Introduction
Respiratory syncytial virus (RSV), a single-stranded
negative-sense RNA virus (1), not
only causes severe lower respiratory tract infections in infants
worldwide but also leads to hospitalization of infants (2–4). As
one of the main targets for RSV infection, airway epithelial cells
play a very important role in host defense system against RSV
infection. Recently, cultures of human respiratory epithelium have
been applied to study the mechanisms of RSV infection (5). After RSV infection, airway epithelial
cells produce a large number of immune-active molecules, including
cytokines, chemokines and reactive oxygen species (ROS) to promote
the occurrence of infantile bronchiolitis and pneumonia (6).
Oxidative stress is an invariable feature of human
lung epithelial cells, resulting in a large amount of ROS
productions that modifies and disrupts cellular biomolecules during
immune-inflammatory responses to viral infection (7). This injury is mainly the results of
additional ROS produced by further increased oxidative stress
(8). It has been reported that RSV
could induce the RelA activation mediated by ROS signaling
(9). The elevation of ROS and IRF3
signals caused by RSV infection in airway epithelial cells could be
blocked by anti-oxidants (10).
The formation of ROS produces an imbalance between the antioxidant
defenses and oxidant molecules such as hydroxy radicals, hydrogen
peroxide, and superoxide anion radicals. ·OH provides strong
evidence of increased oxidative stress and is an effective
initiator of highly reactive lipid peroxidation (11,12).
In addition, nitric oxide (NO) is a key mediator for airway
inflammation, which promotes the migration of inflammatory cells to
the airway (12,13). ROS and free radicals have been
shown to act as cellular signaling molecules participating in
various molecular and biochemical processes, including
pro-inflammatory mediations such as chemokines and cytokines
expressions (14). RSV-induced
intracellular ·OH and NO may therefore modulate the expression of
pro-inflammatory mediators, and oxidative stress may represent an
important mechanism for RSV-induced lung pathogenesis. On this
basis, Mastronarde et al (15) proposed that antioxidants might be
able to block IL-8 production following RSV infection in
vivo. In contrast, the activities of antioxidant enzymes (e.g.
SOD, glutathione peroxidase, catalase, and glutathione
S-transferase) are very important for cellular defense against RSV
infection in A549 cells (6). Our
group previously revealed that RSV-intranasally-inoculated mice can
react to oxidative stress by increasing the malondialdehyde (MDA),
NO and ·OH levels, and reducing SOD and GSH activities in lung
tissues. The application of melatonin with anti-oxidant and
anti-inflammatory functions reversed the pathophysiology by
reducing lung inflammation and ameliorated clinical presentations
in RSV-infected mice (16),
suggesting that oxidative stress is involved in RSV infection.
Toll-like receptors (TLRs) play a fundamental role
in human innate anti-microbial immunity and inflammations by
recognizing the conserved pathogen-associated molecular patterns
(PAMPs) (17,18). Among them, toll-like receptor 3
(TLR3) and TLR7 are considered as the main mediators of
viral-induced signal transductions. TLR3, for example, is able to
identify double-strand viral genomic RNA and the replicative
intermediates of RSV (19),
suggesting that TLR3 plays a role in resisting RSV infection in
human respiratory system (20).
The reaction of TLR3 with dsRNA activates intracellular signaling
and promotes the biosynthesis and secretion of cytokines and other
inflammatory mediators. Dou et al (21) reported that RSV induced gene
expression of TLR3 and TNF-α both in vitro and in mouse
lungs. Once TLR3 is activated, its downstream signaling pathway
will lead to the activation of nuclear factor-κB (NF-κB) and
interferon regulatory factor-3 (IRF3) (22). NF-κB has been shown to modulate the
production of pro-inflammatory cytokines, such as IL-1β, TNF-α and
the neutrophil chemoattractant IL-8 (23), which are strongly associated with
the outcome of inflammatory disease. Whereas IRF3 was shown to
regulate the type I interferon (IFN) expressions (24). Recently, an increase in TLR3
expression was observed in airway epithelial cells of patients with
acute respiratory distress syndrome under airway exposure to
hyperoxic conditions (25),
enhanced TLR3 responses to oxidative stress have also been found in
airway epithelial cells (26).
These phenomena suggest that oxidative stress may participate in
the regulation of TLR3 expression.
N-acetyl-L-cysteine (NAC) is a thiol compound that
directly used as a free radical scavenger and a reduced glutathione
(GSH) precursor (27), which allow
it to be used as an antioxidant in a broad spectrum (28). On the contrary, hydrogen peroxide
(H2O2) induces oxidative and inflammatory
responses in epithelial cells and it can be used as an oxidant
(29).
To our knowledge, by now, there is no study
reporting that whether RSV infection increases TLR3 signaling in
airway epithelial cells through oxidative stress induction. In
order to understand the relationship between TLR3 expression and
oxidative stress modulation during RSV infection in A549 cells, we
studied the intervening effects of oxidative stress agonist
hydrogen peroxide (H2O2) and inhibitor
N-acetyl-L-cysteine (NAC) on TLR3 expression. Besides, we proposed
that oxidative stress induced by RSV infection might serve as one
of the key events in the process of TLR3 activation We hoped that
our study would provide a potential new pharmacological method to
improve RSV-induced acute lung inflammation.
Materials and methods
Cells, viruses and reagents
The human lung adenocarcinoma alveolar basal
epithelial cell line A549 (ATCC® CCL-185®;
American Type Culture Collection, Manassas, VA, USA) was gifted by
Professor Hai-Ming Wei, Institute of Immunology, University of
Science and Technology of China (USTC, Hefei, Anhui, China) and
maintained in Dulbecco's modified Eagle's medium (DMEM; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and
1% penicillin (100 U/ml)-streptomycin (100 µg/ml) at 5%
CO2 and 37°C.
The laryngeal epithelial carcinoma HEp-2 cell line
was maintained in our laboratory. Though it has been reported being
contaminated by HeLa cells, an epithelium-like cell from a cervical
adenocarcinoma (iclac.org/databases/cross-contaminations/),
Nevertheless, HEp-2 cells are still a good substrate for RSV
(30). Additionally, RSV used in
this study was harvested from culture supernatant and was further
purified by density gradient centrifugation; therefore, either
HEp-2 or HeLa cells would have no intervening effect on the result
interpretation of the analysis.
The RSV Long strain was also gifted by Professor
Hai-ming Wei, Institute of Immunology, University of Science and
Technology of China (USTC, Hefei, Anhui, China) and was
multiplicated in HEp-2 cells. Then, the culture supernatant was
precipitated by polyethylene glycol 4000, followed by
centrifugation on 35–65% discontinuous sucrose gradients. Purified
virus suspension was aliquoted, quickly frozen, and stored in
liquid nitrogen. The viral titer of purified RSV reached
7×106 PFU/ml as measured by a methylcellulose plaque
assay in HEp-2 cells (31).
The UV-inactivated RSV was prepared as follows: A
one ml RSV pool was transferred to a culture plate. The plate was
placed under a germicidal lamp TUV-15 W/G15 T8 (Philips, The
Netherlands) and irradiated at a distance of 10 cm in 3-min
intervals, with swirling between the intervals, for a total of 30
min. UV-inactivated virus titers were also determined by plaque
assays on HEp-2 cells to confirm the inactivation effect, then the
virus stock was stored in liquid nitrogen until use.
H2O2 and NAC were purchased
from Sigma Corporation (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). Commercial assay kits for measuring ·OH, NO and SOD
activities were purchased from Nanjing Jiancheng Bioengineering
Institute, (Nanjing, China).
Experimental design and sample
collection
Seven experimental groups were assigned: normal
groups (untreated and uninfected with RSV);
H2O2 control groups (pretreated with
H2O2 without RSV infection); NAC control
groups (pretreated with NAC without RSV infection); RSV infection
control (cells infected with RSV but no pretreatment); NAC+RSV
groups (pretreated with NAC at 5 mM prior to RSV infection),
H2O2 +RSV groups (pretreated with
H2O2 at 150 µM prior to RSV infection) and
inactivated RSV groups (cells infected with inactivated RSV but no
pretreatment). The pretreatment time with
H2O2 or NAC for the corresponding group was 1
h. Cells in the corresponding groups were infected with RSV at
MOI=1 in serum-free mediums for 2 h before fresh mediums were
added. The culture supernatants and trypsinized cells were
separately collected by centrifugation at 4, 8, 12 and 24 h post
infection (pi) and the samples were assessed by the following
experiments.
Measurement of ·OH and NO in culture
supernatants
·OH concentration was determined using a commercial
·OH assay kit based on the Fenton reaction method according to the
manufacturer's protocol (Nanjing Jiancheng Bioengineering
Institute) (32). Briefly, the
reaction mixtures were added to a quartz capillary tube, then
Griess chromogenic reagent was added to form a colored substance
for another 20 min at room temperature (RT) (16). The color depth of the substance is
proportional to the amount of ·OH (33). The absorbance values at 550 nm were
recorded, and the data was expressed as units/ml.
