Introduction
Aniridia (MIM #106210) is a congenital
disorder of complete or partial iris hypoplasia (1,2). The
prevalence of aniridia ranges from 1:50,000 to 1:100,000 live
births (3). Aniridia can occur as
an isolated or a syndromic form (4). Approximately two thirds of all cases
are familial following an autosomal dominant mode of inheritance
with high penetrance, while the remaining cases are sporadic
(3,5).
Classic aniridia is a panocular condition caused by
PAX6 heterozygous mutations and it affects the iris, cornea,
lens, retina and optic nerve. It can be accompanied by foveal
hypoplasia, strabismus and optic nerve hypoplasia, generally
leading to impaired visual acuity, while late-onset manifestations
can include nystagmus, glaucoma, cataract and corneal pannus
(5,6). Patients may also display non-ocular
sensory and neurological abnormalities, such as reduced olfaction
and hearing difficulties (7), and
a range of neuroanatomical abnormalities, including hypoplasia of
the anterior commissure, the pineal glands and the optic chiasm
(8,9). PAX6 point mutations are
responsible for classic aniridia in approximately 90% of patients
(10,11). PAX6 maps to chromosomal
region 11p13 and encodes a transcription factor, which plays a
crucial role in early ocular morphogenesis. It is also involved in
the development of the central nervous system, gut and pancreas
(12,13). Furthermore, deletions at 11p13
involving PAX6 or the regulatory region upstream of
PAX6 leaving its coding region intact are thought to be rare
causes of classic aniridia (14).
A small number of patients develop aniridia as part of the WAGR
syndrome (Wilms tumor, Aniridia, Genital anomalies, mental
Retardation) caused by a contiguous gene deletion encompassing
PAX6 and WT1 (4).
Finally, about 10% of cases display aniridia-like phenotypes that
result from mutations in other genes, such as FOXC1 and
PITX2 (15–17). The purpose of this study was to
analyze the PAX6 gene in a group of aniridia patients of
Cypriot ancestry and describe their clinical features. We
identified three previously reported PAX6 mutations, in
addition to a novel frameshift variant that was identified in one
of our familial cases.
Materials and methods
Patients
A total of 17 affected individuals from six families
were evaluated with a complete ophthalmological examination and
then referred to the Clinical Genetics Clinic at the Cyprus
Institute of Neurology and Genetics to investigate for PAX6
mutations. Informed consent was obtained by study participants or
their guardians if they were younger than 18 year olds.
Array-comparative genomic
hybridization (CGH)
Array CGH analysis or multiplex ligation-dependent
probe amplification assay (MLPA) were initially performed to
investigate for whole gene deletions involving PAX6 and/or
WT1 or deletions downstream of PAX6. Array-CGH was
performed using the Cytochip ISCA array (version 1.0; BlueGnome,
Cambridge, UK) with 180,000 oligos in a 4×180 k format. Fluorescent
ratios were calculated using the Blue Fuse Multi Software (version
4.2; BlueGnome). MLPA was conducted using the SALSA probemix
P219-B3 (MRC-Holland, Amsterdam, Netherlands). Fragment separation
by capillary electrophoresis was performed on an ABI 3130×l genetic
analyzer. MLPA analysis was performed using the Coffalyser.Net
Software (version 1.4; MRC-Holland).
Mutation screening
Genomic DNA was isolated from peripheral blood using
the QIAamp DNA Blood Midi Kit (Qiagen, Inc., Valencia, CA, USA).
All coding exons of PAX6, FOXC1 and PITX2 were
amplified by PCR using primers designed with Primer3 (http://frodo.wi.mit.edu/) and they are available upon
request. Sequencing products were analyzed by a 3130×l Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The cDNA sequence of the most common human PAX6
transcript (NM_000280) is used for the variant nomenclature. Direct
sequencing of PITX2 and FOXC1 was performed in
patients who did not carry any PAX6 mutations only.
Results
Seventeen patients from six families were recruited
for this study, including eight males and nine females. One patient
was a sporadic case while the remaining were familial ones.
Patients from family 1 presented with isolated bilateral aniridia.
