the other AML samples at
selection cut-off fold change >2 and false discovery rate
<0.05. The lncRNAs differentially expressed in a certain AML
subtype in both TARGET and TCGA projects were considered
subtype-specific lncRNAs.
Statistical analysis
After excluding the subtype-specific lncRNAs, the
remaining subtype independent lncRNAs were evaluated in the
prediction of patient event-free survival (EFS) by univariable cox
regression analysis in the TARGET project. The univariable Cox
regression analysis was calculated between lncRNA gene expression
presented as log2 (RPKM+1) and patient EFS as days.
LncRNAs were considered significantly correlated with patient
survival at a threshold of P<0.05. Random survival forests
variable hunting (RSFVH) algorithm was carried out to minimize the
lncRNAs selected (10). The error
rate in the RSFVH model was calculated by 1,000 permutation runs.
Then, the selected lncRNAs were subjected to multivariate Cox
regression analysis and the risk score was constructed by estimated
regression coefficients in the multivariable Cox regression and
expression of lncRNAs. The median risk score was selected in the
training set as a cut-off dividing the patients into low- and
high-risk groups. The Kaplan-Meier method was used to compare the
survival difference between the low- and high-risk groups in
training and validation sets. Multivariate Cox analysis and
Kaplan-Meier survival analysis of cytogenetic stratified AML
patients was used to identify the three lncRNAs expression
signature as an independent prognostic factor. The sensitivity and
specificity of the lncRNA expression signature was evaluated by the
area under the receiver operating characteristic (AUROC) of five
years EFS. Cox regression, Kaplan-Meier survival analysis and ROC
were performed using Statistical software R (version 3.3.3;
www.r-project.org/) and survival package,
survival ROC package, time ROC package, survminer package based on
software R (11,12).
Co-expression network construction and
investigation of cis/trans/ceRNA regulation
We computed the Pearson correlation coefficient
between the mRNA and lncRNA expression levels. The mRNA-lncRNA
pairs with absolute Pearson correlation coefficient >0.5 and
P<0.001 in both TCGA and TARGET projects were selected for
co-expression network construction.
We identified the genomic distance of the
co-expressed lncRNA-mRNA pairs, and the pairs located at the same
chromosome within 300 kb were identified as potentially
cis-regulated.
For trans prediction, we focused on the idea that
lncRNA may interact with TFs and trans-regulate the expression
levels of the target gene of the TF. We used iRegulon plugin within
cytoscape software to predict the potential associated TF from the
co-expressed gene set of the given lncRNA (13). The target set of TFs with
Normalized Enrichment Score (NES) >5 were considered as
significantly enriched.
For ceRNA network analysis, we searched the
lncRNA-mRNA pairs for the same miRNA seed sequence binding site.
The miRNA binding sites of lncRNAs were predicted by miRcode
(www.mircode.org/) and the miRNA-mRNA
relationships were predicted by Targetscan (www.targetscan.org/).
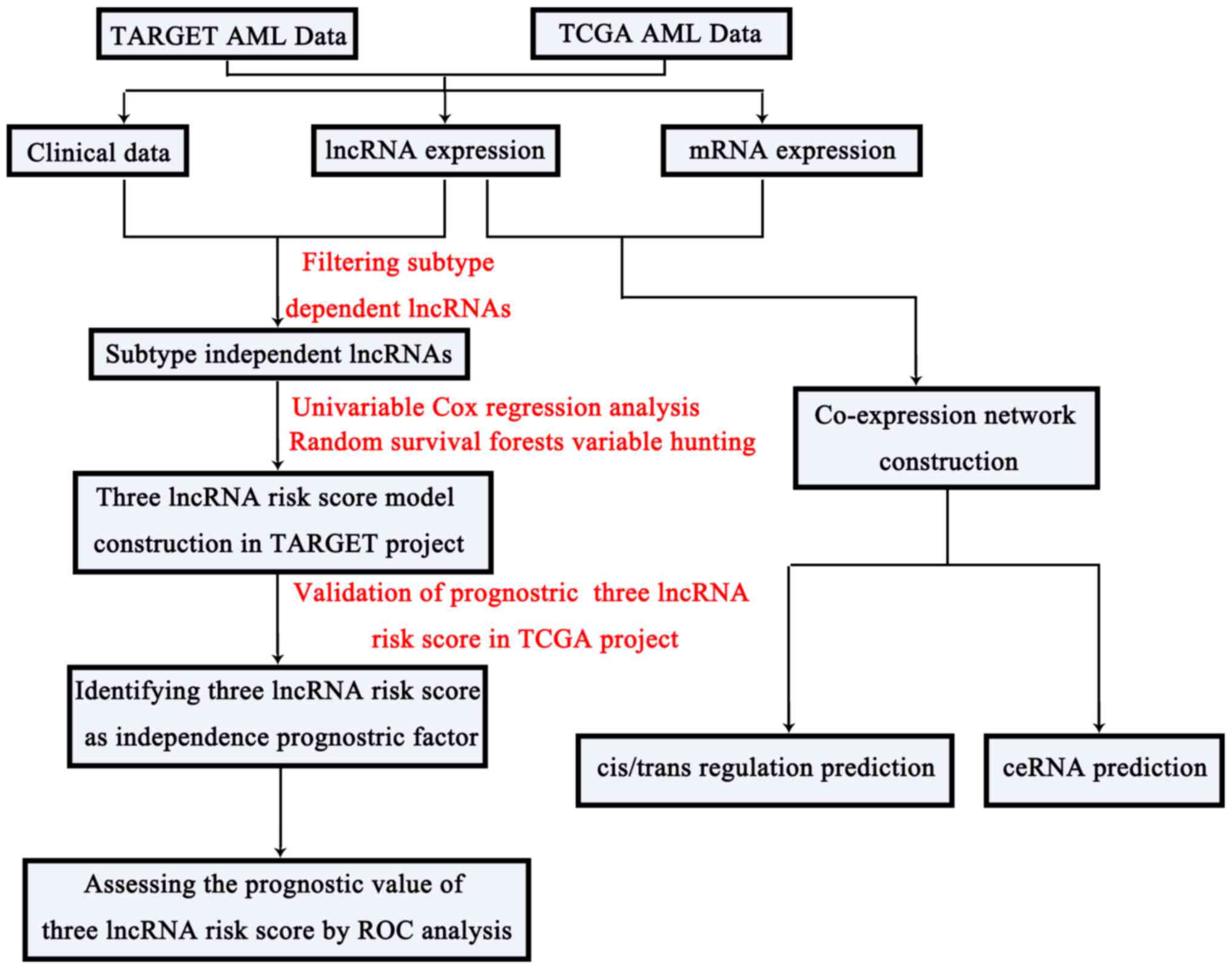
Results
In the present study, we identified subtype
independent prognostic lncRNAs, constructed a lncRNA risk score and
explored the potential lncRNA functional mechanisms in
cis/trans/ceRNA regulation based on a co-expression network. The
framework of this study was presented in Fig. 1.
