Introduction
Activating transcription factor 4 (ATF4) is a
transcription factor that belongs to the C/EBP transcription factor
family that binds the cAMP response element (CRE) (1,2).
ATF4 is a master transcription factor for which temporal expression
and activity are under tight cellular control. The translation of
ATF4 is regulated by eukaryotic translation initiation factor 2α
(eIF2α) (3). Under normal
conditions, ATF4 protein is quickly degraded by the proteasome
contributing to its short half-life. Under stress conditions, the
phosphorylation of eIF2α leads to general inhibition of
translation, but it results in translational upregulation of
specific mRNAs including ATF4 (4,5).
ATF4 is involved in the regulation of many
biological processes including cellular amino acid metabolism,
osteoblast differentiation, and the oxidative stress response
(2,6–8).
In vivo evidence has shown that ATF4 plays an important role
in glucose metabolism, insulin sensitivity, and lipid metabolism
(9–12). Liver injury is a common initiating
process of many liver diseases, including hepatitis, cirrhosis, and
hepatoma (13). There are many
common risk factors which can induce liver injury, such as
hepatitis virus, alcohol, and drugs. Although there are numbers of
pathways reported to mediate liver injury (14,15),
the precise mechanisms behind liver injury remain largely
unknown.
In our current study, we observed that ATF4 protein
is highly expressed in mouse livers. The liver ATF4 protein levels
decreased upon carbon tetrachloride (CCl4) and
lipopolysaccharide/D-galactosamine (LPS/D-GalN) induced liver
injury. Furthermore, we show that suppressing ATF4 using
CRISPR-Cas9 plasmids enhanced CCl4 and LPS/D-GalN induced liver
injury in mice, while ATF4 overexpression attenuated CCl4 and
LPS/D-GalN induced liver injury.
Materials and methods
Chemicals and antibodies
Tunicamycin was purchased from Tocris (Minneapolis,
MN, USA). CCl4 was purchased from Guoyao (Beijing, China). LPS and
D-GalN were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Antibodies against ATF4, p-eIF2α and Bip were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA). Antibodies
against eIF2α and GAPDH were purchased from Santa Cruz
Biotechnology, Inc. (Heidelberg, Germany).
Animals and treatments
Male C57BL/6 mice (10 weeks, 20–22 g) were purchased
from the Model Animal Research Center of Nanjing University
(Nanjing, China). The use of animals was approved by the Ethics
Committee of Southwest Medical University on Animal Care (Sichuan,
China).
Plasmid hydrodynamic injection
Hydrodynamic injection was performed as described in
the report of Chen and Calvisi (16). In brief, 10 µg ATF4-targeting
CRISPR-Cas9 plasmid, ATF4 overexpression plasmid or empty vector
were diluted in 2 ml saline (0.9% NaCl), filtered through a 0.22 µm
filter and injected into the lateral tail vein of 10-week-old male
C57BL/6 mice in 5 to 7 sec.
CCl4-induced liver injury model
Male C57BL/6 mice (6 mice per group) were injected
intraperitoneally with CCl4 (4 ml/kg, 5% w/v dissolved in olive
oil) three times a week for 2 weeks as previously described
(17). The mice were killed 24 h
after the final injection of CCl4, and liver tissues were harvested
for analysis.
LPS/D-GalN-induced liver injury
model
Male C57BL/6 mice (6 mice per group) were injected
intraperitoneally with LPS (50 µg/kg) and D-GalN (800 mg/kg,
phosphate buffer saline as control) and killed 6 h after LPS/D-GalN
injection (18).
Histological analysis
Liver tissues of mice were fixed in 4% formalin at
room temperature for at least 24 h, embedded in paraffin and cut
into 5 µm sections. Liver sections were deparaffinized and stained
with hematoxylin and eosin (H&E) for morphologic analysis.
Sirius red staining was performed according to the usual method and
the positive area was quantified with Image J software.
Semi-quantitative (sq)- and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated with TRIzol reagent
(Invitrogen; Thermo Fischer Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's instructions. The reverse
transcription reactions were carried out using the M-MLV reverse
transcriptase (Promega Corporation, Madison, WI, USA) according to
the manufacturer's protocol. sqPCR was performed by running the
products on a 1% (for ATF4 and 18S) or 4% (for XBP1) agarose gel.
RT-qPCR analyses were performed using SYBR Premix Ex Taq (Takara
Bio, Inc., Otsu, Japan) as previously described (17). Results were normalized with 18S and
quantified using the 2−∆∆Cq method (19). The primers used are as follows:
Mouse ATF4-forward: 5′-TCCTGAACAGCGAAGTGTTG, andmouse ATF4-reverse:
5′-AGAGCTCATCTGGCATGGTT-3′; mouse XBP1-forward:
5′-TGCTGAGTCCGCAGCAGGTG-3′, and mouse XBP1-reverse:
5′-ACTAGCAGACTCTGGGGAAG-3′; mouse 18S-forward:
5′-CGGCTACCACATCCAAGGAA-3′, and mouse 18S-reverse:
5′-GCTGGAATTACCGCGGCT-3′.
Western blot analysis
Mouse tissues were lysed in Triton lysis buffer (20
mM Tris, pH 7.4, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM
EDTA, 1 mM PMSF, 10 mM NaF, 5 mg/ml aprotinin, 20 mM leupeptin, and
1 mM sodium orthovanadate) and centrifuged at 4°C, 12,000 × g for
15 min. Protein concentrations of the supernatant were measured
using the BCA assay. Protein samples were denatured with 4×
SDS-loading buffer (200 mM Tris, pH 6.8, 8% SDS, 400 mM DTT, 0.4%
bromophenol blue, 40% glycerol) at 100°C for 5 min and subjected to
standard SDS-PAGE and western blot analysis as previously described
(17).
Statistical analysis
Results are expressed as the mean ± standard
deviation. Statistical analysis was performed using Student's
t-test and Excel software (version 2010; Microsoft Corporation,
Redmond, WA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
ATF4 protein is highly expressed in
the mouse liver
To investigate the expression of ATF4 in
vivo, we evaluated both protein and mRNA levels of ATF4 in
mouse tissues, including liver, heart, kidney, lung, stomach,
spleen, and small and large intestine. Interestingly, the western
blot results showed ATF4 protein is highly expressed in mouse
liver, while being almost nondetectable in other tissues (Fig. 1A). However, the RNA levels of ATF4
in these tissues are comparably high (Fig. 1B).