NO concentration was determined using a commercial
NO assay kit based on the Griess reaction method according to the
manufacturer's instructions (Nanjing Jiancheng Bioengineering
Institute) (34). First, 50 µl of
Griess reagent I was added to 100 µl of the supernatant from A549
cultures. Second, 50 µl of Griess reagent II was added. Third, the
reaction mixture was incubated for 10 min at RT, and 160 µl of the
supernatant was used for detection after centrifugation at 3500 rpm
for 15 min. Last, the absorbance at 550 nm was recorded, and the
results were expressed as µmol/l. All samples were tested in
triplicate.
Measurement of total SOD activity in
A549 cells
Proteins from A549 cells were prepared as a method
previously described (16). First,
the cells were washed with PBS before the treatment with
Versene-Trypsin PBS solution. Then, the trypsin-digested cells were
centrifuged at 1,400 g for 10 min and the pellet was re-suspended
in a lysis buffer supplemented with protease inhibitors. Next,
after 30 min of incubation on the ice, the extraction mixture was
centrifuged at 12,000 g at 4°C for 30 min. Last, the supernatant
was transferred to a fresh tube, and its protein concentration was
measured by the Lowry method.
Superoxide dismutase (SOD), an important enzyme in
the antioxidant system, is able to convert
O2− to hydrogen peroxide
(H2O2) and eventually change them to water.
The mechanism for total SOD assay was based on its ability to
inhibit the oxyamine oxidation within the xanthine/xanthine oxidase
system (35). SOD activity was
determined using a commercial WST-1 assay kit (Nanjing Jiancheng
Bioengineering Institute). The absorbances at 450 nm at the
reaction endpoint were read by a microplate reader (ELX800UV;
BioTek Instruments, Inc., Winooski, VT, USA). The SOD activity of
each sample was calculated using a previously described equation
(36). All samples were tested for
three times, and the results were expressed as units per mg
protein.
TLR3, NF-κB p65, IRF3 and superoxide dismutase 1
(SOD1) mRNA semi-quantification by reverse transcription polymerase
chain reaction (RT-PCR). The kinetics of gene expression of
TLR3, NF-κB p65, IRF3 and SOD1 were analyzed
by semi-quantitative RT-PCR. Total RNA was extracted from A549
cells with TRI Reagent™ (Sigma-Aldrich; Merck KGaA) following the
manufacturer's instructions. Then, the total RNA was treated with
RNase-free DNase I (Invitrogen; Thermo Fisher Scientific, Inc.) to
remove genomic DNA contamination and reverse-transcribed into cDNA
using Thermo Scientific Revert Aid First Strand cDNA Synthesis kit
(Fermentas; Invitrogen; Thermo Fisher Scientific, Inc.) following
the manufacturer's instructions. Next, the synthesized first strand
cDNA was amplified by PCR with the primers for TLR3, NF-κB p65,
IRF3, or SOD1 genes. The PCR primers (Table I) for TLR3, NF-κB p65, IRF3, SOD1
and β-actin genes were designed using the Primer Express software,
as previously described (37). The
reaction conditions and cycle numbers used for the PCR of each gene
were shown in Table II. PCR
products and a DNA molecular weight marker (DL2000; Takara
Biotechnology Co., Ltd., Dalian, China) were electrophoresed in a
1.5% agarose gel and visualized under UV light. The density of the
bands was quantified by densitometry using Labworks software, and
the expression levels were expressed as the fold-increase compared
to β-actin controls.
 | Table I.Primer sequence and length for
polymerase chain reaction products of each human gene. |
Table I.
Primer sequence and length for
polymerase chain reaction products of each human gene.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Size (bp) |
|---|
| TLR3 |
GATCTGTCTCATAATGGCTTG |
GACAGATTCCGAATGCTTGTG | 304 |
| NF-κB p65 |
CACAAGGAGACATGAAACAG |
CCAGAGACCTCATAGTTGT | 187 |
| SOD1 |
ATGGCGACGAAGGCC |
TTATTGGGCGATCCC | 465 |
| IRF3 |
GACCTCACGACCCACATAA |
ACCCCACCAGCCGCAGGCCC | 377 |
| β-actin |
GGCTTTGAGTAATGAGAATTTCGA |
ATCAGTTGCAATCAAGAAGTGTTG | 520 |
| Table II.Polymerase chain reaction
thermocycling conditions and cycle numbers for each gene. |
Table II.
Polymerase chain reaction
thermocycling conditions and cycle numbers for each gene.
| Gene | Initial
Denature | Denature | Annealing | Extension | No. of cycles |
|---|
| TLR3 | 94°C for 5 min | 94°C for 40
sec | 55°C for 45
sec | 72°C for 1 min | 32 |
| NF-κB p65 | 94°C for 5 min | 94°C for 40
sec | 57.7°C for 45
sec | 72°C for 1 min | 32 |
| SOD1 | 94°C for 5 min | 94°C for 40
sec | 57°C for 45
sec | 72°C for 1 min | 32 |
| IRF3 | 94°C for 5 min | 94°C for 40
sec | 49.4°C for 45
sec | 72°C for 1 min | 32 |
| β-actin | 94°C for 5 min | 94°C for 40
sec | 55°C for 45
sec | 72°C for 1 min | 32 |
TLR3 and p-NF-κB protein
quantification by western blot
The protein level changes of TLR3 and p-NF-κB were
analyzed by western blot assay. First, the cells in each group were
washed with PBS solution and lysed in a lysis buffer containing
phenylmethylsulfonyl fluoride (PMSF). All samples were incubated on
ice for 30 min and centrifuged at 11,000 g for 5 min. Second, the
protein concentrations in each group of centrifuged supernatant
were determined by the Lowry method. Third, fifty µg of protein was
run on a 10% SDS-PAGE and then transferred to a polyvinylidene
fluoride (PVDF) membrane (sc-296042; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). Fourth, nonspecific binding sites were
blocked with 5% nonfat milk in Tris-buffered saline with Tween 20
(TBST) for 2 h at RT. Fifth, the membranes were incubated with
rabbit anti-TLR3 antibody (sc-28999; Santa Cruz Biotechnology Inc,
Santa Cruz, CA, USA) and anti-p-NF-κB antibody (sc-33020; Santa
Cruz Biotechnology Inc.) separately at a 1:1,000 dilution overnight
at 4°C. After washing with TBST for 3×10 min, the membranes were
incubated with a horseradish peroxidase-conjugated anti-rabbit IgG
(1:10,000) (sc-2004; Santa Cruz Biotechnology Inc.) for 2 h at RT.
Last, the blots were washed, and the antigens were visualized using
an enhanced chemiluminescence western blot detection system
(SuperSignal West Femto kit; Thermo Scientific, Inc.) and recorded
by a Tanon 4500 automatic digital gel image analysis system (Tanon
4500; Tanon, Shanghai, China).
A β-actin monoclonal antibody (1:1,000; TA-09;
OriGene Technologies, Inc., Beijing, China) and horseradish
peroxidase-conjugated sheep anti-mouse IgG (1:10,000; sc-2005;
Santa Cruz Biotechnology Inc.) were used as the internal control,
The molecular weights of TLR3, NF-κB and β-actin in the PAGE gel
were 104, 65 and 43 KDa, respectively. The density of the bands on
the blot was quantified using Labworks imaging software. Compared
with β-actin control group, densitometry in each group was
expressed as the fold-increase.
Statistical analysis
The resulting data are recounted as the mean ±
standard error mean. One-way analysis of variance analysis and the
Fisher post hoc test were performed to determine statistical
significance among each group using SPSS v.17.0 software (SPSS
Inc., Chicago, IL, USA). P<0.05 for the null hypothesis was
considered to indicate a statistically significant difference.
Results
The releasements of ·OH and NO after
different treatments
Oxidative stress can be induced from either excess
ROS generation or impaired antioxidant capacity, which produced
free radicals and reactive oxygen molecules in activated cells
(11). Because that oxidative
stress-mediated events were correlated with the releasement of ·OH
and NO, therefore, ·OH generation was considered as a marker of
free oxygen species due to the extreme instability of ROS. In this
study, the NO and ·OH in A549 cells from inactivated-RSV control,
NAC control and H2O2 control groups, at
different time points, had no significant changes compared to the
normal control group within 24 h (Fig.
1). However, in both the RSV-infected group and the
H2O2+RSV-treated group (Fig. 1A), the ·OH concentration increased
dramatically compared to the normal cell control group (P<0.01)
Nevertheless, in cell group treated with NAC+RSV (Fig. 1A), although the ·OH levels also
increased with an increasing time of RSV infection, they were
significantly lower than those in the RSV-infected group at the
corresponding time points. Thus, the ·OH elevations were associated
with the extended time duration of RSV infection, suggesting ·OH in
A549 cells was up-regulated in a time-dependent manner after RSV
infection. On the results of NO concentration, its variation had a
similar change pattern to that of ·OH concentration (Fig. 1B), which also demonstrated that the
RSV infection induced a significant elevation of oxidative stress
in A549 cells. Notably, UV-inactivated RSV did not elevate the ·OH
and NO levels compared with the normal group (data not shown),
suggesting that the alteration of oxidative stress is dependent on
virus replication.