Identification of subtype dependent
lncRNAs
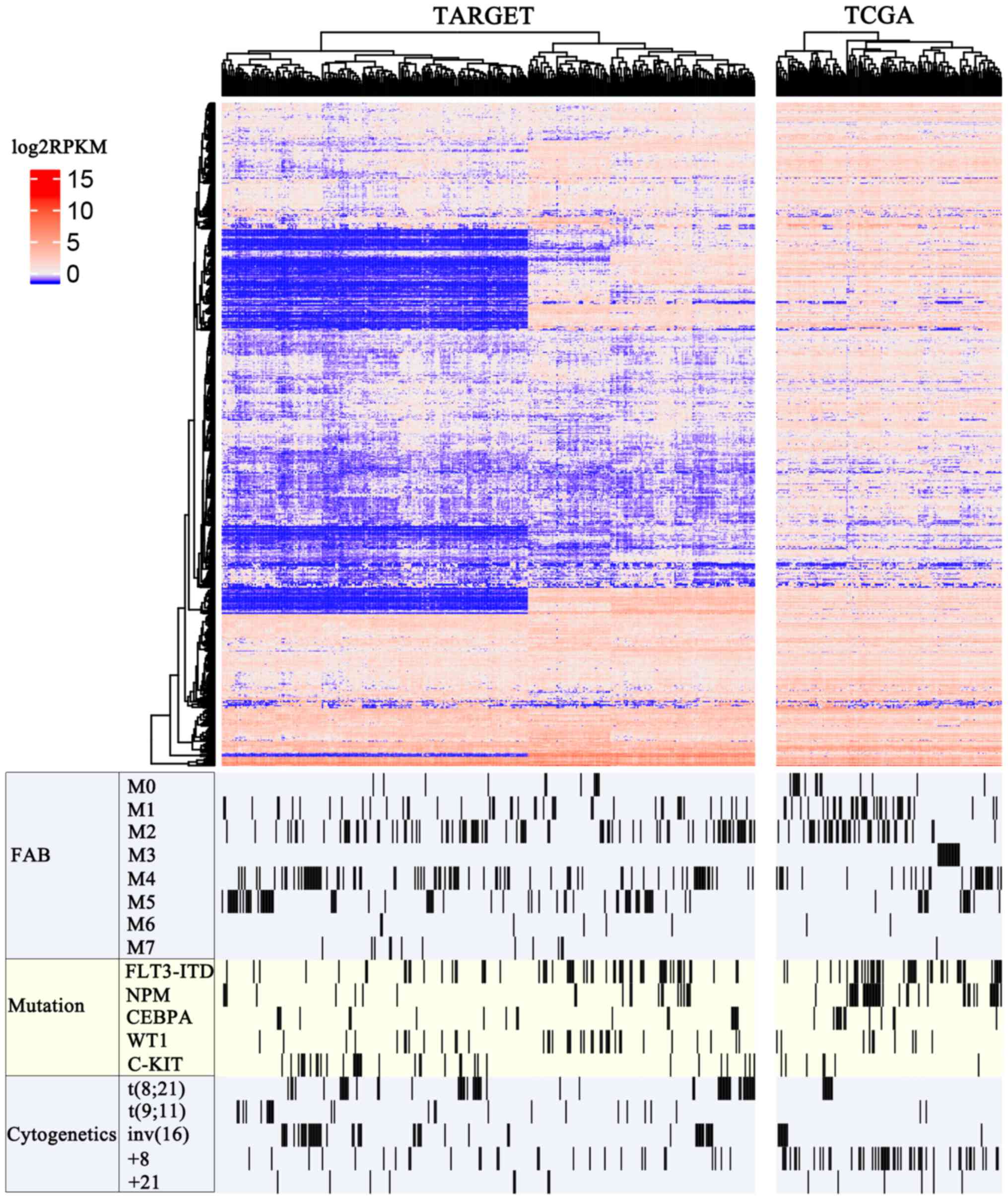
A total number of 15778 lncRNAs were recorded in
current human GENCODE release (version 27). A total of 655 of these
lncRNAs had stable expression profiling in both the TCGA and TARGET
AML datasets (RPKM >1 and read count >200 in >50%
patients). The heatmap of these lncRNAs in the TARGET and TCGA
projects are shown in Fig. 2. To
work out subtype independent prognostic lncRNAs, we first
identified subtype-specific lncRNAs according to the
French-American-British (FAB) system, gene mutations and
cytogenetic abnormality.
It was revealed that CRNDE, AP002761.1, AC009506.1,
LINC01637, AL391832.2 and LINC02081 were highly expressed in the
FAB M5 subtype (Fig. 3A and
B).
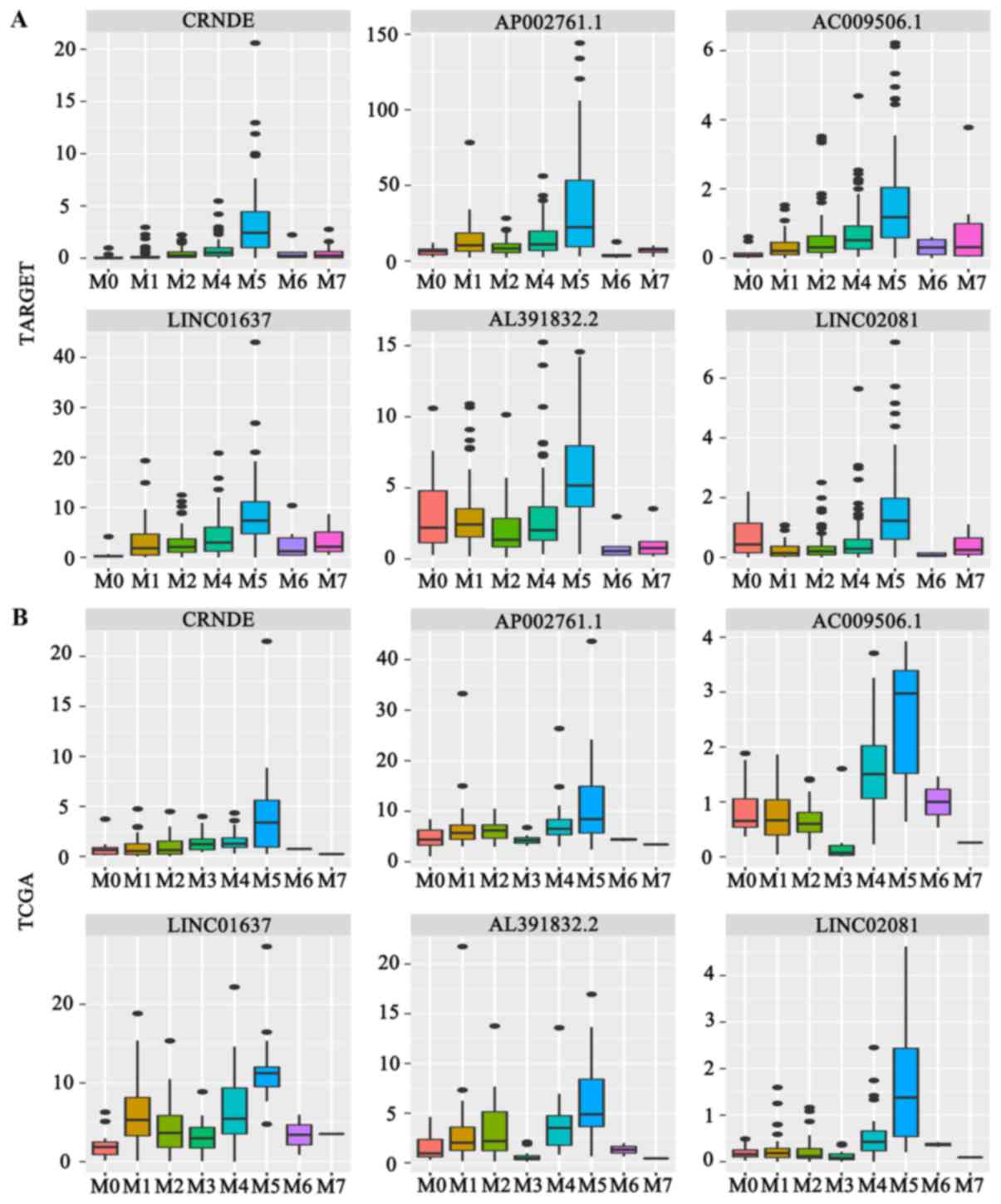 | Figure 3.Box plot graphics were employed to
illustrate the comparisons in lncRNAs expression between subgroups.