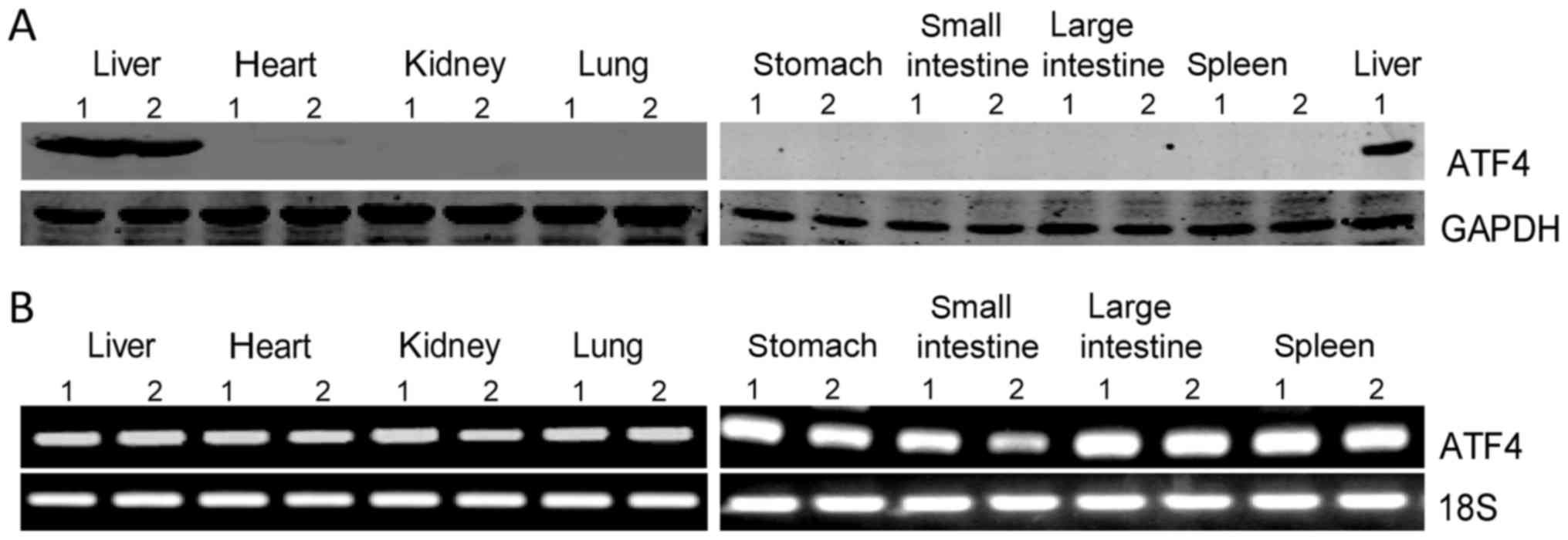 | Figure 1.ATF4 protein is highly expressed in
mouse liver. (A) Western blot analysis of ATF4 protein expression
in mouse liver, heart, kidney, lung, stomach, small and large
intestine, and spleen. (B) Levels of ATF4 mRNA in mouse liver,
heart, kidney, lung, stomach, small and large intestine and spleen
were analyzed by semi-quantitative polymerase chain reaction assay.
ATF4, activating transcription factor 4. |
High levels of ATF4 protein in the
liver are independent of ER stress or eIF2α
Considering that ATF4 is conventionally regulated by
eIF2α, we analyzed the phosphorylation level of eIF2α in mouse
tissues by western blotting. Our results showed that phospho-eIF2α
levels are very low in the tissues tested (Fig. 2A), which seemed contradictory with
the high protein level of ATF4 in the liver. To clarify whether
ATF4 can be upregulated by ER stress in the mouse liver, we treated
mice with tunicamycin. As shown in Fig. 2B, tunicamycin treatment caused XBP1
mRNA splicing, suggesting the induction of the unfolded protein
response. Next, we determined the eIF2α/ATF4 signal in mouse liver
and lung upon tunicamycin treatment. The results showed that
tunicamycin significantly promoted eIF2α phosphorylation, and
increased ATF4 and Bip protein levels in mouse lungs (Fig. 2C). In the liver tissue,
phosphorylation of eIF2α and expression of Bip were increased upon
tunicamycin administration as expected. However, the ATF4 protein
level decreased in a time-dependent manner after tunicamycin
treatment (Fig. 2C). These results
indicated that the high levels of liver ATF4 protein present in the
liver are independent of eIF2α or ER stress.
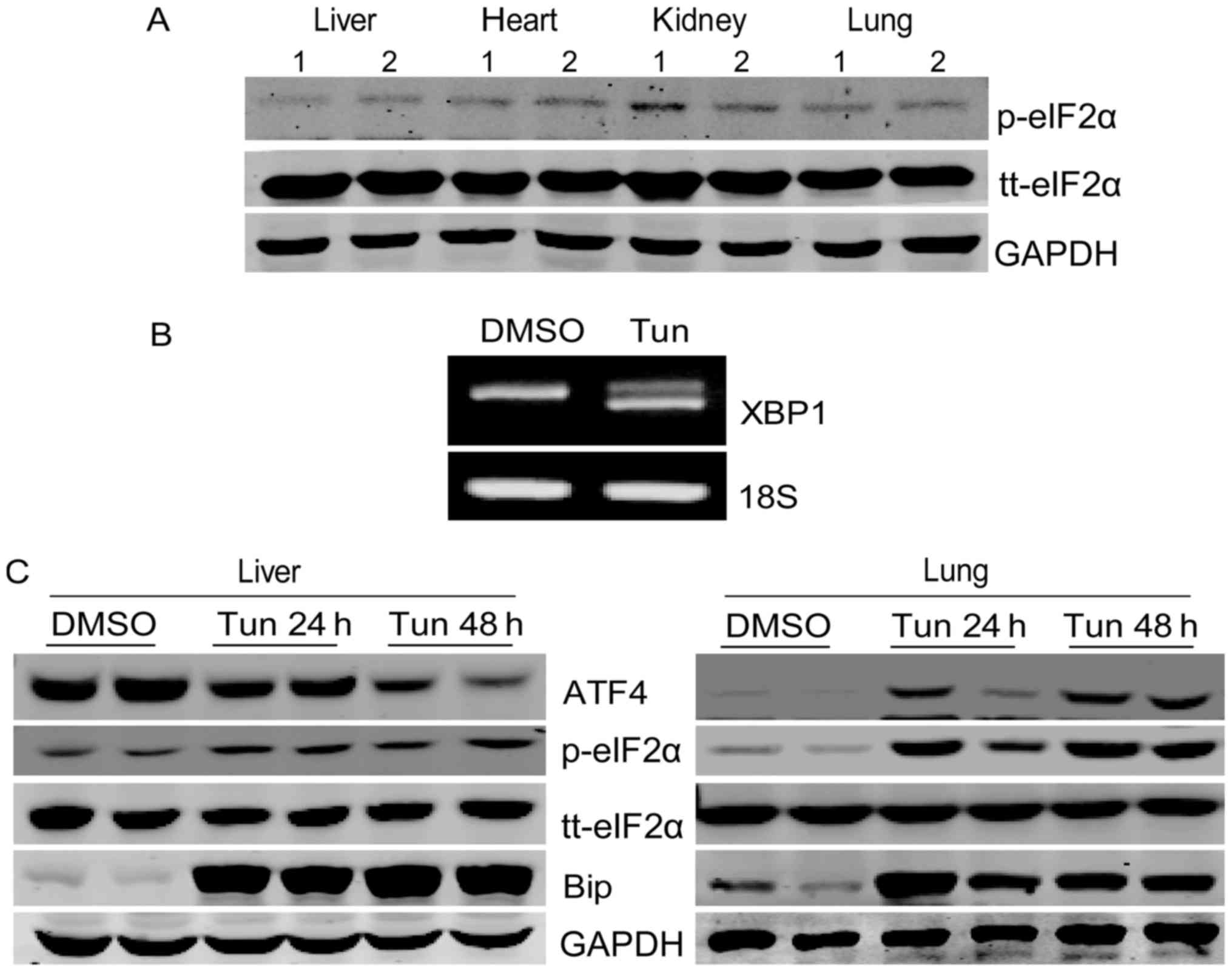 | Figure 2.Liver ATF4 protein expression is not
upregulated by endoplasmic reticulum stress or eIF2α activation.