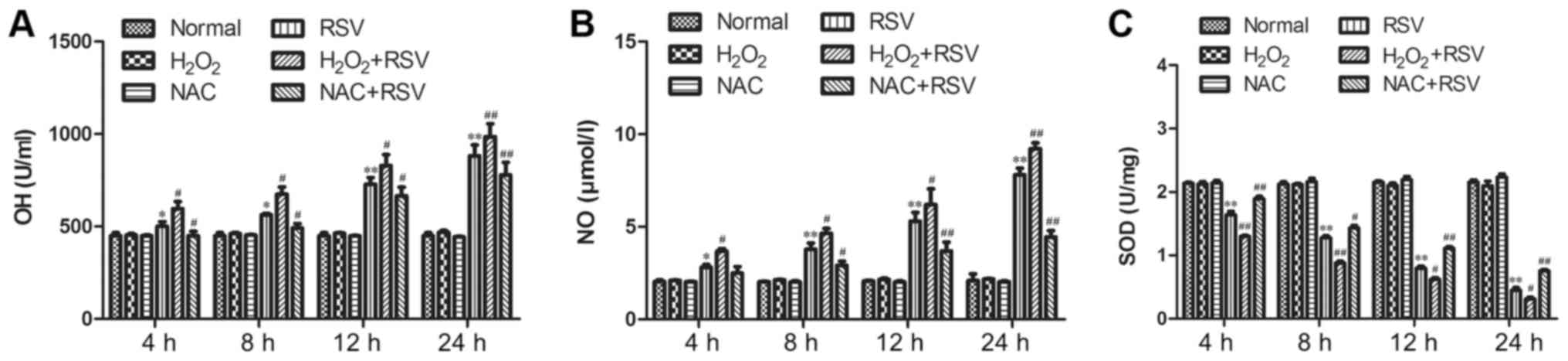 | Figure 1.Production of ·OH and NO in culture
supernatants, and total SOD activity in cell lysates in each group
following different treatments.·OH was measured by (A) Fenton
reaction, and NO was determined by (B) Griess reaction in each
group at various time points. The concentration of ·OH was
significantly different among the RSV-treated group and
H2O2+RSV group or NAC+RSV group at 4 h pi.
The level of NO was also significantly different among the
RSV-treated group and H2O2+RSV group or
NAC+RSV group at 8 h pi. (C) The total SOD activity was assessed by
a commercial assay kit for the determination of total SOD based on
the hydroxylamine method. When compared with the normal cell group,
the total SOD activity in the RSV-treated group was substantially
decreased at 4, 8, 12 and 24 h pi. A significant decrease was also
observed in the H2O2+RSV-treated group
compared with the RSV-treated group. However, SOD activity was
markedly reversed in the NAC+RSV-treated group when compared with
the RSV-treated group and H2O2+RSV-treated
group. Data are expressed as the mean ± standard error mean of
three independent experiments. *P<0.05 and **P<0.01 vs.
normal cell control group; #P<0.05 and
##P<0.01 vs. RSV-treated group. ·OH, hydroxyl free
radical; NO, nitric oxide; pi, post infection; SOD, superoxide
dismutase; RSV, respiratory syncytial virus; NAC,
N-acetyl-L-cysteine; H2O2, hydrogen
peroxide. |
The total SOD activity after different
treatments
The protein level and activity of SOD are important
indicators for the cellular antioxidant stress capacity, and
increased SOD expression can contribute to antioxidant functions
in vivo (16). As shown in
Fig. 1C, the SOD levels were
significantly decreased in a time-dependent manner in the
RSV-infected group compared with those in the normal group at all
time points tested (P<0.01; Fig.
1C). In contrast, although pretreatment of A549 cells with
inactivated-RSV or NAC or H2O2 alone had no
impact on SOD release within 24 h, the administration of NAC+RSV,
but not H2O2+RSV, was able to markedly
improve SOD activity during infection compared with RSV-infected
group (Fig. 1C). Thus, the
antioxidant capacity of RSV-infected A549 cells was enhanced by the
pretreatment of NAC, which suggested that oxidative stress damage
indeed occurred after RSV infection.
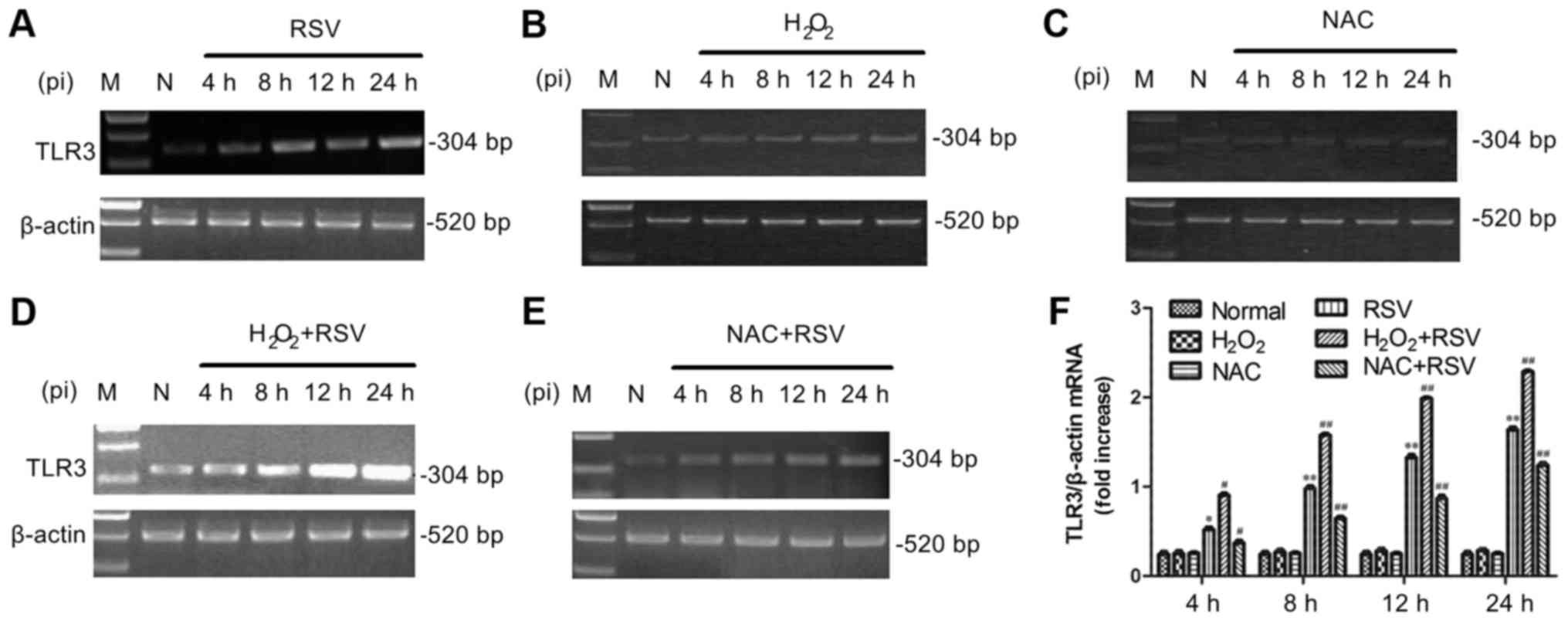
The mRNA levels of TLR3, NF-κB p65,
IRF3 and SOD1 genes after different treatments
TLR3 activation and the mRNA expression levels of
its downstream molecules, including NF-κB p65, IRF3 and SOD1, were
analyzed by semi-quantitative RT-PCR in this study. Figs. 2 and 3 showed the mRNA expression of TLR3,
NF-κB p65, SOD1 and IRF3 in A549 cells with RSV infection (Figs. 2A and 3A) and cultures pretreatment of
H2O2 (Figs.
2D and 3B) or NAC (Figs. 2E and 3C). The results showed that adding
inactivated-RSV (data not shown) or H2O2
(Fig. 2B) or NAC (Fig. 2C) alone has no effect on TLR3 mRNA
expression in A549 cells. However, the RSV infection treatment and
H2O2+RSV treatment significantly elevated
both TLR3 (Fig. 2F) and NF-κB p65
(Fig. 3D) mRNA levels at all time
points tested. Conversely, the mRNA expression of TLR3 and NF-κB
was decreased in the NAC+RSV group compared to the RSV group, which
suggested that NAC played an inhibitory role in RSV-induced TLR3
and NF-κB activation.