(A) Expression of 6 FAB-M5 associated lncRNAs in the TARGET
project. (B) Expression of 6 FAB-M5 associated lncRNAs in TCGA
project. (C) Expression of 3 FLT3-itd associated lncRNAs, 2 NPM1
mutation associated lncRNAs and a CEBPA mutation associated lncRNA
in the TARGET project. (D) Expression of 3 FLT3-itd associated
lncRNAs, 2 NPM1 mutation associated lncRNAs and a CEBPA mutation
associated lncRNA in TCGA project. (E) Expression of 3 inv
(16) associated lncRNAs in the
TARGET project. (F) Expression of 3 inv (16) associated lncRNAs in TCGA project.
TARGET, Therapeutically Available Research to Generate Effective
Treatments; TCGA, The Cancer Genome Atlas; lncRNA, long non-coding
RNA; FAB, French-American-British system; wt, wild type; mut,
mutation; FLT3-itd, tyrosine kinase 3-internal tandem duplication;
NPM, nucleophosmin; CEBPA, CCAAT/enhancer binding protein-α; WT1,
Wilms tumor 1. |
Gene mutations revealed that LINC00982, WT1-AS and
LINC0147 were highly expressed in Flt3-itd mutation for AML;
HOTAIRM1 and HOXB-AS3 were highly expressed in NPM1 mutation for
AML; LINC01252 was highly expressed in CEBPA mutation for AML
(Fig. 3C and D).
As for cytogenetic abnormalities, we identified that
AP003774.3, AC021915.2 and AC129507.1 were highly expressed in AML
with inv16 (Fig. 3E and F).
Construction of subtype independent
prognostic model based on lncRNA expression
As there were more AML patients in the TARGET
database than the TCGA, TARGET AML samples were classified the
training set and TCGA AML samples as validation set (14). After filtering out subtype specific
lncRNAs, we identified 25 potential prognostic factors that were
significantly correlated with the patients' EFS (P<0.05) in the
TARGET project.
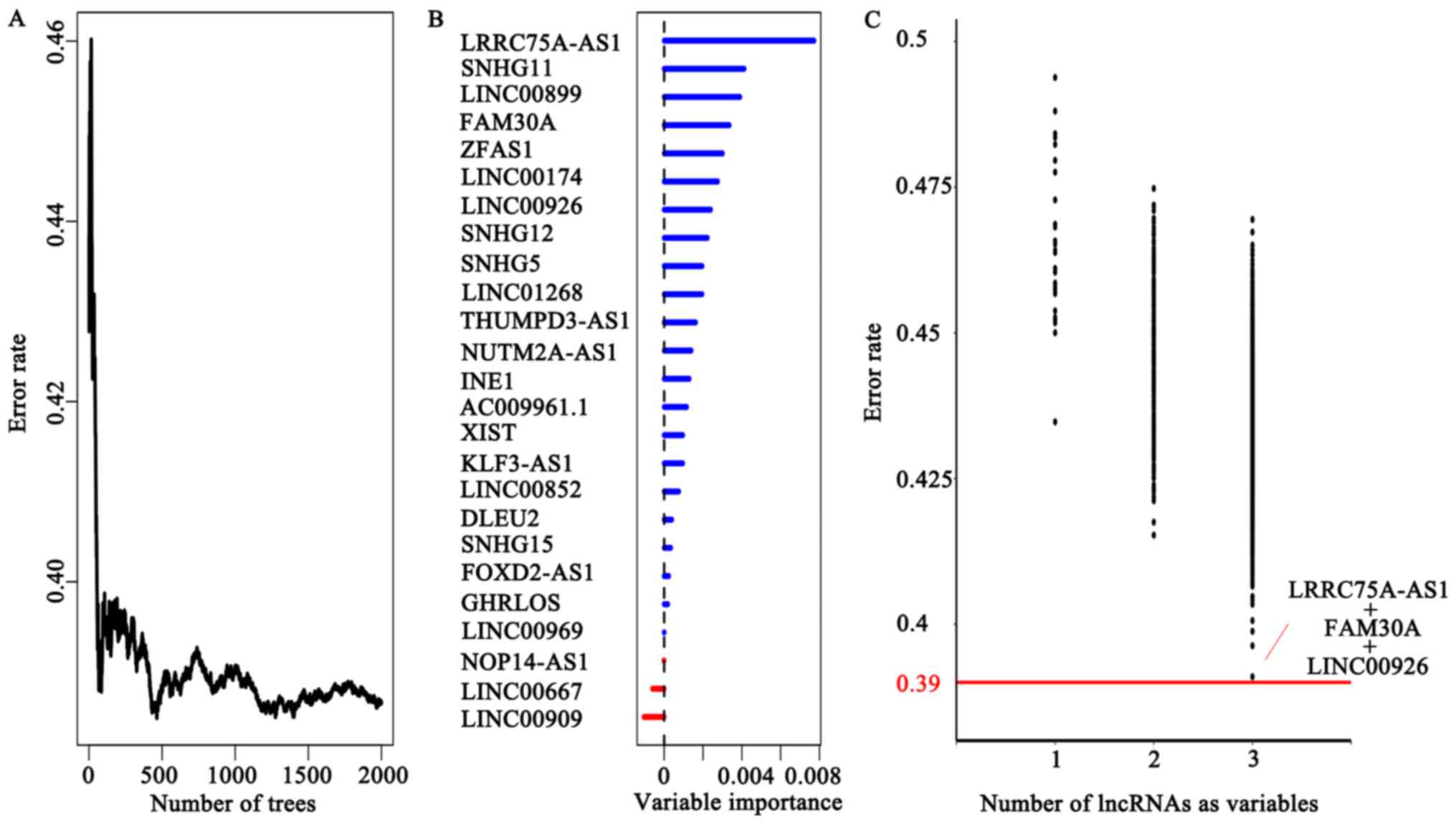
To build a more practical model for prediction, we
performed the RSFVH algorithm. First, we obtained a prediction with
a 0.39 error rate using all 25 potential prognostic lncRNAs as
variables (Fig. 4A), and then the
variable importance was shown in Fig.
4B.
Then, we calculated the error rate of using smaller
number of lncRNAs as variables and found that the error rate was
relatively high when using one or two lncRNAs. When using three
lncRNA as variables, the lowest error rate combination
(LINC00926+LRRC75A-AS1+FAM30A) reached 0.391 which was close to
0.39 (Fig. 4C). Following this, we
subjected the three lncRNAs to multivariate Cox regression model
for EFS outcome of 340 TARGET AML patients (Table I). The lncRNAs (LINC00926,
LRRC75A-AS1) with negative coefficients suggested that higher
expressions were associated with favorable survival and the
remaining lncRNA (FAM30A) with a positive coefficient suggested
higher expression was related to poor survival.
 | Table I.Multivariable Cox regression of three
long non-coding RNAs risk score in Therapeutically Available
Research to Generate Effective Treatments acute myeloid leukemia
set. |
Table I.