(A) The phosphorylation levels of eIF2α and tt-eIF2α in mouse
liver, heart, kidney and lung were analyzed by western blotting.
(B) Male 8-week-old C57BL/6 mice were injected intraperitoneally
with tunicamycin (1 mg/kg) and assessed 24 h later. The liver mRNA
was subjected to semi-quantitative polymerase chain reaction to
detect spliced (active form) and unspliced (inactive form) XBP1
mRNA. (C) Mice were injected intraperitoneally with tunicamycin (1
mg/kg) or with DMSO as the control and then assessed following the
indicated incubation time. The protein levels of ATF4, p-eIF2α,
tt-eIF2α and Bip in mouse liver and lung were analyzed by western
blotting. ATF4, activating transcription factor 4; eIF2α,
eukaryotic translation initiation factor 2α; XBP1, X-box binding
protein 1; p-, phosphorylated; tt-, total; Bip, binding
immunoglobulin protein. |
ATF4 protein was decreased in CCl4 and
LPS/D-GalN induced mouse liver injury
It was interesting to find that ATF4 protein
displayed high levels of expression in the mouse liver and was
nonconventionally regulated. We therefore investigated the role of
ATF4 in liver injury. Animal models of liver injury are commonly
used in research, for example CCl4 is a classical hepatotoxicant,
which is used to induce chronic liver injury and liver fibrosis
(20). Similarly, LPS plus D-GalN
is a well-known acute liver injury model (21). Thus, CCl4 was used to establish
chronic liver injury while LPS/D-GalN was used to induce acute
liver injury. The western blot assay demonstrated that ATF4 protein
was decreased significantly following repeated CCl4 exposure, while
the mRNA level of ATF4 was not significantly changed (Fig. 3A and B). In addition, the ATF4
protein decreased markedly after 6 h of LPS/D-GalN treatment
(Fig. 3C). In contrast, the mRNA
of ATF4 did not change significantly (Fig. 3D). These results suggested that
ATF4 protein is downregulated in response to both chronic and acute
liver injury.
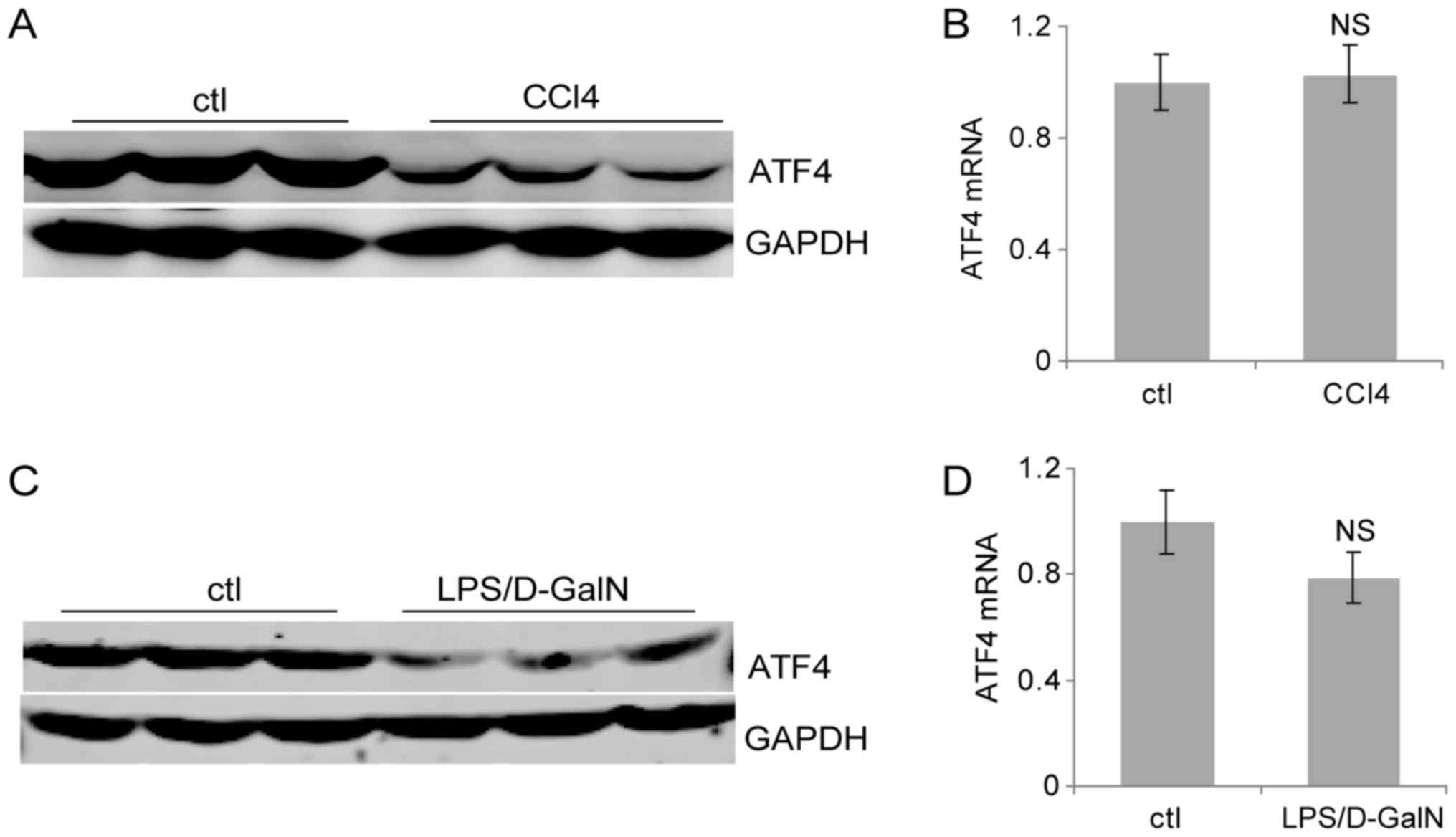 | Figure 3.Liver ATF4 protein expression
decreases in liver injury. (A) Mice were injected intraperitoneally
with CCl4 (4 ml/kg) or olive oil as the control for 2
weeks. Liver ATF4 protein levels were evaluated by western
blotting. (B) Following the administration of CCl4 (4