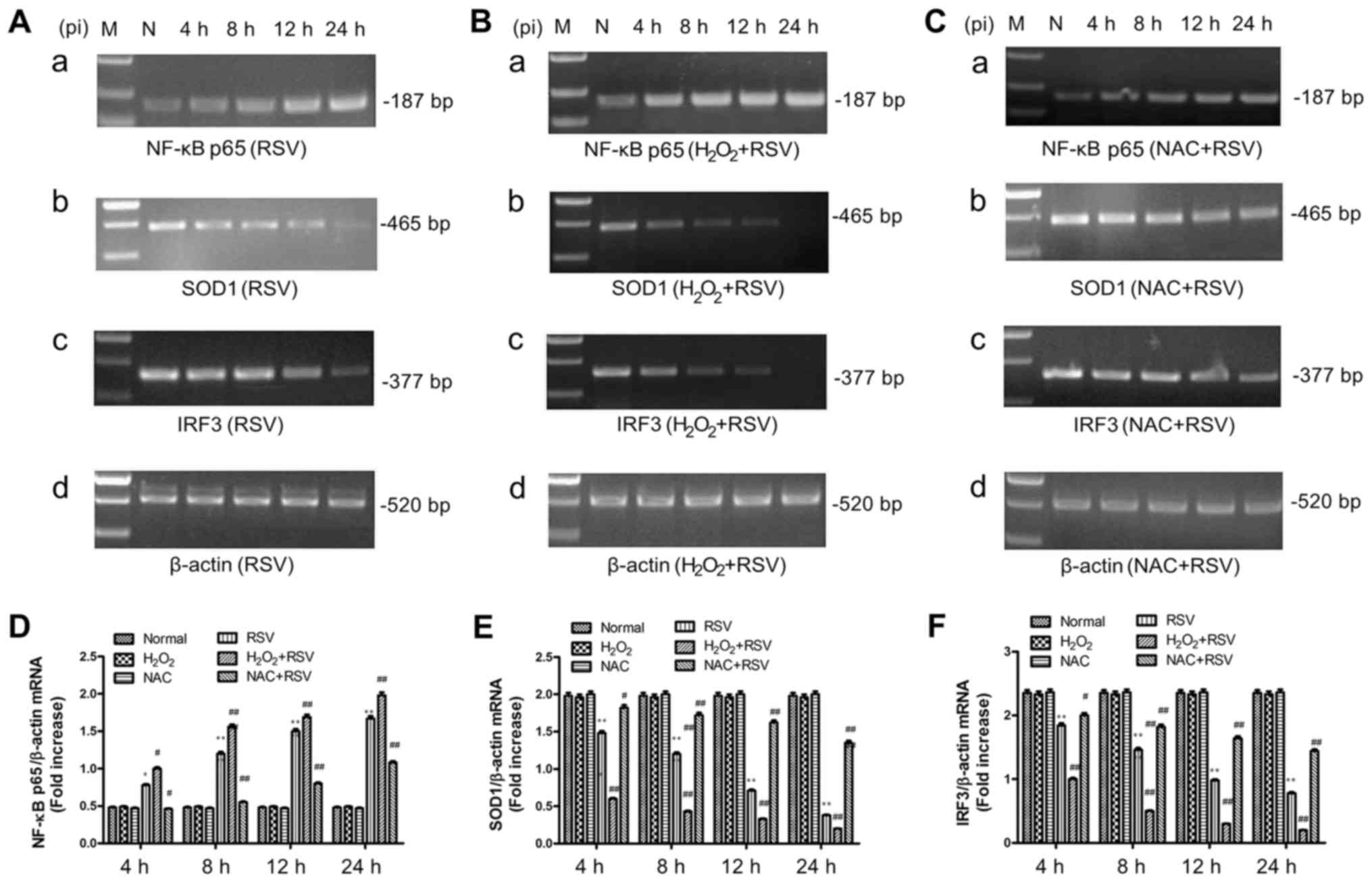 | Figure 3.mRNA expression levels of NF-κB p65,
SOD1 and IRF3 in each group were measured by semi-quantitative
verse transcription-polymerase chain reaction assay. Differences in
the mRNA expression of NF-κB p65, SOD1, IRF3 and β-actin genes were
measured in the (A) RSV-treated group, (B) the
H2O2+RSV-treated group and (C) the
NAC+RSV-treated group, respectively. The relative content of mRNA
levels for (D) NF-κB p65, (E) SOD1 and (F) IRF3 genes were
quantified and calculated using Labworks software. The unit of
densitometry was expressed as fold increase. Data are expressed as
the mean ± standard error mean of three different experiments
groups. *P<0.05 and **P<0.01 vs. normal cell control group;
#P<0.05 and ##P<0.01 vs. RSV-treated
group. NF-κB p65, nuclear factor-κB p65; SOD, superoxide dismutase;
IRF3, interferon regulatory factor-3; pi, post infection; RSV,
respiratory syncytial virus; H2O2, hydrogen
peroxide; NAC, N-acetyl-L-cysteine. |
In RSV-treated and
RSV+H2O2-treated groups, the levels of IRF3
and SOD1 mRNA were markedly decreased with statistically
significant differences, compared to the normal cell group
(Fig. 3). This reduction could be
reversed by the NAC+RSV treatment, but the mRNA levels of IRF3 and
SOD1 in NAC+RSV-treated group were still less than that shown in
the RSV-treated group.
Therefore, the RSV infection in A549 cells enhanced
the mRNA expression of TLR3 and NF-κB p65 but suppress the mRNA
expression of IRF3 and SOD1, indicating that the oxidative stress
inhibitor and agonist altered TLR3 activation and the expression of
relative downstream signaling molecules at the transcriptional
level. These data are in line with the results described above with
the determination of ·OH and NO concentration and total SOD
activity.
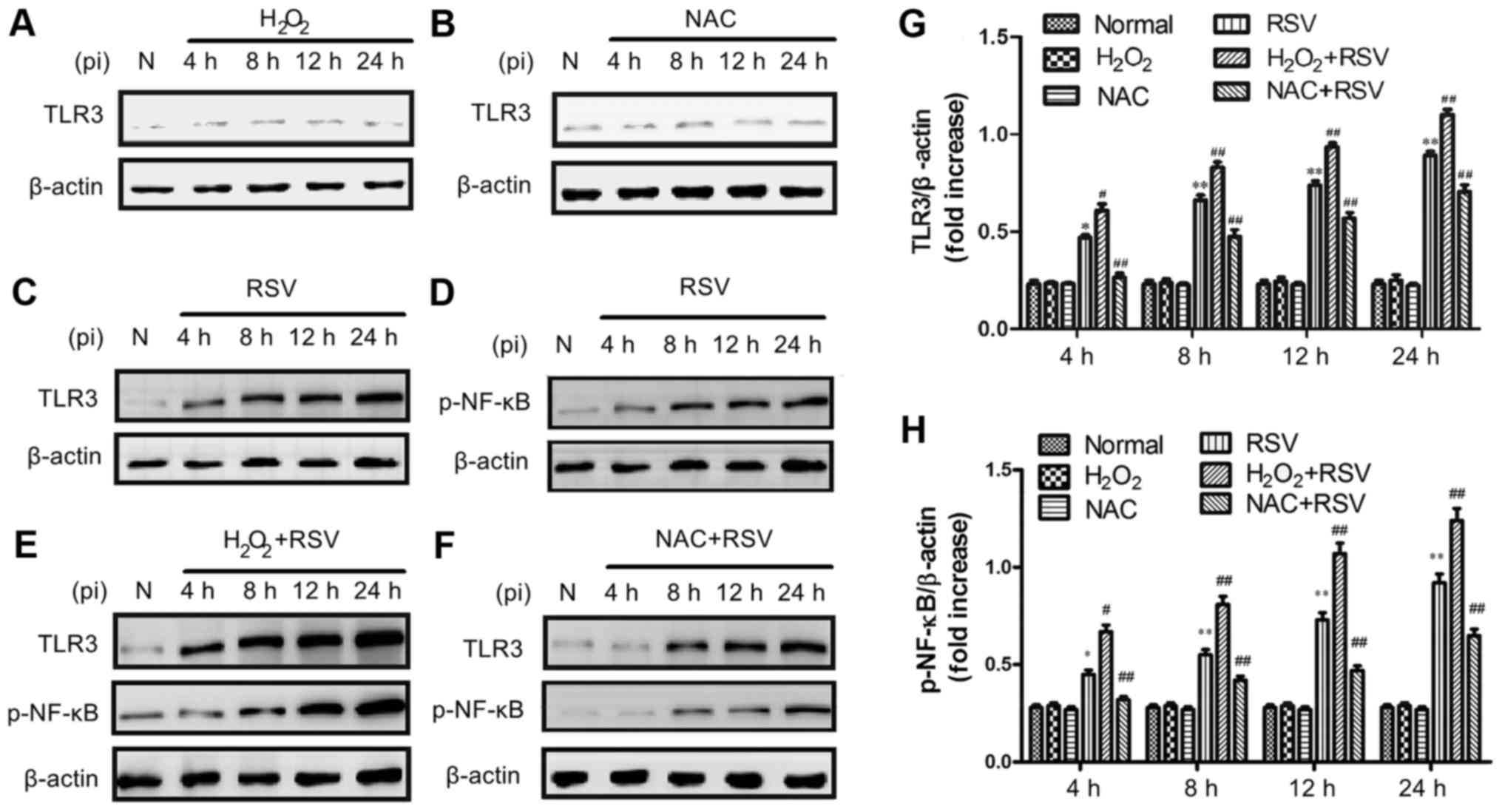
The protein expression of TLR3 and
p-NF-κB after different treatments
Because that TLR3 was recently reported to be able
to recognize the viral dsRNA intermediates produced during RSV
replication and activate NF-κB (20,22),
the expression of both TLR3 and p-NF-κB proteins were studied by
western blot assay. It was found that there were no significant
differences in TLR3 and p-NF-κB changes in inactivated-RSV control
or H2O2 control or NAC control groups at the
indicated concentrations (Fig. 4),
therefore the use of inactivated-RSV or H2O2
or NAC alone had no effect on the experimental results. As shown in
Fig. 4, TLR3 and p-NF-κB protein
were significantly increased in the RSV-treated and the
H2O2+RSV-treated groups compared to the
normal cell control group. However, the treatment with NAC+RSV
attenuated the RSV-induced elevation of TLR3 and p-NF-κB protein,
suggesting that NAC may inhibit RSV-induced TLR3 activation
(Fig. 4F).