The proband of family 2 presented with bilateral aniridia,
cataracts, glaucoma of the left eye, nystagmus and reduced visual
acuity. She also had pseudophakia of the left eye. Fundoscopy
revealed small, hypoplastic discs, macular hypoplasia, mild foveal
hypoplasia and optic nerve hypoplasia. Her mother had aniridia with
loss of vision in the right eye and developed retinal detachment in
the left. The proband of family 3 had bilateral aniridia,
nystagmus, cataracts, glaucoma and photophobia. She also had a
history of osteoporosis and hypercholesterolemia. Her mother had
relapsing remitting multiple sclerosis in addition to aniridia. The
proband of family 4 had bilateral aniridia, atrial septal defect
and thyroid nodules. Patients from family 5 exhibited bilateral
aniridia and cataracts. Finally, our sporadic patient from family 6
had bilateral aniridia, nystagmus, cataracts and glaucoma. He had a
history of Peters anomaly and he showed markedly abnormal anterior
segment. Non-ocular abnormalities included antenatal bilateral
hydronephrosis, vesicoureteric obstruction, bilateral megaureter,
bipolar disorder and hypertension. His parents were thought to be
distantly related. All probands were negative for deletions
spanning PAX6 and/or WT1, excluding WAGR
syndrome.
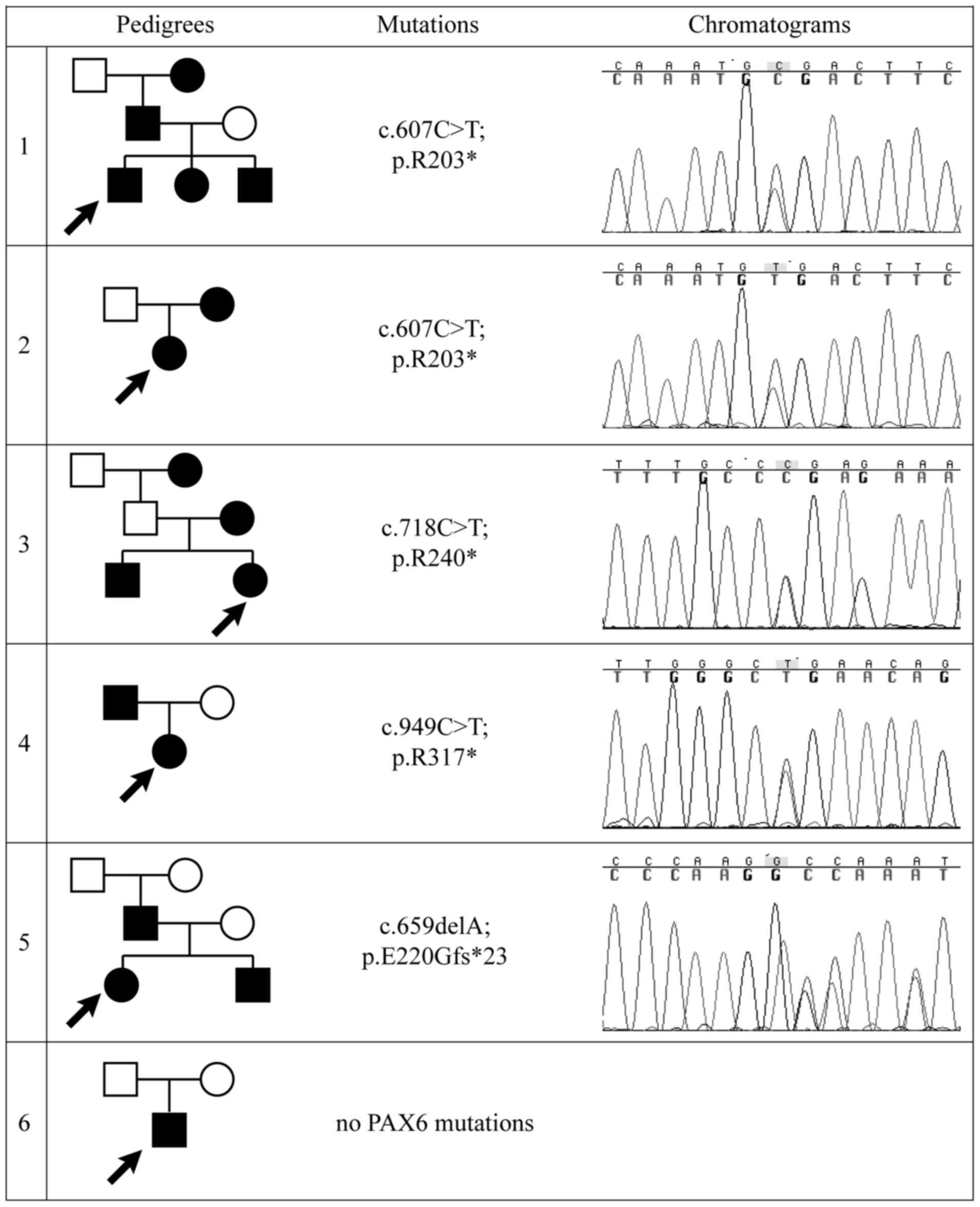
Sequencing results
Four different heterozygous PAX6 mutations
were identified in five out of six families with aniridia, yielding
a diagnostic rate of approximately 84%. Three stop-gain mutations
identified in four familial cases (p.R203*, p.R240* and p.R317*)
have been reported elsewhere, while the frameshift mutation
c.659delA (p.E220Gfs*23) is novel and was found to occur de
novo in the affected father (Fig.