Multivariable Cox regression of three
long non-coding RNAs risk score in Therapeutically Available
Research to Generate Effective Treatments acute myeloid leukemia
set.
| Gene symbol | P-value | Hazard ratio | Variable
importance | Relative
importance |
|---|
| LINC00926 | 0.00699 | 0.8678 | 0.0162 | 0.514 |
| FAM30A | 0.00347 | 1.0749 | 0.0063 | 0.199 |
| LRRC75A-AS1 | 0.00022 | 0.7586 | 0.0316 | 1.000 |
A three-lncRNA signature predicts the
survival of AML patients in the training set
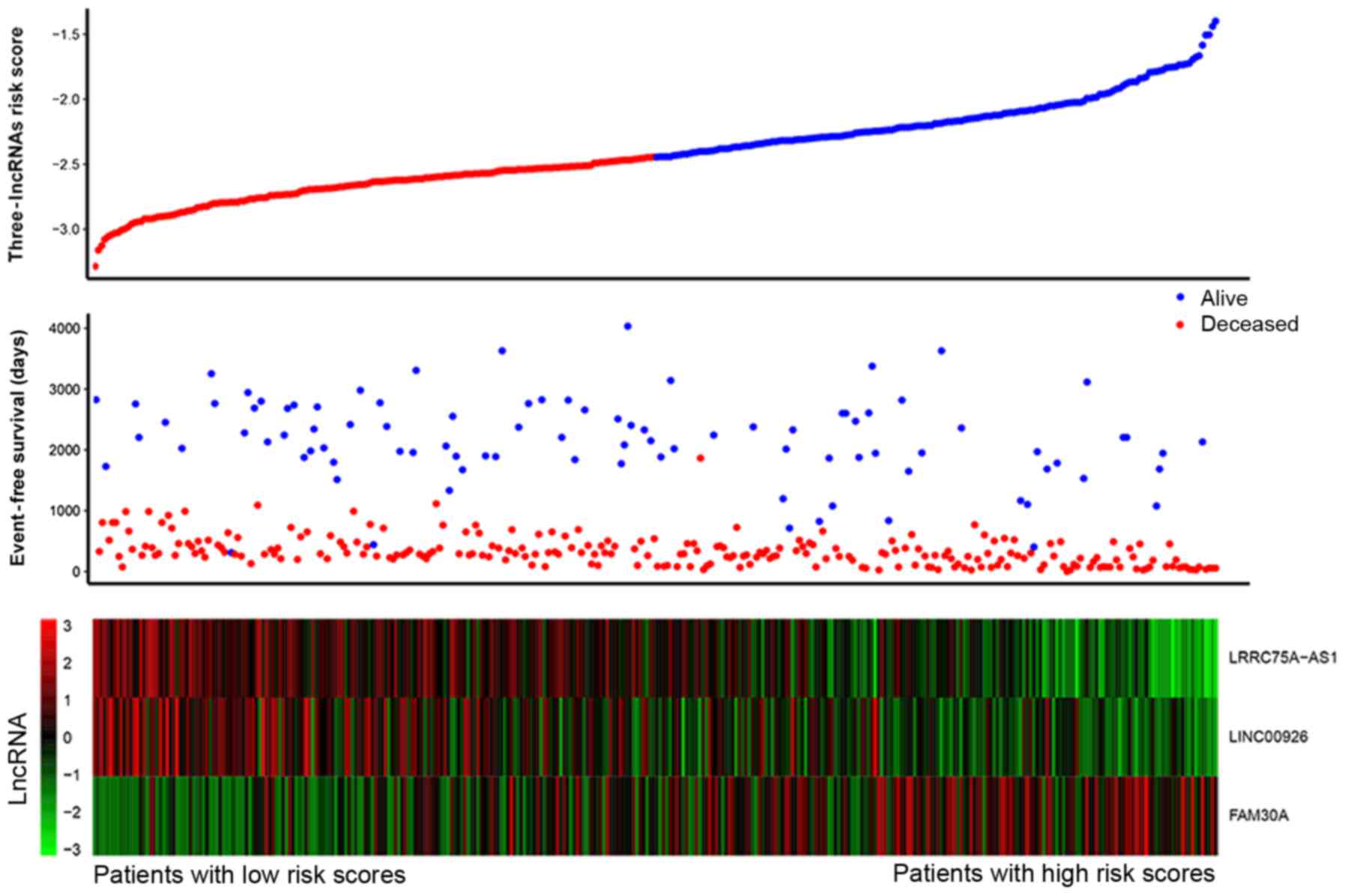
We constructed a formula according to the expression
level of three lncRNA for EFS outcome in 340 TARGET AML samples
using the multivariate Cox regression model as follows: (−0.2763 ×
expression level of LRRC75A-AS1) + (0.0822 × expression level of
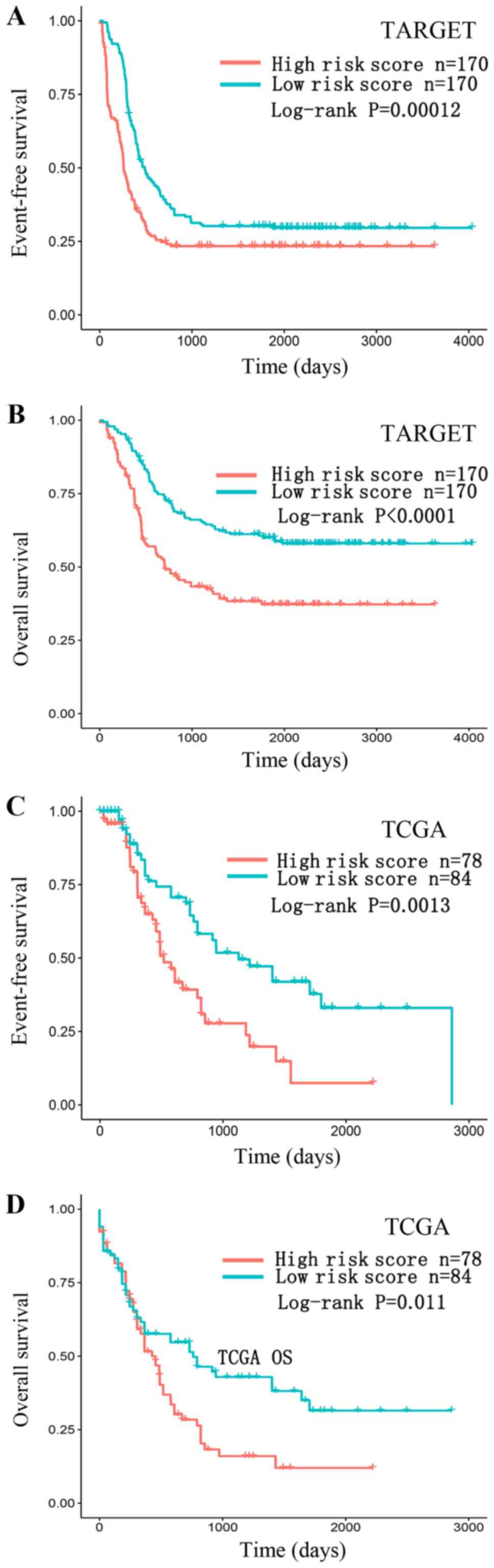
FAM30A) + (−0.1418 × expression level of LINC00926). Fig. 5 shows that patients with low-risk
scores tended to express high levels of protective lncRNAs
(LRRC75A-AS1, LINC00926), whereas patients with high-risk scores
tended to express high levels of risky lncRNA (FAM30A). Using the
median of risk score (−2.380) as the cut-off point, patients were
divided into a high-risk group (score >-2.380, N=170) and a
low-risk group (score ≤-2.380, N=170). We observed that AML
patients with high-risk scores had lower EFS rates (log-rank,
P=0.00012) and OS rates (log-rank, P<0.0001) compared with those
with low-risk scores. To validate our findings, we also classified
patients in the TCGA AML set into a high-risk group (N=78) and a
low-risk group (N=84) using the same cut-off value. Consistent with
the findings described above, patients in the high-risk group
suffered significantly poorer EFS (log-rank, P=0.0013) and OS
(log-rank, P=0.011) compared with those in the low-risk group
(Fig. 6).