These results demonstrate that the elevated TLR3
expression is consistent with the increasing of p-NF-κB protein
expression during RSV infection, and the TLR3 activation can
enhance the up-regulation of its downstream signaling proteins,
including NF-κB.
Discussion
RSV is a highly pathogenic virus that can lead to
severe respiratory diseases in newborns, children, the elderly and
individuals with immune impairment (38,39).
It has been reported that airway inflammation plays a crucial role
in the disease outcome in RSV-infected hosts (40). Although the pathogenesis of RSV
infection remains largely unknown, previous studies have suggested
that a relative overload of oxidants in response to RSV infection
in human airway epithelial cells may have an important impact on
lung injury (41).
Oxidative stress was reported to induce the
production of reactive oxygen species (ROS), causing oxidative
damage in tissue during RSV infection in vivo (11,42).
Also, the decrease of SOD total activity can result in an excess
availability of superoxide and generate hydroxyl radicals that are
associated with the initiation and propagation of lipid
peroxidation (43). Though a study
has reported that RSV infection of A549 cells was able to induce a
significant decrease in SOD1, SOD3, GST and catalase gene
expressions along with the increase of SOD2; however, the total SOD
activity would decrease initially, followed by a subsequent
increase after 24 h (41). The
latter observations are consistent with our findings in the current
study, namely that total SOD activity is decreased within 24 h of
RSV infection.
The activation of TLR2, TLR3, TLR4, and TLR7/8 in
the innate immune system can lead to the strong up-regulation of
SOD2 gene expression in macrophages during microbial infection
(44). To et al (45) had demonstrated that influenza A
virus-induced TLR7 activation enhanced the oxidative burst of
NOX2-oxidase dependence in macrophages, suggesting the occurring of
acute lung injury after influenza A virus infections. Although
oxidative stress was recently found to augment the response of TLR3
to dsRNA in airway epithelial cells through NF-κB pathway (26), to date, there is no study described
the relationship between RSV infection with both TLR3 activation
and oxidative stress generation, therefore, we performed the
experiments and firstly reported that the elevated expression of
TLR3 could be modulated by oxidative stress during RSV
infection.
TLR3 gene expression may be regulated by
RIG-I-induced IFN-β secretion in the early response of host cells
to RSV infection and TLR3 indeed mediates epithelial responses to
RSV infection (46). Our group
previously found that RSV infection induced TLR gene transcription
by recognizing the viral dsRNA genome, and it is likely that RSV
infection promotes a rapid activation of innate responses via the
increased expression of TLR3 (47). However, the molecular mechanism for
RSV-induced TLR3 up-regulation was unclear (48,49).
In the present study, RSV infection was shown to up-regulate both
mRNA and protein expression levels of TLR3 in A549 cells,
furthermore, it was also shown that RSV-induced, TLR3-mediated
early signal events could lead to the activation of NF-κB and IRF3,
both of which were two key transcriptional factors for the
expression of inflammatory cytokines and chemokines in airway
epithelial cells. Pretreatment with H2O2
before RSV infection increases TLR3 expression and TLR3-mediated
NF-κB activity, whereas pretreatment with antioxidant NAC inhibits
the activation of TLR3 pathways, including pNF-κB expression
(Figs. 2–4). These results are in agreement with
our previous finding that RSV can induce TLR3 expression and
NF-κB/RelA subunit phosphorylation and transcriptional activation
(30).
Matsukura et al (50) reported that dsRNAs such as poly
(I:C) bound to TLR3 that distributed on the cell surface and
induced some chemokines and cytokines gene expressions through
activation of NF-κB and IRF3. Their results indicated that dsRNA
could increase the expression of inflammatory cytokines and
chemokines via TLR3-NF-κB and TLR3-IRF3 signaling in airway
epithelial cells (50,51). Liu et al (46) showed that the TLR3 knockdown
mediated by siRNA significantly reduced NF-κB/RelA transcriptional
levels by blocking the activating phosphorylation of NF-κB/RelA at
serine residue 276. It was also demonstrated that NADPH oxidase 2
(NOX2) mediates ROS production in RSV-infected human airway
epithelial cells and that NOX2 acts upstream of the phosphorylation
of both IκBα at Ser32 and of p65 at Ser536 in
RSV-infected A549 cells and in human bronchial epithelial cells
(52). Jamaluddin et al
(9) demonstrated the RSV-induced
ROS induced activation of RelA and RSV-induced ROS formation also
resulted in STAT activation and IRF gene expression induction
(10). Unanimously, our current
study revealed that oxidative stress was involved in RSV-augmented
NF-κB and IRF activation in airway epithelial cells and lead to an
increase of TLR3 expression.
It is worth noting that preferential induction of
TLR3 is also observed in human astrocytes after its exposure to
H2O2 to induce oxidative stress (53) and that H2O2,
but not poly (I:C), appears to activate NF-κB and nuclear
translocation of p65 in human SH-SY5Y neuroblastoma cells (54). ROS may potentiate TLR3 expression
through NOX2 signaling by RSV infection (55).
As far as the investigations are concerned, we
further confirmed that there were no significant differences in
TLR3 signaling in A549 cells pretreated with either
H2O2 or NAC without RSV infection (Fig. 2B, C and Fig. 4A, B), as previously described
(26,56). Geiler et al (56) reported that there was no
significant decrease in virus titer produced in A549 cells as a
result of NAC treatment (5 mM) at 48 h pi with influenza A virus;
thus, we chose to use NAC at 5 mM in the current study.
Unfortunately, this concentration of NAC did not fully recover SOD
activity to the normal levels, nor did we detect the effect of
H2O2 on virus titer, both of which are the
limitations of the present study.
The transcriptional activators of the NF-κB family
participated in the modulation of cell proliferation,
differentiation, apoptosis, inflammation, immunity and cytokine
expression in response to various types of stimulation (57). In the inactive state, the NF-κB is
mainly located in the cytoplasm, forming complexes with IκB. Once
it is stimulated by extracellular stimuli such as viruses, IκB can
be rapidly phosphorylated and degraded, allowing the release of
NF-κB p65 and subsequent NF-κB p65 translocation to the nucleus
with the increase of NF-κB regulated gene expression (58). IRF3 acts as an important
transcriptional regulator in antiviral immune responses, viral
infection can also induce the IRF3 phosphorylation and its
translocation to the nucleus (50). The binding of IRF3 to
IFN-stimulated response element (ISRE) in the promoters of type I
IFN genes is thought to activate the transcription of these genes
(59). However, non-structural
proteins (NS1 and NS2) of RSV have been demonstrated to inhibit the
induction of IFN-α/β in A549 cells and human macrophages (60,61).
It was proposed that RSV NS1 protein limits IRF3 nuclear
translocation through blocking its phosphorylation by decreasing
the levels of 2 kinases upstream of IRF3 activation, TRAF3 and IKKε
(62). RSV NS2 protein was also
proposed to act on downstream molecules such as TRAF3 and
ultimately inhibit IFN-β expression. Our results found that RSV
infection indeed inhibits early IRF3 activation at 24 h pi, which
is consistent with Hosakote's study (63). Moreover, oxidant
H2O2 pretreatment further inhibits the early
activation of IRF3 (Fig. 3F),
indicating that an RSV-mediated redox-sensitive pathway probably
inhibits IRF3 activation. Further studies can be done to
investigate the mechanisms of interaction between NF-κB and IRF
under conditions of RSV infection.
This study was conducted using A549 cells derived
from human lung cancer tissue, which is similar to previous studies
(41,56,64–66).
A549 cells have also been widely used in the research of oxidative
stress by RSV infection (41,64,67)
or the alteration of TLR3 expression by RSV (21,68).
Therefore, we believe that A549 cells, as an adenocarcinomic
alveolar basal epithelial cell line, will have no intervening
effect on the analysis and conclusion of the study. However, it
will be interesting to investigate whether
H2O2 and NAC have the same effects on
RSV-induced TLR3, NF-κB and IRF3 expression in non-carcinomic
primary epithelial cells in the future.