ml/kg) or vehicle control for 2 weeks, the mouse liver mRNA levels
were quantified by RT-qPCR. (C) Mice were injected
intraperitoneally with LPS (50 µg/kg) and D-GalN (800 mg/kg). Liver
ATF4 protein expression was evaluated by western blotting following
6 h. (D) Mice were injected intraperitoneally with LPS (50 µg/kg)
and D-GalN (800 mg/kg), then sacrificed following 6 h. Liver mRNA
levels were quantified by RT-qPCR. Data are presented as the means
± standard deviation. NS, not significant; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; ctl, control;
CCl4, carbon tetrachloride; D-GalN, D-galactosamine;
ATF4, activating transcription factor 4; LPS, lipopolysaccharide;
NS, not significant. |
ATF4 suppression aggravated CCl4 and
LPS/D-GalN induced liver injury
Next, to investigate effects of ATF4 on liver
injury, ATF4 targeting a CRISPR-Cas9 plasmid (ATF4-cri) was
constructed and injected through the tail vein to knockdown the
expression of ATF4 in the liver. As shown in Fig. 4A and B, the ATF4-cri plasmid
efficiently lowered the liver ATF4 expression at both mRNA and
protein levels. After injection with ATF4-cri plasmid or control
plasmid, mice were challenged with CCl4 or LPS/D-GalN. Serum
transaminase analysis revealed that knockdown of ATF4 by ATF4-cri
significantly increased CCl4-induced levels of AST and ALT compared
with controls (Fig. 4B). Similar
results were obtained in the LPS/D-GalN model (Fig. 4C). These data suggested ATF4
inactivation sensitizes mice to CCl4 and LPS/D-GalN induced liver
injury, indicating a protective role for ATF4 in the liver.
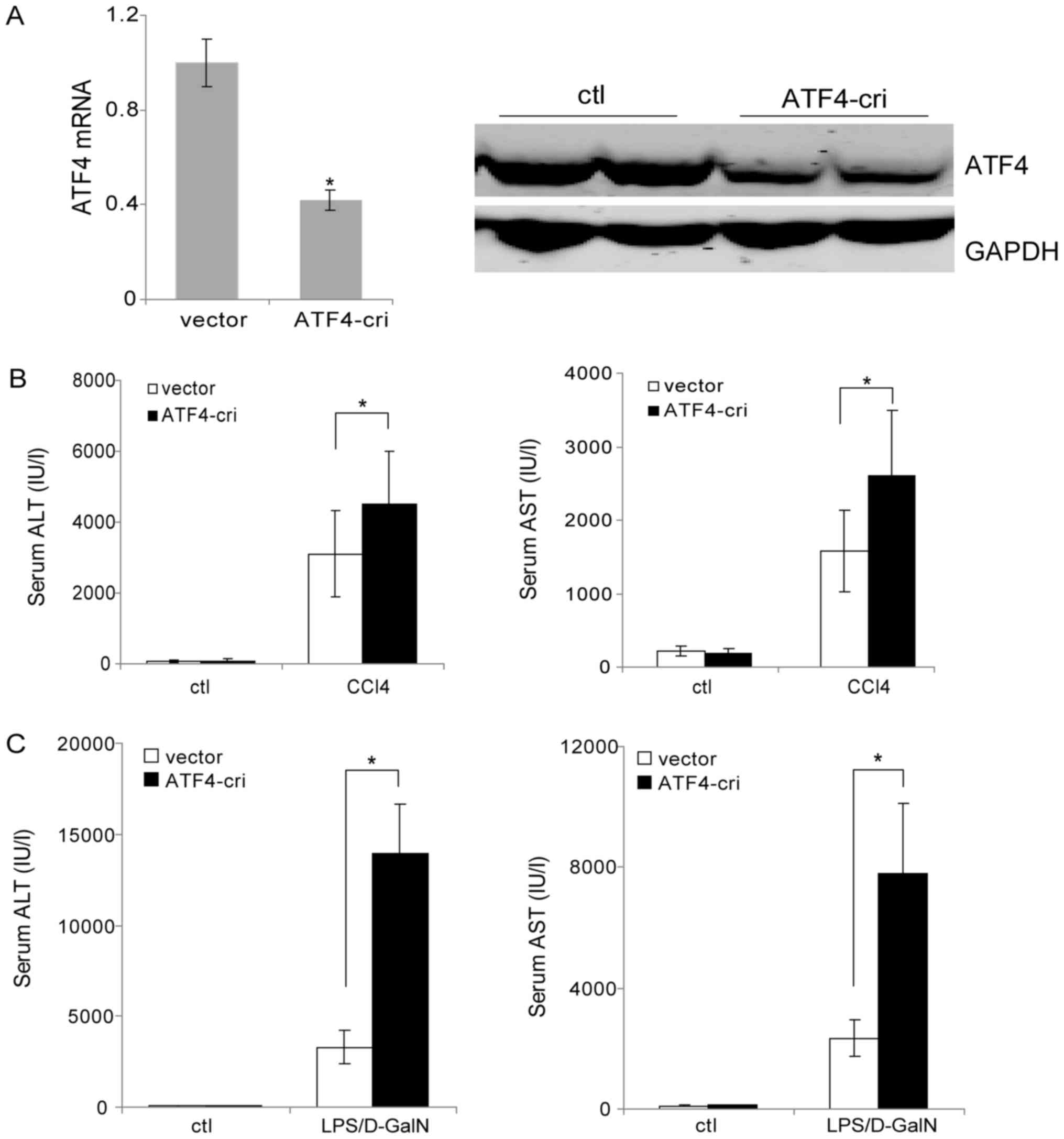 | Figure 4.Suppression of ATF4 promotes
CCl4 and LPS/D-GalN-induced liver injury. (A) Mice were
injected with the ATF4-cri or control plasmid through the tail vein
and were sacrificed 72 h post-injection. The liver ATF4 mRNA levels
were analyzed by reverse transcription-quantitative polymerase
chain reaction and protein levels were analyzed by western
blotting. (B) Mice were injected with ATF4-cri plasmid or empty
vector once a week via the tail vein. Two days following the first
plasmid injection, the mice were injected intraperitoneally with
CCl4 (4 ml/kg) or vehicle control for 2 weeks. Serum AST
and ALT levels were determined at 24 h following the last