A three-lncRNA signature is
independent of the cytogenetics risk group
Before evaluating whether the survival prediction
based on the three-lncRNA signature was independent of clinical
factors, the correlation between clinical factors and the patients'
EFS was examined using univariable Cox regression model in TCGA and
TARGET. As presented in Table II,
we found that only the cytogenetics risk group was closely
associated with EFS in all clinical factors. Additionally,
multivariate Cox regression analysis revealed that the three-lncRNA
signature and cytogenetics risk group were both independent factors
in prognosis prediction in the TCGA AML sets, TARGET AML sets and
merged sets as presented in Table
III.
| Table II.Univariable Cox regression of the
three-lncRNA risk score and clinical prognostic factors in TCGA and
TARGET AML data sets. |
Table II.
Univariable Cox regression of the
three-lncRNA risk score and clinical prognostic factors in TCGA and
TARGET AML data sets.
| A, TCGA set |
|---|
|
| Univariable cox
model |
|---|
|
|
|
|---|
| Variables | HR | 95% CI of HR | P-value |
|---|
| Three-lncRNA | 3.689 | 2.230–6.103 |
3.7×10−7 |
| Cytogenetics risk
group | 2.179 | 1.629–2.914 |
1.6×10−7 |
| Sex |
|
|
|
|
Female | 1.002 | 0.6799–1.477 | 0.991 |
| Male | 0.998 | 0.677–1.471 | 0.991 |
| Initial WBC | 1.0041 | 0.9997–1.009 | 0.068 |
| Bone marrow
leukemic blast cell percentage | 1.0007 | 0.9946–1.007 | 0.807 |
| FAB subgroup |
|
|
|
|
AML-M0 | 0.9728 | 0.5063–1.869 | 0.934 |
|
AML-M1 | 1.0244 | 0.634–1.655 | 0.921 |
|
AML-M2 | 0.9891 | 0.6171–1.586 | 0.964 |
|
AML-M4 | 1.0620 | 0.6757–1.669 | 0.794 |
|
AML-M5 | 1.5604 | 0.8858–2.749 | 0.124 |
|
AML-M6 | 2.4879 | 0.6072–10.19 | 0.205 |
|
AML-M7 | 0.7425 | 0.7582–7.677 | 0.136 |
| Not
classified | 2.8980 | 0.235–6.25 | 0.062 |
|
| B, TARGET
set |
|
|
| Univariable cox
model |
|
|
Variables | HR | 95% CI of
HR | P-value |
|
| Three-lncRNA | 2.6390 | 1.786–3.900 |
1.1×10−6 |
| Cytogenetics risk
group | 1.6510 | 1.387–1.965 |
1.7×10−8 |
| Sex |
|
|
|
|
Female | 1.1872 | 0.9247–1.524 | 0.178 |
|
Male | 0.8423 | 0.6561–1.081 | 0.178 |
| Initial WBC | 1.001 | 0.9997–1.002 | 0.131 |
| Bone marrow
leukemic blast cell percentage | 1.005 | 1.0001–1.0108 | 0.057 |
| FAB subgroup |
|
|
|
|
AML-M0 | 1.3073 | 0.48–3.805 | 0.269 |
|
AML-M1 | 0.6137 | 0.4026–1.2354 | 0.232 |
|
AML-M2 | 0.8256 | 0.6201–1.099 | 0.189 |
|
AML-M4 | 1.1479 | 0.8618–1.529 | 0.346 |
|
AML-M5 | 1.2127 | 0.8672–1.696 | 0.260 |
|
AML-M6 | 1.3316 | 0.549–3.23 | 0.526 |
|
AML-M7 | 0.7425 | 0.4329–1.274 | 0.279 |
|
NOS | 1.4514 | 0.846–2.49 | 0.176 |
| Not
classified | 1.0976 | 0.6144–1.961 | 0.753 |
| Table III.Multivariate Cox regression of
three-lncRNA risk score and cytogenetics risk status in TCGA,
TARGET and merged data sets. |
Table III.
Multivariate Cox regression of
three-lncRNA risk score and cytogenetics risk status in TCGA,
TARGET and merged data sets.
| A, TCGA set |
|---|
|
| Multivariable cox
model |
|---|
|
|
|
|---|
| Variables | HR | 95% CI of HR | P-value |
|---|
|
|---|
| Three-lncRNA | 2.503 | 1.423–4.403 | 0.0015 |
| Cytogenetics risk
group | 1.671 | 1.196–2.336 | 0.0026 |
|
| B, TARGET
set |
|
|
| Multivariable
cox model |
| B, TARGET
set |
|
Variables | HR | 95% CI of
HR | P-value |
|
|
| Three-lncRNA | 2.1371 | 1.438–3.176 | 0.00017 |
| Cytogenetics risk
group | 1.5318 | 1.282–1.830 |
2.6×10−6 |
|
| C, TCGA+TARGET
set |
|
|
| Multivariable
cox model |
|
|
Variables | HR | 95% CI of
HR | P-value |
|
|
| Three-lncRNA | 2.1591 | 1.539–3.028 |
8.2×10−6 |
| Cytogenetics risk
group | 1.3519 | 1.153–1.586 | 0.00021 |
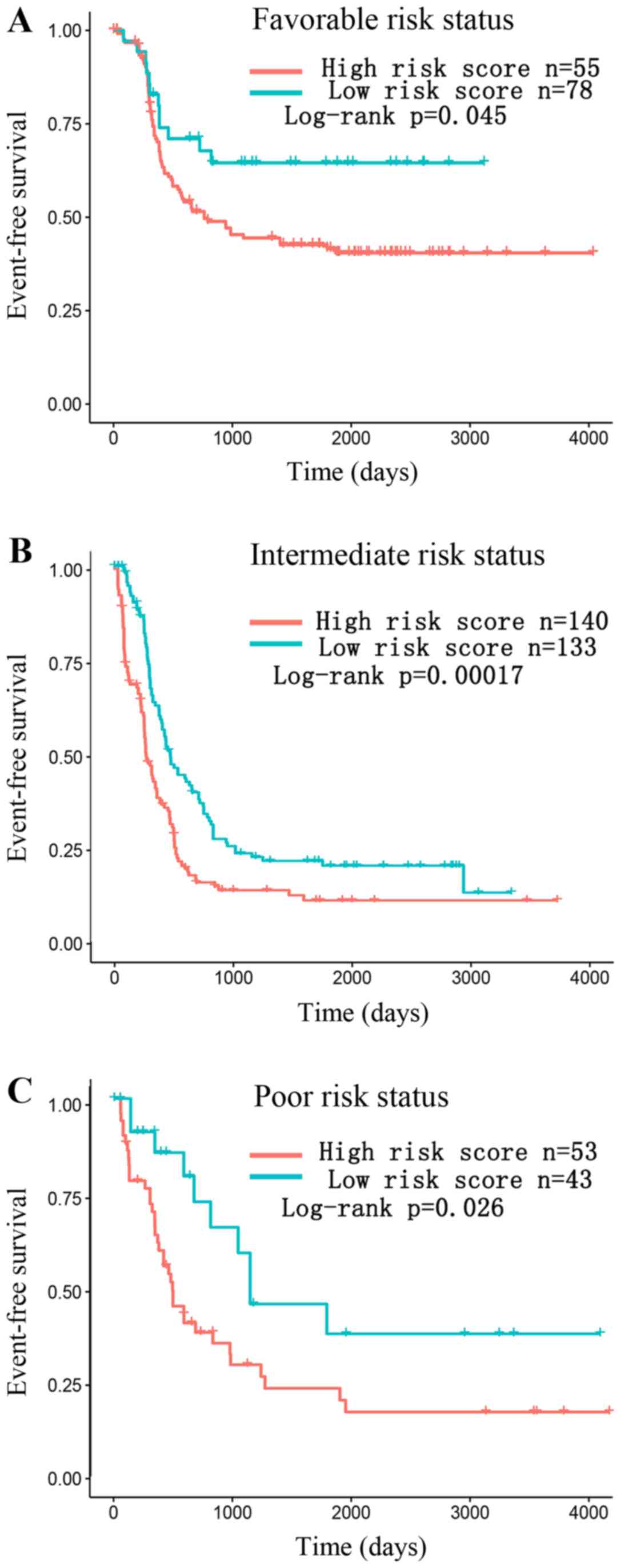
In the stratified analysis, the merged data set of
TCGA and TARGET was divided into favorable, intermediate and poor
group according to the cytogenetics risk group and the result
showed that the three-lncRNA risk score may further divide AML
patients into high-risk and low-risk subgroup within each group
(Fig. 7).