For production of type I IFN, RSV is generally
considered as a poor inducer of IFN-α and IFN-β, comparing with
other RNA viruses (69,70). However, RSV is previously reported
to induce high levels of IFN-β expression in cultures of various
types of human fibroblasts, respiratory epithelial cells, and
mesenchymal stem cells (MSCs) (71–73).
Besides, RSV is also reported to induce high levels of IFN-α
expression in different subsets of dendritic cells (DC) (74–77).
Furthermore, RSV treatment is also reported to induce a type I IFN
response in both human cord blood-derived mast cell (CBMCs) and
peripheral blood derived mast cells (78,79).
Therefore, the results on the type I IFN production in responding
to RSV infection are controversial and it seems that many different
factors can determine the type I IFN production under the infection
of RSV, such as different cell types and different viral strains
(71,72,74–77,80).
On these basis, we tentatively thought that the response of IFN-α
and IFN-β to the infection of RSV long strain in A549 cells, which
was related to the experiments in this study, might have various
possible results. Thus, for the reason that the inducement of type
I IFN in A549 cells may not be representative, we consider that it
is not necessary to detect IFN-α and IFN-β in this study.
Taken together, we investigated the relationship
between TLR3 expression and oxidative stress modulation in
RSV-infected A549 cells in this study. We used oxidative stress
agonist H2O2 and inhibitor NAC (equivalent to
oxidant and antioxidant) to interfere with RSV infection from both
positive and negative sides to determine the effect of oxidative
stress on TLR3 expression and TLR3-mediated inflammatory and immune
pathways. Our results showed that, in RSV-infected A549 cells, the
production of hydroxyl free radical (·OH) and nitric oxide (NO)
were induced, while the superoxide dismutase (SOD) activity was
reduced. On the variation of gene expression, our results showed
that both of mRNA and protein expression levels of TLR3 and NF-κB
were up-regulated. Pretreatment of H2O2 plus
RSV infection enhanced RSV-induced TLR3 and NF-κB expression,
whereas Pretreatment of NAC plus RSV infection reduced them. These
results indicated that oxidative stress was a critical regulator of
TLR3 activation in RSV infection and suggested that oxidative
stress might potentiate increasing the TLR3 expression in A549
cells after RSV infection, which might partly explain the
enhancement of the associated downstream signaling pathway,
including NF-κB activity. The elevated TLR3 and NF-κB activities
might be key factors in interpreting oxidative stress effects
induction. These findings may contribute to the study of RSV
pathogenesis and the development of RSV prevention and control.
To further confirm the role of oxidative stress
discovered in this study, we plan to knock-out TLR3 and p-NF-κB
genes using the CRISPR/Cas9 technique in RSV future work. First, we
will synthesize three high-grade small-guide RNAs (sgRNAs) that
could specifically identify TLR3 and p-NF-κB genes and inserted
them into lenti CRISPRv2 plasmid. Second, 293T cells will be
transfected with the recombinant sgRNA-lenti CRISPRv2 plasmid to
yield subsequent sgRNA-Cas9 lentivirus prior to its further
infection of A549 cells. Third, positive A549 cells will be
screened using puromycin and be validated by PCR and western blot.
Last, sequencing analysis will be adopted to confirm the mutation
site of the obtained genes-knockout A549 cells.
Besides, we also plan to over-express TLR3 and
p-NF-κB genes by lentiviral transfection to verify the function of
oxidative stress. First, we will amplify TLR3 and p-NF-κB genes by
RT-PCR and insert them into the lentiviral vector pLENTI-cGFP using
the homologous recombination method. Second, the constructed
recombinant vector will be confirmed by DNA sequencing and be
co-transfected with psPAX2 and pMD2. G helper plasmids into HEK293T
packaging cells to produce the lentiviral particles on the ratio of
3:2:1 by PEI transfection reagent. Third, the lentiviral particles
will be transduced into A549 cells and the infection efficiency
will be measured by Fluorescence Microscopy. Last, the
GFP-tag-expressed cells will be sorted by the Flow Cytometer (FCM)
after 2 weeks and the protein expressions of TLR3 and p-NF-κB and
GFP in the stable cell lines will be confirmed by western blot.
Once the TLR3 and p-NF-κB gene deletion and
over-expression are achieved, the established cell lines will be
subjected to molecular biological tests in the cases of RSV
infection and H2O2 or NAC intervention. We
hope that these experiments will facilitate further investigation
of the molecular mechanism of oxidative stress modulation of the
TLR3 expression in RSV infection.
Acknowledgements
The authors would like to thank Professor Hai-ming
Wei (University of Science and Technology of China, Hefei, Anhui,
China) for kindly providing the A549 cells and RSV Long strain. The
authors are also grateful to Ms. Xiao-yan Zhang (Department of
Microbiology, Anhui Medical University, Hefei, Anhui, China) for
her technical assistance, and Mr. Hai-yang Yu (Department of
Microbiology, Anhui Medical University) for his helpful discussions
and suggestions during the preparation of this manuscript.
Funding
The present study was supported by grants from the
Natural Science Foundation of China (grant no. 81371797), the
Natural Science Foundation of Anhui Province of China (grant no.
1308085MH129) and the Key Project of Natural Science Research of
Anhui Education Department (grant no. KJ2012A152).
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors' contributions
MMW and SHH conceived and planned the study. XW
performed the statistical analysis and JXC analyzed the data. WWL
purified the RSV Long strain, and TS and HQ performed RT-PCR and
western blot experiments. MMW, ML and CLZ carried out the chemical
detection experiments. MMW, ML and CLZ wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
dsRNA
|
double stranded RNA
|
|
GSH
|
glutathione
|
|
H2O2
|
hydrogen peroxide
|
|
IFN
|
interferon
|
|
IRF3
|
interferon regulatory factor-3
|
|
ISRE
|
IFN-stimulated response element
|
|
MDA
|
malondialdehyde
|
|
NAC
|
N-acetyl-L-cysteine
|
|
NF-κB
|
nuclear factor-κB
|
|
NO
|
nitric oxide
|
|
NS
|
nonstructural protein
|
|
·OH
|
hydroxyl free radical
|
|
PAMPs
|
pathogen-associated molecular
patterns
|
|
PMSF
|
phenylmethylsulfonyl fluoride
|
|
PVDF
|
polyvinylidene fluoride
|
|
ROS
|
reactive oxygen species
|
|
RT-PCR
|
reverse transcription polymerase
chain reaction
|
|
RSV
|
respiratory syncytial virus
|
|
SOD
|
superoxide dismutase
|
|
TLRs
|
Toll-like receptors
|
|
TLR3
|
Toll-like receptors 3
|
|
pi
|
post infection
|
|
RT
|
room temperature
|
References
|
1
|
Nair H, Verma VR, Theodoratou E, Zgaga L,
Huda T, Simões EA, Wright PF, Rudan I and Campbell H: An evaluation
of the emerging interventions against Respiratory Syncytial Virus
(RSV)-associated acute lower respiratory infections in children.
BMC Public Health. 11 Suppl 3:S302011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Piedimonte G: Respiratory syncytial virus
and asthma: Speed-dating or long-term relationship? Curr Opin
Pediatr. 25:344–349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mejías A, Chávez-Bueno S, Gómez AM, Somers
C, Estripeaut D, Torres JP, Jafri HS and Ramilo O: Respiratory
syncytial virus persistence: Evidence in the mouse model. Pediatr
Infect Dis J. 27 10 Suppl:S60–S62. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wedzicha JA: Role of viruses in
exacerbations of chronic obstructive pulmonary disease. Proc Am
Thorac Soc. 1:115–120. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCutcheon KM, Jordan R, Mawhorter ME,
Noton SL, Powers JG, Fearns R, Cihlar T and Perron M: The
interferon type I/III response to respiratory syncytial virus
infection in airway epithelial cells can be attenuated or amplified
by antiviral treatment. J Virol. 90:1705–1717. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hosakote YM, Komaravelli N, Mautemps N,
Liu T, Garofalo RP and Casola A: Antioxidant mimetics modulate
oxidative stress and cellular signaling in airway epithelial cells
infected with respiratory syncytial virus. Am J Physiol Lung Cell
Mol Physiol. 303:L991–L1000. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bakunina N, Pariante CM and Zunszain PA:
Immune mechanisms linked to depression via oxidative stress and
neuroprogression. Immunology. 10–Jan;2015.(Epub Ahead of Print).
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Antonio AM and Druse MJ: Antioxidants
prevent ethanol-associated apoptosis in fetal rhombencephalic
neurons. Brain Res. 1204:16–23. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jamaluddin M, Tian B, Boldogh I, Garofalo
RP and Brasier AR: Respiratory syncytial virus infection induces a
reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required
for cytokine expression. J Virol. 83:10605–10615. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu T, Castro S, Brasier AR, Jamaluddin M,
Garofalo RP and Casola A: Reactive oxygen species mediate
virus-induced STAT activation: Role of tyrosine phosphatases. J
Biol Chem. 279:2461–2469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miller AL, Bowlin TL and Lukacs NW:
Respiratory syncytial virus-induced chemokine production: Linking
viral replication to chemokine production in vitro and in vivo. J
Infect Dis. 189:1419–1430. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reiter RJ, Tan DX, Terron MP, Flores LJ
and Czarnocki Z: Melatonin and its metabolites: New findings
regarding their production and their radical scavenging actions.