CCl4 injection. (C) Mice were injected with ATF4-cri
plasmid or empty vector through the tail vein. Two days following
plasmid injection, the mice were injected intraperitoneally with
LPS (50 µg/kg) and D-GalN (800 mg/kg). Serum AST and ALT levels
were determined following 6 h. Data are presented as the means ±
standard deviation. *P<0.05, as indicated. ATF4, activating
transcription factor 4; LPS, lipopolysaccharide; CRISPR, Clustered
Regularly Interspaced Short Palindromic Repeats; ATF4-cri,
ATF4-targeting CRISPR/CRISPR associated protein 9 plasmid;
CCl4, carbon tetrachloride; D-GalN, D-galactosamine;
AST, aspartate transaminase; ALT, alanine aminotransferase; ctl,
control. |
Reduced expression of ATF4 enhanced
JNK activation after CCl4 and LPS/D-GalN treatment
To reveal the basis for the increased liver injury
by ATF4 inactivation, mouse liver sections were subjected to
histopathological examination. Hematoxylin and eosin (H&E)
staining results revealed more serious hepatocellular necrosis and
morphological alterations in the ATF4-cri group after CCl4
treatment (Fig. 5A). In addition,
we found enhanced liver fibrosis in the ATF4-cri group mice as
evidenced by increased intensity of Sirius red staining (Fig. 5B). H&E staining of LPS-treated
liver sections showed markedly more hemorrhage, necrosis and
inflammatory cell infiltration in ATF4-cri-treated mice livers
(Fig. 5C). These data demonstrated
that ATF4 suppression augmented hepatocyte damage and the
inflammatory response in both the CCl4 and LPS/D-GalN models. The
c-Jun-N-terminal kinase (JNK) is a mitogen-activated protein kinase
family member that plays important roles in the regulation of cell
death, survival, and inflammation (22). We therefore explored a possible
role for JNK in our model. The results showed that both CCl4 and
LPS/D-GalN treatment lead to the activation of JNK (Fig. 5D). More importantly, ATF4
suppression increased the activation of JNK induced by CCl4 and
LPS/D-GalN (Fig. 5D). These
results suggested that ATF4 plays a protective role of in the
liver, in part, through regulating JNK signaling.
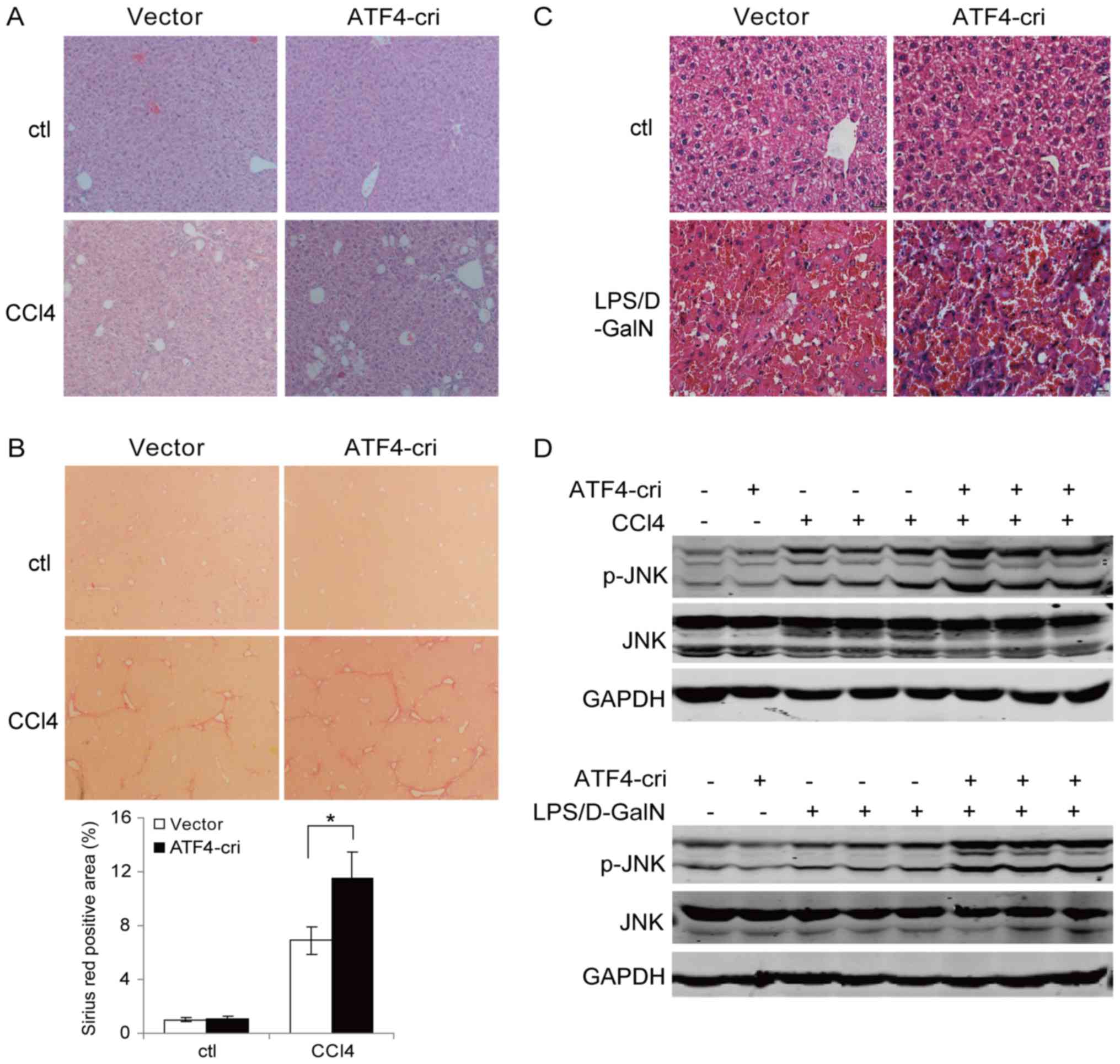 | Figure 5.ATF4 inhibition promotes
CCl4 and LPS/D-GalN mediated JNK activation. (A) Mice