Evaluation of the three-lncRNA
signature performance by ROC curve analysis
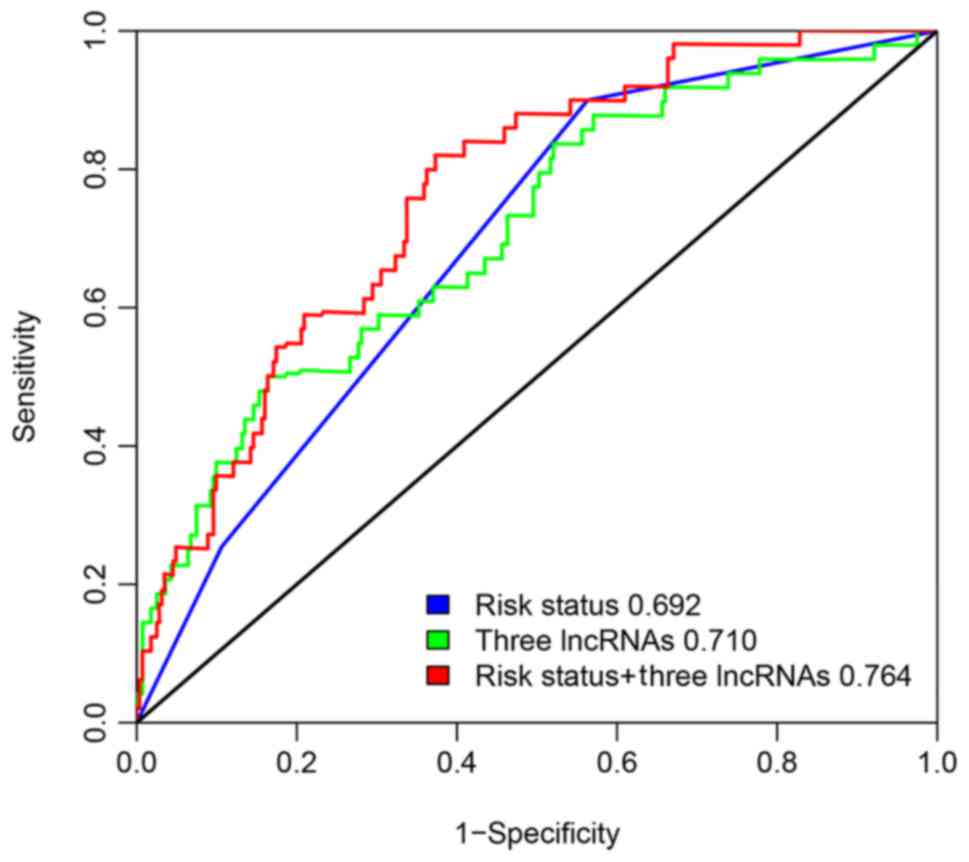
To compare the sensitivity and specificity of the
three-lncRNA signature in AML EFS outcome, time dependent ROC
curves analysis was performed for the three-lncRNA signature and
cytogenetics risk group. The AUROC was determined and compared
between these two prognostic factors for five-year EFS in the
merged set of TCGA and TARGET. Fig.
8 shows that the AUROC of the three-lncRNA signature was 0.710,
and the AUROC of the cytogenetics risk group was 0.692. There was
no significant difference between the AUROC of the three-lncRNA
risk score with risk status based on validated cytogenetics and
molecular abnormalities (P=0.8574). However, we observed that the
combination of lncRNA risk score and cytogenetics risk group was a
more effective prognostic prediction than each individually
(AUROC=0.764, P=0.0069).
LncRNAs target prediction and
potential regulation mechanisms
At present, the functions of most lncRNAs have not
yet been elucidated. To predict the potential target of the three
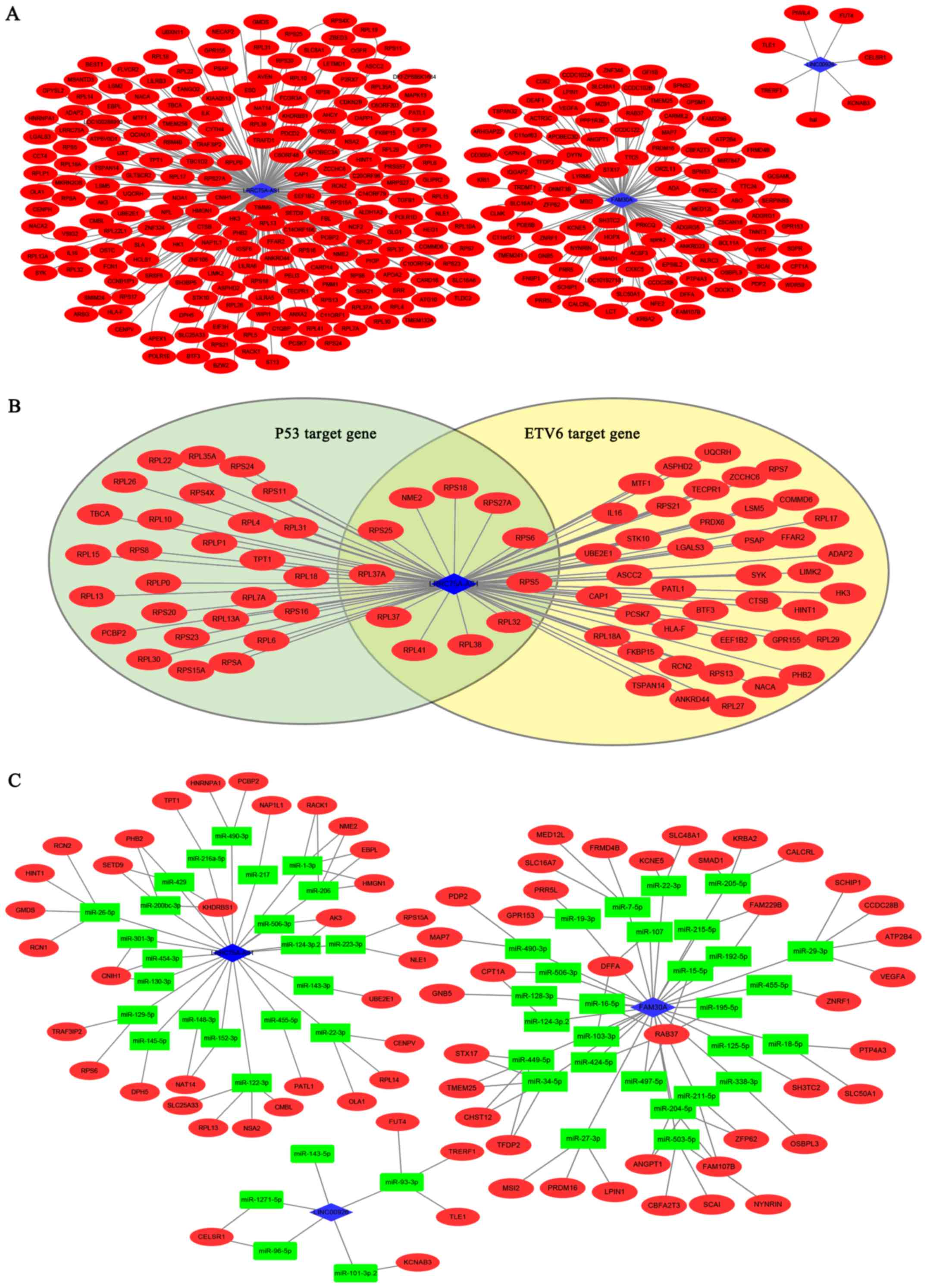
lncRNAs, we constructed a co-expression network based on the
Pearson correlation between the mRNAs and lncRNAs of the given
lncRNAs in both TARGET and TCGA projects (Fig. 9A). The red nodes in Fig. 8 represent the mRNAs and the blue
diamonds indicate the lncRNAs.