1). Mutations were located in exons 8, 9 and 11. All mutations
are summarized in Table I.
 | Table I.Summary of PAX6 mutational
spectrum in Cypriot families. |
Table I.
Summary of PAX6 mutational
spectrum in Cypriot families.
| Protein change | Times reported in
LOVD | Exon | Protein domain | Predicted
effect |
|---|
| p.R203* | 40 | 8 | Linker domain | NMD |
| p.E220Gfs*23 | 0 | 8 | Linker domain | NMD |
| p.R240* | 51 | 9 | Homeodomain | NMD |
| p.R317* | 41 | 11 | PST domain | NMD |
Direct sequencing of the coding exons and flanking
intronic sites of PITX2 and FOXC1 in the proband of
family 5 revealed no pathogenic mutations.
Discussion
In this study, we analyzed 17 patients from six
different Cypriot families with aniridia and identified five
different PAX6 pathogenic variants in five out of six
probands, yielding a diagnostic rate of 84% that is comparable to
other studies in other populations (18–21).
This is the first study on PAX6 molecular analysis in
aniridia patients of Cypriot ancestry. Although, deletions spanning
the 3′regulatory region of PAX6 or the PAX6 coding
region are less common than PAX6 mutations in aniridia
patients, we have performed array-CGH or MLPA first as it was
recommended by Hingorani et al due to the clinical
importance of detecting WT1 deletions, which requires
surveillance for Wilms tumor (7).
None of our patients carried a deletion and therefore PAX6
mutation screening was subsequently performed.
To date, 472 unique PAX6 variants have been
recorded in the Human PAX6 Allelic Variant Database
(http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6).
Over 50% of these variants comprise frameshift or stop-gain
variants resulting in a premature termination codon, which usually
leads to the degradation of the truncated mRNA via
nonsense-mediated decay. Therefore, these mutations result in 50%
reduction in protein levels supporting haploinsufficiency as the
main mechanism underlying aniridia (22,23).
All of the identified variants in our study, as well, are predicted
to lead to nonsense-mediated decay. Three of the identified
PAX6 variants in our study account for 16% of patients
included in the PAX6 mutation database (http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6).
The first identified mutation (p.R203*) is currently reported in 40
patients, the second one (p.R240*) is currently reported in 51
patients and the third one (p.R317*) is currently reported in 41
patients. These are the most recurrent mutations found in
PAX6 in aniridia patients to date.
The PAX6 protein consists of a paired domain at the
N-terminus, a homeodomain and a proline-serine-threonine rich
transactivation (PST) domain at the C-terminus (24–26).
In our study, mutations were found in exons encoding the linker
domain (p.R203* and p.E220Gfs*23), the homeodomain (p.R240*) and
the PST domain (p.R317*). No correlation between the location of
the mutation and the associated phenotypes was observed. Even
though all patients had truncating mutations, not all patients had
cataract, glaucoma, nystagmus or foveal hypoplasia. In addition,
the impairment of visual acuity varied between patients carrying
the same PAX6 mutation, as previously observed in other
studies (27). The commonest
additional ocular features seen in our group of patients were
nystagmus, cataracts and glaucoma.
However, no copy number variants or mutations were
detected in PAX6, FOXC1 or PITX2 in the proband of
family 6 with a sporadic form of classical aniridia. Although,
classical aniridia is primarily caused by PAX6 mutations,
its phenotypic presentation may overlap with aniridia-like
phenotypes. The abnormal anterior segment seen in our patient
prompted us to directly sequence FOXC1 and PITX2,
because mutations in these genes are more commonly associated with
anterior segment dysgenesis even though they can also cause
isolated aniridia. FOXC1 mutations are more commonly
associated with isolated ocular, heart and/or hearing defects and
PITX2 are more commonly associated with ocular, dental and
umbilical anomalies. However, both genes account for approximately
40% of cases with Axenfeld-Rieger syndrome only (28,29).
Therefore, our molecular findings confirm the genetic heterogeneity
that underlies aniridia-like phenotypes as other recent reports
have suggested (19,30).