Acta Biochim Pol. 54:1–9. 2007.PubMed/NCBI
|
|
13
|
Stark JM, Khan AM, Chiappetta CL, Xue H,
Alcorn JL and Colasurdo GN: Immune and functional role of nitric
oxide in a mouse model of respiratory syncytial virus infection. J
Infect Dis. 191:387–395. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chtourou Y, Aouey B, Kebieche M and Fetoui
H: Protective role of naringin against cisplatin induced oxidative
stress, inflammatory response and apoptosis in rat striatum via
suppressing ROS-mediated NF-κB and P53 signaling pathways. Chem
Biol Interact. 239:76–86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mastronarde JG, Monick MM and Hunninghake
GW: Oxidant tone regulates IL-8 production in epithelium infected
with respiratory syncytial virus. Am J Respir Cell Mol Biol.
13:237–244. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang SH, Cao XJ, Liu W, Shi XY and Wei W:
Inhibitory effect of melatonin on lung oxidative stress induced by
respiratory syncytial virus infection in mice. J Pineal Res.
48:109–116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marshak-Rothstein A and Rifkin IR:
Immunologically active autoantigens: The role of toll-like
receptors in the development of chronic inflammatory disease. Annu
Rev Immunol. 25:419–441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Meylan E and Tschopp J: Toll-like
receptors and RNA helicases: Two parallel ways to trigger antiviral
responses. Mol Cell. 22:561–569. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tatematsu M, Nishikawa F, Seya T and
Matsumoto M: Toll-like receptor 3 recognizes incomplete stem
structures in single-stranded viral RNA. Nat Commun. 4:18332013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alexopoulou L, Holt AC, Medzhitov R and
Flavell RA: Recognition of double-stranded RNA and activation of
NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dou Y, Zhao Y, Zhang ZY, Mao HW, Tu WW and
Zhao XD: Respiratory syncytial virus infection induces higher
Toll-like receptor-3 expression and TNF-α production than human
metapneumovirus infection. PLoS One. 8:e734882013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rot A and von Andrian UH: Chemokines in
innate and adaptive host defense: Basic chemokinese grammar for
immune cells. Annu Rev Immunol. 22:891–928. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Majumder S, Zhou LZ, Chaturvedi P, Babcock
G, Aras S and Ransohoff RM: p48/STAT-1alpha-containing complexes
play a predominant role in induction of IFN-gamma-inducible
protein, 10 kDa (IP-10) by IFN-gamma alone or in synergy with
TNF-alpha. J Immunol. 161:4736–4744. 1998.PubMed/NCBI
|
|
25
|
Murray LA, Knight DA, McAlonan L,
Argentieri R, Joshi A, Shaheen F, Cunningham M, Alexopolou L,
Flavell RA, Sarisky RT and Hogaboam CM: Deleterious role of TLR3
during hyperoxia-induced acute lung injury. Am J Respir Crit Care
Med. 178:1227–1237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Koarai A, Sugiura H, Yanagisawa S,
Ichikawa T, Minakata Y, Matsunaga K, Hirano T, Akamatsu K and
Ichinose M: Oxidative stress enhances toll-like receptor 3 response
to double-stranded RNA in airway epithelial cells. Am J Respir Cell
Mol Biol. 42:651–660. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cotgreave IA: N-acetylcysteine:
Pharmacological considerations and experimental and clinical
applications. Adv Pharmacol. 38:205–227. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jaspers I, Ciencewicki JM, Zhang W,
Brighton LE, Carson JL, Beck MA and Madden MC: Diesel exhaust
enhances influenza virus infections in respiratory epithelial
cells. Toxicol Sci. 85:990–1002. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Song JJ, Lim HW, Kim K, Kim KM, Cho S and
Chae SW: Effect of caffeic acid phenethyl ester (CAPE) on H2O2
induced oxidative and inflammatory responses in human middle ear
epithelial cells. Int J Pediatr Otorhinolaryngol. 76:675–679. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Collins PL, Chanock RM and Murphy BR:
Respiratory syncytial virus, in fields virology. 4th Edition. Knipe
D and Howley P: Lippincott Williams & Wilkins; Philadelphia:
pp. 1443–1485. 2001
|
|
31
|
Huang SH, Cao XJ and Wei W: Melatonin
decreases TLR3-mediated inflammatory factor expression via
inhibition of NF-kappa B activation in respiratory syncytial
virus-infected RAW264.7 macrophages. J Pineal Res. 45:93–100. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lloyd RV, Hanna PM and Mason RP: The
origin of the hydroxyl radical oxygen in the Fenton reaction. Free
Radic Biol Med. 22:885–888. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wink DA, Wink CB, Nims RW and Ford PC:
Oxidizing intermediates generated in the Fenton reagent: Kinetic
arguments against the intermediacy of the hydroxyl radical. Environ
Health Perspect. 102 Suppl 3:S11–S15. 1994. View Article : Google Scholar
|
|
34
|
Kalaivani P, Saranya S, Poornima P,
Prabhakaran R, Dallemer F, Padma Vijaya V and Natarajan K:
Biological evaluation of new nickel(II) metallates: Synthesis,
DNA/protein binding and mitochondrial mediated apoptosis in human
lung cancer cells (A549) via ROS hypergeneration and depletion of
cellular antioxidant pool. Eur J Med Chem. 82:584–599. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang H, Wei W, Shen YX, Dong C, Zhang LL,
Wang NP, Yue L and Xu SY: Protective effect of melatonin against
liver injury in mice induced by Bacillus Calmette-Guerin plus
lipopolysaccharide. World J Gastroenterol. 10:2690–2696. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou JY and Prognon P: Raw material
enzymatic activity determination: A specific case for validation
and comparison of analytical methods-the example of superoxide
dismutase (SOD). J Pharm Biomed Anal. 40:1143–1148. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Balenger SL, McClure CJ and Hill GE:
Primer design and transcript quantification of a highly multiplexed
RT-PCR for a nonmodel avian species. Mol Ecol Resour. 12:116–122.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huo X, Fang B, Liu L, Yu H, Chen H, Zheng
J, Zhang Y, Xu Z, Klena JD, Varma JK, et al: Clinical and
epidemiologic characteristics of respiratory syncytial virus
infection among children aged <5 years, Jingzhou City, China,
2011. J Infect Dis. 208 Suppl 3:S184–S188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Collins PL and Graham BS: Viral and host
factors in human respiratory syncytial virus pathogenesis. J Virol.
82:2040–2055. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Segovia J, Sabbah A, Mgbemena V, Tsai SY,
Chang TH, Berton MT, Morris IR, Allen IC, Ting JP and Bose S:
TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux
activates NLRP3/ASC inflammasome during respiratory syncytial virus
infection. PLoS One. 7:e296952012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hosakote YM, Liu T, Castro SM, Garofalo RP
and Casola A: Respiratory syncytial virus induces oxidative stress
by modulating antioxidant enzymes. Am J Respir Cell Mol Biol.
41:348–357. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Roy J, Pallepati P, Bettaieb A, Tanel A
and Averill-Bates DA: Acrolein induces a cellular stress response
and triggers mitochondrial apoptosis in A549 cells. Chem Biol
Interact. 181:154–167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao H, Liu J, Pan S, Sun Y, Li Q, Li F,
Ma L and Guo Q: SOD mRNA and MDA expression in rectus femoris
muscle of rats with different eccentric exercise programs and time
points. PLoS One. 8:e736342013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rakkola R, Matikainen S and Nyman TA:
Proteome analysis of human macrophages reveals the upregulation of
manganese-containing superoxide dismutase after toll-like receptor
activation. Proteomics. 7:378–384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
To EE, Broughton BR, Hendricks KS, Vlahos
R and Selemidis S: Influenza A virus and TLR7 activation potentiate
NOX2 oxidase-dependent ROS production in macrophages. Free Radic
Res. 48:940–947. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu P, Jamaluddin M, Li K, Garofalo RP,
Casola A and Brasier AR: Retinoic acid-inducible gene I mediates
early antiviral response and Toll-like receptor 3 expression in
respiratory syncytial virus-infected airway epithelial cells. J
Virol. 81:1401–1411. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang S, Wei W and Yun Y: Upregulation of
TLR7 and TLR3 gene expression in the lung of respiratory syncytial
virus infected mice. Wei Sheng Wu Xue Bao. 49:239–245.