In the network, LRRC75A-AS1 regulated most mRNAs
(203 mRNAs), followed by FAM30A (99 mRNAs) and then LINC00926 (7
mRNAs). In the result of multivariate Cox regression, these three
lncRNAs were independent prognostic factors for AML. No common
target was found among the three lncRNAs in this network.
Based on the co-expression network, we explored the
potential regulatory mechanism of the lncRNA-mRNA pairs in
cis/trans/ceRNA regulation.
A nearby gene that is located <300 kb upstream or
downstream from the given lncRNA may be the potential cis target.
However, none of the three lncRNAs were significantly co-expressed
with a nearby gene (data not shown).
As for trans-regulation, it is reported that lncRNA
can interact with TFs thereby influencing the expression of the TF
target genes. We applied iRegulon to study whether the co-expressed
gene sets of the three lncRNAs were overlapping with TF target
genes. There were two significant TF enrichments of LRRC75A-AS1
co-expressed genes, TP53 and ETV6. The top scored TF for
LRRC75A-AS1 was TP53 with an NES of 7.270 with 38 direct targets,
followed by ETV6 (NES=6.473, 52 direct targets) (Fig. 9B). No significant TF enrichments of
FAM30A and LINC00926 were identified (Fig. 9B).
In the ceRNA hypothesis, lncRNAs can compete with
the mRNAs containing the same miRNA response elements that bind the
miRNAs, thereby influencing the expression of the miRNA target
genes. We constructed ceRNA networks based on lncRNA/miRNA and
miRNA/mRNA interactions. The lncRNA-mRNA pairs that had positively
correlated expression profiles and shared at least one common miRNA
target were selected for ceRNA network construction. We identified
33 mRNAs as the ceRNA of LRRC75A-AS1, 39 mRNAs as the ceRNA of
FAM30A and 6 mRNAs as the ceRNA of LINC00926 (Fig. 9C).
Discussion
AML is a highly heterogeneous hematopoietic
malignancy with a poor outcome. Despite the advances in the risk
classification for AML, some patients eventually relapse, even with
lack of adverse risk factors. This may result from undiscovered
prognostic factors which may refine disease classification. In this
study, we first identified the subtype-specific lncRNAs through
different expression analysis of clinical and genetic subtypes of
AML. Among the subtype-specific lncRNAs, HOTAIRM1 and HOXB-AS3 were
reported in previous studies to be upregulated in NPM1-mutated AML,
in addition to WT1-AS in AML with Flt3-itd (7,15).
Following exclusion of the subtype-specific lncRNAs,
the present study investigated the associations between lncRNA
expression and clinical characters and identified three-lncRNAs
(LINC00926, FAM30A, LRRC75A-AS1) significantly associated with AML
patient survival in the training set (TARGET). A prognostic risk
score based on the three-lncRNA expression signature was
constructed and was efficiently able to divide patients into a
different prognostic subgroup. The model was further validated in
the testing set (TCGA). It was then confirmed to be an independent
prognostic predictor for patients with AML. ROC analysis result
showed that the three-lncRNA risk score was competitive for
survival prediction. Furthermore, the combination of the
three-lncRNA expression signature and the cytogenetics risk group
was more informative than each of them individually.
AML patients in TARGET were treated in three
clinical trials: CCG2961 (NCT00003790) (n=45); AAML03P1
(NCT00070174) (n=57); AAML0531 (NCT01407757) (n=238) (NCT01723657).
Briefly, the induction regimen of CCG2961 was 5-drug combination
chemotherapy consisting of dexamethasone, cytarabine, thioguanine,
etoposide, and rubidomycin (daunomycin) (DCTER) or IdaDCTER which
replaced rubidomycin with IDA at 4:1 ratio in the 5-drug
combination while in AAML03P1 and AAML0531, it consisted of
standard chemotherapy with or without gemtuzumab ozogamicin.
Patients with an unfavorable lncRNA score had lower CR rates in
AAML03P1 (P=0.0022, OR=0.0952) and AAML0531 (P=5.375e-11,
OR=0.1422). There was no significant different CR rates in CCG2961
in the comparison between favorable and unfavorable patients
(P=0.6641, OR=0.6933).
Among the TCGA AML patients, therapeutic regimen
consisted of a seven-day continuous infusion of cytarabine plus 3
days of an anthracycline (7+3) (n=58) contained the enough patients
for analysis of CR rate and no significant difference in the
comparison between favorable and unfavorable patients was observed
(P=0.6006, OR=0.7563).
It was reported that lncRNAs function through cis,
trans and ceRNA regulatory mechanisms to impact the transcription
of protein-coding genes (8,16).
We identified two significant TF enrichments of LRRC75A-AS1
co-expressed genes, TP53 and ETV6.
TP53 is a well-known tumor suppressor participating
in the DNA damage response. TP53-deficient hematopoietic stem cells
exhibited high levels of apoptosis and subsequently induction of
cancer development under conditions of DNA damage. In a recent
study, AML patients with TP53 mutations were defined as an
additional AML subgroup and exhibited a worse prognosis (17). The ETV6 gene encodes a
transcriptional repressor that plays a critical role in
hematopoiesis. Loss of function of ETV6 resulting from mutations or
deregulated expression can contribute to leukemogenesis (18).
According to the ceRNA hypothesis, lncRNA acts as a
molecular sponge for miRNA thereby regulates the target genes of
the miRNA. We constructed the ceRNA network based on the
co-expression network. VEGFA was a VEGF family member that was
associated with poor prognosis in AML through VEGFA signaling
regulating both normal hematopoiesis and AML (19,20).
The lncRNA FAM30A may promote VEGFA expression as a ceRNA targeting
miR-29-3p.
Previous works have reported some prognostic lncRNAs
in AML, such as CRNDE (6),
LINC01268 (LOC285758) (21),
HOTAIRM1 (15,22), H19 (23). LINC01268 was presented in 25
potential prognostic factors which was significantly correlated
with the patients' EFS (Fig. 4).
CRNDE and HOTAIRM1 showed a subtype specific expression pattern
that they might be more suitable for disease classification than
prognosis. H19 was reported as an independent prognostic marker and
significantly upregulated in patients with AML-M2. The reason why
H19 cannot be validated in TARGET database might be the different
between populations and ages. The AML patients in TARGET database
was based on pediatric Hispanic or Latino, whereas the AML patients
in work of Zhao et al (23)
were Chinese adult.