In conclusion, a high diagnostic yield (84%) was
obtained in this study, which was the first one to be conducted in
Cyprus for aniridia patients. We have identified a novel frameshift
mutation in one of our families thus expanding the number of
PAX6 mutations that cause aniridia. Mutation screening of
PAX6 plays a crucial role in determining whether the
affected individual has isolated aniridia or WAGR syndrome. The
identification of a PAX6 mutation eliminates the need for
surveillance by renal ultrasound and improves genetic counseling as
well as the accuracy of prognosis and recurrence risk.
Acknowledgements
The authors would like to thank Dr Irene Savvidou
and Neophyta Pantelidou (both working at the Archbishop Makarios
III Hospital, Cyprus) for their help gathering the clinical
information and all of the families who participated in the
study.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AS performed mutation screening of the patients,
analyzed the results and prepared the manuscript. NN also performed
mutation screening of the patients and was a major contributor in
writing and editing the manuscript. AA and IP performed array-CGH
or MLPA in the participants. MN supervised and assisted AS with the
Sanger sequencing and the interpretation of the results. EL was the
ophthalmologist who contributed clinical data regarding the
patients. CS supervised the performance of the array-CGH and MLPA,
contributed to the interpretation of the results and provided the
sequencing facility. SM contributed to the design of this project,
the analysis and interpretation of the data and he ensured that the
questions related to the accuracy of this work were appropriately
investigated and resolved. VCA and GAT were the pediatricians and
clinical geneticists who examined the families and requested
genetic testing, they each contributed to the conception of the
research project and ensured that questions related to the study
were adequately and accurately resolved. GAT was also the principal
investigator of the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Participants of this study underwent PAX6
screening as part of the diagnostic workup that was performed at
the Cyprus Institute of Neurology and Genetics to facilitate the
clinical diagnosis, an application to the Cyprus National Bioethics
Committee was not necessary for this. Written informed consent for
participation in the study was obtained from all participants or
their legal guardians prior to their inclusion.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nelson LB, Spaeth GL, Nowinski TS, Margo
CE and Jackson L: Aniridia. A review. Surv Ophthalmol. 28:621–642.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hingorani M and Moore A:
AniridiaGeneReviews® [Internet]. Pagon RA, Adam MP,
Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N,
Mefford HC, Smith RJH and Stephens K: University of Washington
Seattle; WA: 1993
|
|
3
|
Grønskov K, Olsen JH, Sand A, Pedersen W,
Carlsen N, Jylling Bak AM, Lyngbye T, Brøndum-Nielsen K and
Rosenberg T: Population-based risk estimates of Wilms tumor in
sporadic aniridia. A comprehensive mutation screening procedure of
PAX6 identifies 80% of mutations in aniridia. Hum Genet. 109:11–18.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fischbach BV, Trout KL, Lewis J, Luis CA
and Sika M: WAGR syndrome: A clinical review of 54 cases.
Pediatrics. 116:984–988. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Valenzuela A and Cline RA: Ocular and
nonocular findings in patients with aniridia. Can J Ophthalmol.
39:632–638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee H, Khan R and O'Keefe M: Aniridia:
Current pathology and management. Acta Ophthalmol. 86:708–715.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hingorani M, Hanson I and van Heyningen V:
Aniridia. Eur J Hum Genet. 20:1011–1017. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sisodiya SM, Free SL, Williamson KA,
Mitchell TN, Willis C, Stevens JM, Kendall BE, Shorvon SD, Hanson
IM, Moore AT and van Heyningen V: PAX6 haploinsufficiency causes
cerebral malformation and olfactory dysfunction in humans. Nat
Genet. 28:214–216. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grant MK, Bobilev AM, Pierce JE, DeWitte J
and Lauderdale JD: Structural brain abnormalities in 12 persons
with aniridia. Version 2. F1000Res. 6:2552017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jordan T, Hanson I, Zaletayev D, Hodgson
S, Prosser J, Seawright A, Hastie N and van Heyningen V: The human
PAX6 gene is mutated in two patients with aniridia. Nat Genet.