were injected with ATF4-cri plasmid or empty vector once a week via
the tail vein. Two days following the first plasmid injection, the
mice were injected intraperitoneally with CCl4 (4 ml/kg)
or vehicle control for 2 weeks. Liver sections were subjected to
hematoxylin and eosin staining (magnification, ×100). (B) Sirius
red staining of liver sections (magnification, ×100) from (A) and
quantification using Image J software. Data are presented as means
± standard deviation. *P<0.05, as indicated. (C) Mice were
injected with ATF4-cri plasmid or empty vector through the tail
vein. Two days following plasmid injection, the mice were injected
intraperitoneally with LPS (50 µg/kg) and D-GalN (800 mg/kg). Liver
sections were subjected to hematoxylin and eosin staining
(magnification, ×400). (D) Western blot analysis of p-JNK and JNK
in the liver samples shown in (A) and (C). ATF4, activating
transcription factor 4; LPS, lipopolysaccharide; CRISPR, Clustered
Regularly Interspaced Short Palindromic Repeats; ATF4-cri,
ATF4-targeting CRISPR/CRISPR associated protein 9 plasmid;
CCl4, carbon tetrachloride; D-GalN, D-galactosamine;
JNK, c-Jun N-terminal kinase; p-, phosphorylated. |
ATF4 overexpression alleviated CCl4
and LPS/D-GalN induced liver injury
To verify the protective role of ATF4 in the liver,
we investigated the effects of ATF4 overexpression on liver injury
induced by CCl4 and LPS/D-GalN. After injection with ATF4
overexpression plasmid (ATF4-ov) or control plasmid, mice were
challenged with CCl4 or LPS/D-GalN. Serum transaminase analysis
revealed that overexpression of ATF4 significantly decreased CCl4
induced AST and ALT elevation compared with controls (Fig. 6A). Similar results were obtained in
the LPS/D-GalN model (Fig. 6B).
These data thus further confirm the protective role of ATF4 in the
liver.
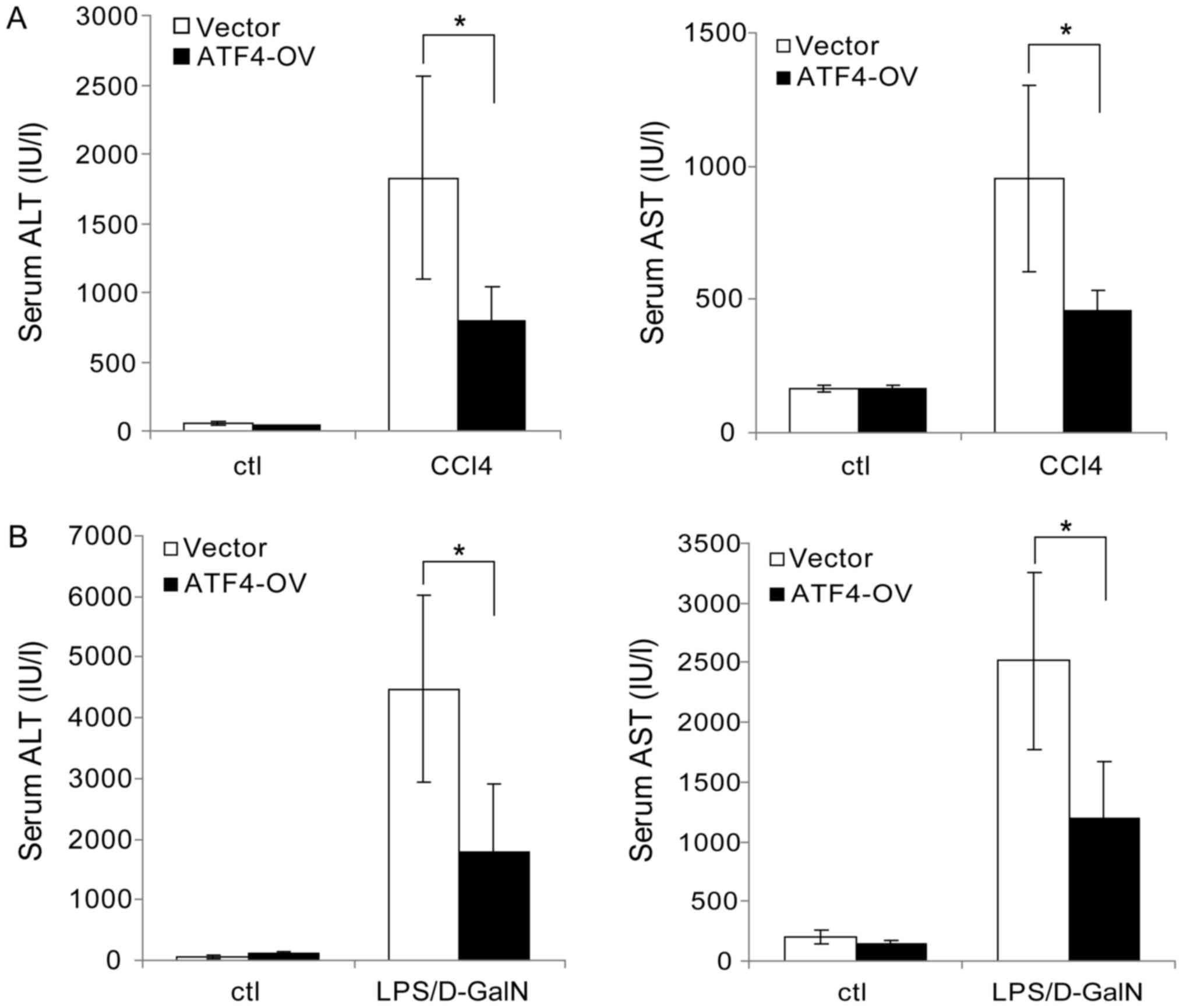 | Figure 6.ATF4 overexpression inhibits
CCl4 and LPS/D-GalN-induced liver injury. (A) Mice were
injected with ATF4-OV plasmid or empty vector once a week through
the tail vein. Two days following the first plasmid injection, the
mice were injected intraperitoneally with CCl4 (4 ml/kg)
or vehicle control for 2 weeks. Serum AST and ALT levels were then
determined at 24 h following the last CCl4 injection.