2009.PubMed/NCBI
|
|
48
|
Groskreutz DJ, Monick MM, Powers LS,
Yarovinsky TO, Look DC and Hunninghake GW: Respiratory syncytial
virus induces TLR3 protein and protein kinase R, leading to
increased double-stranded RNA responsiveness in airway epithelial
cells. J Immunol. 176:1733–1740. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rudd BD, Smit JJ, Flavell RA, Alexopoulou
L, Schaller MA, Gruber A, Berlin AA and Lukacs NW: Deletion of TLR3
alters the pulmonary immune environment and mucus production during
respiratory syncytial virus infection. J Immunol. 176:1937–1942.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Matsukura S, Kokubu F, Kurokawa M,
Kawaguchi M, Ieki K, Kuga H, Odaka M, Suzuki S, Watanabe S,
Takeuchi H, et al: Synthetic double-stranded RNA induces multiple
genes related to inflammation through Toll-like receptor 3
depending on NF-kappaB and/or IRF-3 in airway epithelial cells.
Clin Exp Allergy. 36:1049–1062. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ieki K, Matsukura S, Kokubu F, Kimura T,
Kuga H, Kawaguchi M, Odaka M, Suzuki S, Watanabe S, Takeuchi H, et
al: Double-stranded RNA activates RANTES gene transcription through
co-operation of nuclear factor-kappaB and interferon regulatory
factors in human airway epithelial cells. Clin Exp Allergy.
34:745–752. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fink K, Duval A, Martel A, Soucy-Faulkner
A and Grandvaux N: Dual role of NOX2 in respiratory syncytial
virus- and sendai virus-induced activation of NF-kappaB in airway
epithelial cells. J Immunol. 180:6911–6922. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bsibsi M, Persoon-Deen C, Verwer RW,
Meeuwsen S, Ravid R and Van Noort JM: Toll-like receptor 3 on adult
human astrocytes triggers production of neuroprotective mediators.
Glia. 53:688–695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Larouche A, Berube P, Sarret P and Grignon
S: Subacute H2O2, but not poly(IC), upregulates dopamine D2
receptors in retinoic acid differentiated SH-SY5Y neuroblastoma.
Synapse. 62:70–73. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Grandvaux N, Soucy-Faulkner A and Fink K:
Innate host defense: Nox and Duox on phox's tail. Biochimie.
89:1113–1122. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Geiler J, Michaelis M, Naczk P, Leutz A,
Langer K, Doerr HW and Cinatl J Jr: N-acetyl-L-cysteine (NAC)
inhibits virus replication and expression of pro-inflammatory
molecules in A549 cells infected with highly pathogenic H5N1
influenza A virus. Biochem Pharmacol. 79:413–420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dong QG, Sclabas GM, Fujioka S, Schmidt C,
Peng B, Wu T, Tsao MS, Evans DB, Abbruzzese JL, McDonnell TJ and
Chiao PJ: The function of multiple IkappaB: NF-kappaB complexes in
the resistance of cancer cells to Taxol-induced apoptosis.
Oncogene. 21:6510–6519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ji K, Xing C, Jiang F, Wang X, Guo H, Nan
J, Qian L, Yang P, Lin J, Li M, et al: Benzo[a]pyrene induces
oxidative stress and endothelial progenitor cell dysfunction via
the activation of the NF-κB pathway. Int J Mol Med. 31:922–930.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wang XA, Zhang R, She ZG, Zhang XF, Jiang
DS, Wang T, Gao L, Deng W, Zhang SM, Zhu LH, et al: Interferon
regulatory factor 3 constrains IKKβ/NF-κB signaling to alleviate
hepatic steatosis and insulin resistance. Hepatology. 59:870–885.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Spann KM, Tran KC and Collins PL: Effects
of nonstructural proteins NS1 and NS2 of human respiratory
syncytial virus on interferon regulatory factor 3, NF-kappaB, and
proinflammatory cytokines. J Virol. 79:5353–5362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ling Z, Tran KC and Teng MN: Human
respiratory syncytial virus nonstructural protein NS2 antagonizes
the activation of beta interferon transcription by interacting with
RIG-I. J Virol. 83:3734–3742. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ren J, Liu T, Pang L, Li K, Garofalo RP,
Casola A and Bao X: A novel mechanism for the inhibition of
interferon regulatory factor-3-dependent gene expression by human
respiratory syncytial virus NS1 protein. J Gen Virol. 92:2153–2159.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wright PF, Karron RA, Madhi SA, Treanor
JJ, King JC, O'Shea A, Ikizler MR, Zhu Y, Collins PL, Cutland C, et
al: The interferon antagonist NS2 protein of respiratory syncytial
virus is an important virulence determinant for humans. J Infect
Dis. 193:573–581. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hosakote YM, Brasier AR, Casola A,
Garofalo RP and Kurosky A: Respiratory syncytial virus infection
triggers epithelial HMGB1 release as a damage-associated molecular
pattern promoting a monocytic inflammatory response. J Virol.
90:9618–9631. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hosakote YM, Jantzi PD, Esham DL, Spratt
H, Kurosky A, Casola A and Garofalo RP: Viral-mediated inhibition
of antioxidant enzymes contributes to the pathogenesis of severe
respiratory syncytial virus bronchiolitis. Am J Respir Crit Care
Med. 183:1550–1560. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dave KA, Norris EL, Bukreyev AA, Headlam
MJ, Buchholz UJ, Singh T, Collins PL and Gorman JJ: A comprehensive
proteomic view of responses of A549 type II alveolar epithelial
cells to human respiratory syncytial virus infection. Mol Cell
Proteomics. 13:3250–3269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Mochizuki H, Todokoro M and Arakawa H: RS
virus-induced inflammation and the intracellular glutathione redox
state in cultured human airway epithelial cells. Inflammation.
32:252–264. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Xu X, Zheng J, Zheng K, Hou Y, Zhao F and
Zhao D: Respiratory syncytial virus NS1 protein degrades STAT2 by
inducing SOCS1 expression. Intervirology. 57:65–73. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hall CB, Douglas RG Jr, Simons RL and
Geiman JM: Interferon production in children with respiratory
syncytial, influenza, and parainfluenza virus infections. J
Pediatr. 93:28–32. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Roberts NJ Jr, Hiscott J and Signs DJ: The
limited role of the human interferon system response to respiratory
syncytial virus challenge: Analysis and comparison to influenza
virus challenge. Microb Pathog. 12:409–414. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Garofalo R, Mei F, Espejo R, Ye G,
Haeberle H, Baron S, Ogra PL and Reyes VE: Respiratory syncytial
virus infection of human respiratory epithelial cells up-regulates
class I MHC expression through the induction of IFN-beta and IL-1
alpha. J Immunol. 157:2506–2513. 1996.PubMed/NCBI
|
|
72
|
Jamaluddin M, Wang S, Garofalo RP, Elliott
T, Casola A, Baron S and Brasier AR: IFN-beta mediates coordinate
expression of antigen-processing genes in RSV-infected pulmonary
epithelial cells. Am J Physiol Lung Cell Mol Physiol.
280:L248–L257. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cheung MB, Sampayo-Escobar V, Green R,
Moore ML, Mohapatra S and Mohapatra SS: Respiratory syncytial
virus-infected mesenchymal stem cells regulate immunity via
interferon beta and indoleamine-2,3-dioxygenase. PLoS One.
11:e01637092016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hornung V, Schlender J, Guenthner-Biller
M, Rothenfusser S, Endres S, Conzelmann KK and Hartmann G:
Replication-dependent potent IFN-alpha induction in human
plasmacytoid dendritic cells by a single-stranded RNA virus. J
Immunol. 173:5935–5943. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Schlender J, Hornung V, Finke S,
Günthner-Biller M, Marozin S, Brzózka K, Moghim S, Endres S,
Hartmann G and Conzelmann KK: Inhibition of toll-like receptor 7-
and 9-mediated alpha/beta interferon production in human
plasmacytoid dendritic cells by respiratory syncytial virus and
measles virus. J Virol. 79:5507–5515. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Guerrero-Plata A, Casola A, Suarez G, Yu
X, Spetch L, Peeples ME and Garofalo RP: Differential response of
dendritic cells to human metapneumovirus and respiratory syncytial
virus. Am J Respir Cell Mol Biol. 34:320–329. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang H, Peters N and Schwarze J:
Plasmacytoid dendritic cells limit viral replication, pulmonary
inflammation, and airway hyperresponsiveness in respiratory
syncytial virus infection. J Immunol. 177:6263–6270. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Al-Afif A, Alyazidi R, Oldford SA, Huang
YY, King CA, Marr N, Haidl ID, Anderson R and Marshall JS:
Respiratory syncytial virus infection of primary human mast cells
induces the selective production of type I interferons, CXCL10, and
CCL4. J Allergy Clin Immunol. 136:1346–1354.e1. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kulka M, Alexopoulou L, Flavell RA and
Metcalfe DD: Activation of mast cells by double-stranded RNA:
Evidence for activation through Toll-like receptor 3. J Allergy
Clin Immunol. 114:174–182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hillyer P, Mane VP, Chen A, Dos Santos MB,
Schramm LM, Shepard RE, Luongo C, Le Nouën C, Huang L, Yan L, et
al: Respiratory syncytial virus infection induces a subset of types
I and III interferons in human dendritic cells. Virology.
504:63–72. 2017. View Article : Google Scholar : PubMed/NCBI
|