1:328–332. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robinson DO, Howarth RJ, Williamson KA,
van Heyningen V, Beal SJ and Crolla JA: Genetic analysis of
chromosome 11p13 and the PAX6 gene in a series of 125 cases
referred with aniridia. Am J Med Genet A. 146A:558–569. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simpson TI and Price DJ: Pax6; a
pleiotropic player in development. Bioessays. 24:1041–1051. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van Heyningen V and Williamson KA: PAX6 in
sensory development. Hum Mol Genet. 11:1161–1167. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Crolla JA and van Heyningen V: Frequent
chromosome aberrations revealed by molecular cytogenetic studies in
patients with aniridia. Am J Hum Genet. 71:1138–1149. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Perveen R, Lloyd IC, Clayton-Smith J,
Churchill A, van Heyningen V, Hanson I, Taylor D, McKeown C, Super
M, Kerr B, et al: Phenotypic variability and asymmetry of Rieger
syndrome associated with PITX2 mutations. Invest Ophthalmol Vis
Sci. 41:2456–2460. 2000.PubMed/NCBI
|
|
16
|
Khan AO, Aldahmesh MA and Al-Amri A:
Heterozygous FOXC1 mutation (M161K) associated with congenital
glaucoma and aniridia in an infant and a milder phenotype in her
mother. Ophthalmic Genet. 29:67–71. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ito YA, Footz TK, Berry FB, Mirzayans F,
Yu M, Khan AO and Walter MA: Severe molecular defects of a novel
FOXC1 W152G mutation result in aniridia. Invest Ophthalmol Vis Sci.
50:3573–3579. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bobilev AM, McDougal ME, Taylor WL,
Geisert EE, Netland PA and Lauderdale JD: Assessment of PAX6
alleles in 66 families with aniridia. Clin Genet. 89:669–677. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Primignani P, Allegrini D, Manfredini E,
Romitti L, Mauri L, Patrosso MC, Veniani E, Franzoni A, Del Longo
A, Gesu GP, et al: Screening of PAX6 gene in Italian congenital
aniridia patients revealed four novel mutations. Ophthalmic Genet.
37:307–313. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pérez-Solórzano S, Chacón-Camacho OF,
Astiazarán MC, Ledesma-Gil G and Zenteno JC: PAX6 allelic
heterogeneity in Mexican congenital aniridia patients: Expanding
the mutational spectrum with seven novel pathogenic variants. Clin
Exp Ophthalmol. 45:875–883. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vasilyeva TA, Voskresenskaya AA,
Käsmann-Kellner B, Khlebnikova OV, Pozdeyeva NA, Bayazutdinova GM,
Kutsev SI, Ginter EK, Semina EV, Marakhonov AV and Zinchenko RA:
Molecular analysis of patients with aniridia in Russian Federation
broadens the spectrum of PAX6 mutations. Clin Genet. 92:639–644.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vincent MC, Pujo AL, Olivier D and Calvas
P: Screening for PAX6 gene mutations is consistent with
haploinsufficiency as the main mechanism leading to various ocular
defects. Eur J Hum Genet. 11:163–169. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bhuvanagiri M, Schlitter AM, Hentze MW and
Kulozik AE: NMD: RNA biology meets human genetic medicine. Biochem
J. 430:365–377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Glaser T, Jepeal L, Edwards JG, Young SR,
Favor J and Maas RL: PAX6 gene dosage effect in a family with
congenital cataracts, aniridia, anophthalmia and central nervous
system defects. Nat Genet. 7:463–471. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wilson DS, Guenther B, Desplan C and
Kuriyan J: High resolution crystal structure of a paired (Pax)
class cooperative homeodomain dimer on DNA. Cell. 82:709–719. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu HE, Rould MA, Xu W, Epstein JA, Maas RL
and Pabo CO: Crystal structure of the human Pax6 paired domain-DNA
complex reveals specific roles for the linker region and
carboxy-terminal subdomain in DNA binding. Genes Dev. 13:1263–1275.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yokoi T, Nishina S, Fukami M, Ogata T,
Hosono K, Hotta Y and Azuma N: Genotype-phenotype correlation of
PAX6 gene mutations in aniridia. Hum Genome Var. 3:150522016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Reis LM, Tyler RC, Kloss Volkmann BA,
Schilter KF, Levin AV, Lowry RB, Zwijnenburg PJ, Stroh E, Broeckel
U, Murray JC and Semina EV: PITX2 and FOXC1 spectrum of mutations
in ocular syndromes. Eur J Hum Genet. 20:1224–1233. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Samant M, Chauhan BK, Lathrop KL and
Nischal KK: Congenital aniridia: Etiology, manifestations and
management. Exp Rev Ophthalmol. 11:135–144. 2016. View Article : Google Scholar
|
|
30
|
Ansari M, Rainger J, Hanson IM, Williamson
KA, Sharkey F, Harewood L, Sandilands A, Clayton-Smith J, Dollfus
H, Bitoun P, et al: Genetic analysis of ‘PAX6-negative’ individuals
with aniridia or gillespie syndrome. PLoS One. 11:e01537572016.
View Article : Google Scholar : PubMed/NCBI
|