(B) Mice were injected with ATF4-OV plasmid or empty vector through
the tail vein. Two days following plasmid injection, the mice were
injected intraperitoneally with LPS (50 µg/kg) and D-GalN (800
mg/kg). Serum AST and ALT levels were determined following 6 h.
Data are represented as the mean ± standard deviation. *P<0.05,
as indicated. ATF4, activating transcription factor 4; LPS,
lipopolysaccharide; CCl4, carbon tetrachloride; D-GalN,
D-galactosamine; ATF4-OV, ATF4 overexpression plasmid; AST,
aspartate transaminase; ALT, alanine aminotransferase; ctl,
control. |
Discussion
In this study, we characterized the expression
pattern of ATF4 in vivo at both the protein and mRNA level.
We firstly discovered that ATF4 maintained high protein levels in
the mouse liver under normal conditions. Considering there is no
difference in mRNA levels of ATF4 between the tissues we tested,
the difference in ATF4 protein levels could be due to variation in
translation or stability between tissues. It is well known that
ATF4 protein is usually upregulated by stress conditions and plays
a crucial role in the stress response. Multiple intracellular
stress pathways including endoplasmic reticulum stress, amino acid
deprivation, and oxidative stress can induce the phosphorylation of
eIF2α, which both leads to a general inhibition of protein
synthesis but also the translational upregulation of ATF4 mRNA
(4). Here, we observed high
protein levels of ATF4 in mouse livers but not in other tissues.
However, the phosphorylation levels of eIF2α are uniformly low in
the mouse tissues we tested, inconsistent with the high protein
levels of ATF4 in the liver. We hypothesized that liver ATF4
protein levels are not associated with eIF2α activation. To confirm
this speculation, tunicamycin, an ER stress inducer, was used to
trigger ER stress and eIF2α phosphorylation. Notably, tunicamycin
induced ER stress in mouse liver and lung, as demonstrated by
spliced XBP1 mRNA, increased Bip protein and eIF2α phosphorylation.
The ATF4 protein in the lung was consistently induced by
tunicamycin, indicating a conventional regulation of ATF4 by eIF2α.
Nevertheless, the liver expression of ATF4 protein decreased upon
tunicamycin treatment. This demonstrated a unique regulation
pattern of ATF4 protein in the liver tissue that is not associated
with eIF2α. Another possible mechanism is that the stability of
ATF4 is different between the liver and other tissues. It has been
reported that ATF4 degradation is mediated by the E3 ubiquitin
ligase SCFβTrCP (23).
Additional reports have shown that p300 modulates ATF4 stability
and its transcriptional activity (24). Whether the stability of ATF4
contributes to the difference in tissue ATF4 protein levels
requires further investigation.
We wondered whether the high protein level of ATF4
expression in mouse livers hinted at an important role in the
liver. It has been reported that ATF4 mutations resulted in severe
fetal anemia and fetal liver hypoplasia (25). Additional reports have suggested
that ATF4 plays an important role in hepatic lipid metabolism
(10–12). Liver injury is the most common
liver disorder resulting in aggressive liver diseases. In the
present study, we investigated the role of ATF4 in liver injury
using two models, CCl4-mediated chronic liver injury and
LPS/D-GalN-induced acute liver injury. Intriguingly, we found
decreased ATF4 protein levels in mouse livers following both CCl4
and LPS/D-GalN administration without recognizable mRNA changes.
This indicated posttranscriptional regulation of ATF4 in CCl4 and
LPS/D-GalN models, possibly via regulation of translation or
stability. Our future research will focus on the regulatory
mechanisms of ATF4 in these liver models. However, the question
remained whether the reduction in ATF4 influences liver injury. Our
data showed that inactivation of ATF4 by CRISPR significantly
aggravated CCl4 and LPS/D-GalN induced liver injury, as
demonstrated by elevated serum AST and ALT. In addition, the
overexpression of ATF4 attenuated CCl4 and LPS/D-GalN mediated
liver injury. These results implied a protective role for ATF4
during liver injury. The JNK pathway has been reported to regulate
cellular stress responses, apoptosis, malignant transformation, and
hepatocarcinogenesis (22,26). We demonstrated that ATF4
suppression promoted CCl4 and LPS/D-GalN induced JNK activation.
This may suggest that the inhibition of ATF4 aggravated liver
injury, at least partly, through the upregulation of the JNK
pathway. In a previous study by Masuoka and Townes (25), ATF4 was identified as critical for
normal cellular proliferation, especially for the high-level
proliferation required during fetal-liver hematopoiesis. The liver
is a highly regenerative tissue, as hepatocytes are able to
proliferate in response to injury to restore liver function
(27). Here in our models, a high
level of cell proliferation was required after CCl4 and LPS/D-GalN
treatment. Thus, a reasonable explanation for our results is that
downregulation of ATF4 inhibited compensatory cell proliferation
during liver repair response, resulting in more serious liver
injury. Further studies are needed to investigate the detailed
mechanisms linking ATF4 and liver injury.
In summary, we revealed a nonconventional expression
pattern of ATF4 protein in mouse livers. Chemical-induced liver
injury caused a decrease in liver ATF4 protein. Moreover, we
demonstrated that ATF4 suppression aggravated CCl4 and LPS/D-GalN
induced liver injury, while ATF4 overexpression attenuated CCl4 and
LPS/D-GalN induced liver injury, indicating a hepatoprotective role
for ATF4.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Science and Technology Department of Sichuan Province Foundation
(grant no. 2017JY0134), Health and Family Planning Commission of
Sichuan Province Foundation (grant no. 16PJ539), Southwest Medical
University Foundation (grant no. 2015-YJ007), the National Natural
Science Foundation of China (grant no. 81472312), Innovation Team
of Education Department of Sichuan Province (grant no. 16TD0021),
Luzhou City-Southwest Medical University Foundation (grant nos.