Limitation of this study should be acknowledged.
First, to make more reliable result for prognosis, we filtered the
low expression lncRNAs by RPKM>1 and read count >200 in
>50% patients. Thus, various low expression lncRNAs were
excluded which might be potentially correlated with AML patients'
prognosis. Although TCGA and TARGET projects were both large and
comprehensive cancer genomics datasets, the patients were pediatric
from TARGET and adult from TCGA. It also revealed that the
three-lncRNA risk score was a robust model for survival prediction.
Even through the three lncRNAs signature was verified as a
prognostic biomarker in TCGA and TARGET project. Experimental
studies such as clinical trials were still necessary to avoid the
possibilities of false positives. A series of experiments were
needed to uncover the lncRNA function and mechanism in cancer.
In conclusion, our data demonstrated that risk score
based on three lncRNA expression levels was an independent
prognostic biomarker that refined clinical classification. Further
multivariate Cox regression analysis has shown that the
three-lncRNA signature was an independent prognostic factor.
Furthermore, LRRC75A-AS1 may regulate TP53 and ETV6 target genes in
trans manner. Potential mRNA-lncRNA pairs were identified as ceRNA
based on co-expression network. Practically, the combination of our
three-lncRNA signature and cytogenetic risk group achieved a
significant improvement in AML patient prognostic prediction
compared with either alone.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Ministry of
Science and Technology of China (grant no. 2016YEE0107200), the
National Natural Science Foundation of China (grant no. 81400111)
and the Science and Technology Commission of Shanghai Municipality
(grant nos. 15411968900 and 15XD1503300).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WZ and AL conceived and designed the present study.
FW and XT performed the statistical analyses and drafted the
manuscript. JZ, GW, WY, ZL and ZF performed the experiments and
drafted the manuscript. The final version of the manuscript was
read and approved by all authors.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Döhner H, Estey EH, Amadori S, Appelbaum
FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson
RA, et al: Diagnosis and management of acute myeloid leukemia in
adults: Recommendations from an international expert panel, on
behalf of the European Leukemia Net. Blood. 115:453–474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sill H, Olipitz W, Zebisch A, Schulz E and
Wolfler A: Therapy-related myeloid neoplasms: Pathobiology and
clinical characteristics. Br J Pharmacol. 162:792–805. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Grimwade D, Hills RK, Moorman AV, Walker
H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ and Burnett
AK: Refinement of cytogenetic classification in acute myeloid
leukemia: Determination of prognostic significance of rare
recurring chromosomal abnormalities among 5876 younger adult
patients treated in the United Kingdom Medical Research Council
trials. Blood. 116:354–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Röllig C, Bornhäuser M, Thiede C, Taube F,
Kramer M, Mohr B, Aulitzky W, Bodenstein H, Tischler HJ, Stuhlmann
R, et al: Long-term prognosis of acute myeloid leukemia according
to the new genetic risk classification of the European LeukemiaNet
recommendations: Evaluation of the proposed reporting system. J
Clin Oncol. 29:2758–2765. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Zhou Q and Ma JJ: High expression
of lnc-CRNDE presents as a biomarker for acute myeloid leukemia and
promotes the malignant progression in acute myeloid leukemia cell
line U937. Eur Rev Med Pharmacol Sci. 22:763–770. 2018.PubMed/NCBI
|
|
7
|
Garzon R, Volinia S, Papaioannou D,
Nicolet D, Kohlschmidt J, Yan PS, Mrózek K, Bucci D, Carroll AJ,
Baer MR, et al: Expression and prognostic impact of lncRNAs in
acute myeloid leukemia. Proc Natl Acad Sci USA. 111:18679–18684.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kornienko AE, Guenzl PM, Barlow DP and
Pauler FM: Gene regulation by the act of long non-coding RNA
transcription. BMC Biol. 11:592013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Anders S and Huber W: Differential
expression analysis for sequence count data. Genome Biol.
11:R1062010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ishwaran H and Kogalur UB: Consistency of
random survival forests. Stat Probab Lett. 80:1056–1064. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heagerty PJ, Lumley T and Pepe MS:
Time-dependent ROC curves for censored survival data and a
diagnostic marker. Biometrics. 56:337–344. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Blanche P, Dartigues JF and Jacqmin-Gadda
H: Estimating and comparing time-dependent areas under receiver
operating characteristic curves for censored event times with
competing risks. Stat Med. 32:5381–5397. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Janky R, Verfaillie A, Imrichova H,
Imrichová H, Van de Sande B, Standaert L, Christiaens V, Hulselmans
G, Herten K, Sanchez Naval M, et al: iRegulon: From a gene list to
a gene regulatory network using large motif and track collections.
PLoS Comput Biol. 10:e10037312014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Su Z, Fang H, Hong H, Shi L, Zhang W,
Zhang W, Zhang Y, Dong Z, Lancashire LJ, Bessarabova M, et al: An
investigation of biomarkers derived from legacy microarray data for
their utility in the RNA-seq era. Genome Biol. 15:5232014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Díaz-Beyá M, Brunet S, Nomdedéu J,
Pratcorona M, Cordeiro A, Gallardo D, Escoda L, Tormo M, Heras I,
Ribera JM, et al: The lincRNA HOTAIRM1, located in the HOXA genomic
region, is expressed in acute myeloid leukemia, impacts prognosis
in patients in the intermediate-risk cytogenetic category, and is
associated with a distinctive microRNA signature. Oncotarget.
6:31613–31627. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cesana M, Cacchiarelli D, Legnini I,
Santini T, Sthandier O, Chinappi M, Tramontano A and Bozzoni I: A
long noncoding RNA controls muscle differentiation by functioning
as a competing endogenous RNA. Cell. 147:358–369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Taylor J, Xiao W and Abdel-Wahab O:
Diagnosis and classification of hematologic malignancies on the
basis of genetics. Blood. 130:410–423. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Waalwijk van Doorn-Khosrovani
Barjesteh S, Spensberger D, de Knegt Y, Tang M, Löwenberg B and
Delwel R: Somatic heterozygous mutations in ETV6 (TEL) and frequent
absence of ETV6 protein in acute myeloid leukemia. Oncogene.
24:4129–4137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Santos SC and Dias S: Internal and
external autocrine VEGF/KDR loops regulate survival of subsets of
acute leukemia through distinct signaling pathways. Blood.
103:3883–3889. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim DH, Lee NY, Lee MH, Sohn SK, Do YR and
Park JY: Vascular endothelial growth factor (VEGF) gene (VEGFA)
polymorphism can predict the prognosis in acute myeloid leukaemia
patients. Br J Haematol. 140:71–79. 2008.PubMed/NCBI
|
|
21
|
Lei L, Xia S, Liu D, Li X, Feng J, Zhu Y,
Hu J, Xia L, Guo L, Chen F, et al: Genome-wide characterization of
lncRNAs in acute myeloid leukemia. Brief Bioinform. Feb
15–2017.(Epub ahead of print]).
|
|
22
|
Chen ZH, Wang WT, Huang W, Fang K, Sun YM,
Liu SR, Luo XQ and Chen YQ: The lncRNA HOTAIRM1 regulates the
degradation of PML-RARA oncoprotein and myeloid cell
differentiation by enhancing the autophagy pathway. Cell Death
Differ. 24:212–224. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao TF, Jia HZ, Zhang ZZ, Zhao XS, Zou
YF, Zhang W, Wan J and Chen XF: LncRNA H19 regulates ID2 expression
through competitive binding to hsa-miR-19a/b in acute myelocytic
leukemia. Mol Med Rep. 16:3687–3693. 2017. View Article : Google Scholar : PubMed/NCBI
|