2016LZXNYD-T02, 2015LZCYD-S01-14/15 and 2015LZCYD-S01-8/15) and
Sichuan Province-Luzhou City-Southwest Medical University
Foundation (grant nos. 14JC0082, 14JC0038 and 14ZC0070).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and HZ designed the experiments, and performed
the animal experiments and data analyses. YC performed the western
blot experiments. WY and GL performed the polymerase chain reaction
experiments. CD and FY conducted the histology experiments. BX, CF,
XX, MW and YW participated in data analysis and interpreting the
results. RD and JL designed the experiments, analyzed the data and
wrote the manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Ethics
Committee of Southwest Medical University on Animal Care (Sichuan,
China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Karpinski BA, Morle GD, Huggenvik J, Uhler
MD and Leiden JM: Molecular cloning of human CREB-2: An ATF/CREB
transcription factor that can negatively regulate transcription
from the cAMP response element. Proc Natl Acad Sci USA.
89:4820–4824. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kilberg MS, Shan J and Su N:
ATF4-dependent transcription mediates signaling of amino acid
limitation. Trends Endocrinol Metab. 20:436–443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vattem KM and Wek RC: Reinitiation
involving upstream ORFs regulates ATF4 mRNA translation in
mammalian cells. Proc Natl Acad Sci USA. 101:11269–11274. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rutkowski DT and Kaufman RJ: All roads
lead to ATF4. Dev Cell. 4:442–444. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Y, Alam GN, Ning Y, Visioli F, Dong
Z, Nör JE and Polverini PJ: The unfolded protein response induces
the angiogenic switch in human tumor cells through the PERK/ATF4
pathway. Cancer Res. 72:5396–5406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang X, Matsuda K, Bialek P, Jacquot S,
Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes
TM, et al: ATF4 is a substrate of RSK2 and an essential regulator
of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell.
117:387–398. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lange PS, Chavez JC, Pinto JT, Coppola G,
Sun CW, Townes TM, Geschwind DH and Ratan RR: ATF4 is an oxidative
stress-inducible, prodeath transcription factor in neurons in vitro
and in vivo. J Exp Med. 205:1227–1242. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang C, Li H, Meng Q, Du Y, Xiao F, Zhang
Q, Yu J, Li K, Chen S, Huang Z, et al: ATF4 deficiency protects
hepatocytes from oxidative stress via inhibiting CYP2E1 expression.
J Cell Mol Med. 18:80–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshizawa T, Hinoi E, Jung DY, Kajimura D,
Ferron M, Seo J, Graff JM, Kim JK and Karsenty G: The transcription
factor ATF4 regulates glucose metabolism in mice through its
expression in osteoblasts. J Clin Invest. 119:2807–2817. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Meng Q, Xiao F, Chen S, Du Y, Yu J,
Wang C and Guo F: ATF4 deficiency protects mice from
high-carbohydrate-diet-induced liver steatosis. Biochem J.
438:283–289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang C, Huang Z, Du Y, Cheng Y, Chen S and
Guo F: ATF4 regulates lipid metabolism and thermogenesis. Cell Res.
20:174–184. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao G, Zhang T, Yu S, Lee S,
Calabuig-Navarro V, Yamauchi J, Ringquist S and Dong HH: ATF4
protein deficiency protects against high fructose-induced
hypertriglyceridemia in mice. J Biol Chem. 288:25350–25361. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang DY and Friedman SL:
Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology.
56:769–775. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schwabe RF and Brenner DA: Mechanisms of
Liver Injury. I. TNF-alpha-induced liver injury: Role of IKK, JNK,
and ROS pathways. Am J Physiol Gastrointest Liver Physiol.
290:G583–G589. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Prandota J: Important role of
proinflammatory cytokines/other endogenous substances in
drug-induced hepatotoxicity: Depression of drug metabolism during
infections/inflammation states, and genetic polymorphisms of
drug-metabolizing enzymes/cytokines may markedly contribute to this
pathology. Am J Ther. 12:254–261. 2005.PubMed/NCBI
|
|
16
|
Chen X and Calvisi DF: Hydrodynamic
transfection for generation of novel mouse models for liver cancer
research. Am J Pathol. 184:912–923. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao X, Fu J, Xu A, Yu L, Zhu J, Dai R, Su
B, Luo T, Li N, Qin W, et al: Gankyrin drives malignant
transformation of chronic liver damage-mediated fibrosis via the
Rac1/JNK pathway. Cell Death Dis. 6:e17512015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin X, Zhang S, Huang R, Wei L, Liang C,
Chen Y, Lv S, Liang S, Wu X and Huang Q: Protective effect of
genistein on lipopolysaccharide/D-galactosamine-induced hepatic
failure in mice. Biol Pharm Bull. 37:625–632. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Delire B, Stärkel P and Leclercq I: Animal
models for fibrotic liver diseases: What we have, what we need, and
what is under development. J Clin Transl Hepatol. 3:53–66. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakama T, Hirono S, Moriuchi A, Hasuike S,
Nagata K, Hori T, Ido A, Hayashi K and Tsubouchi H: Etoposide
prevents apoptosis in mouse liver with
D-galactosamine/lipopolysaccharide-induced fulminant hepatic
failure resulting in reduction of lethality. Hepatology.
33:1441–1450. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: Signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lassot I, Ségéral E, Berlioz-Torrent C,
Durand H, Groussin L, Hai T, Benarous R and Margottin-Goguet F:
ATF4 degradation relies on a phosphorylation-dependent interaction
with the SCF (betaTrCP) ubiquitin ligase. Mol Cell Biol.
21:2192–2202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lassot I, Estrabaud E, Emiliani S,
Benkirane M, Benarous R and Margottin-Goguet F: p300 modulates ATF4
stability and transcriptional activity independently of its
acetyltransferase domain. J Biol Chem. 280:41537–41545. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Masuoka HC and Townes TM: Targeted
disruption of the activating transcription factor 4 gene results in
severe fetal anemia in mice. Blood. 99:736–745. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Das M, Garlick DS, Greiner DL and Davis
RJ: The role of JNK in the development of hepatocellular carcinoma.
Genes Dev. 25:634–645. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tao Y, Wang M, Chen E and Tang H: Liver
regeneration: Analysis of the main relevant signaling molecules.
Mediators Inflamm. 2017:42563522017. View Article : Google Scholar : PubMed/NCBI
|














