Introduction
Alagille syndrome (OMIM no. 118450), also known as
arteriohepatic dysplasia, is a rare and complex autosomal dominant
disorder characterized by developmental abnormalities of the heart,
liver, vertebrae, eyes and face. The incidence of ALGS was
estimated to be at least 1:70,000 live births (1,2).
Traditionally, the diagnosis of ALGS is based on finding of
interlobular bile duct paucity in liver biopsy, accompanied by at
least 3 of 5 major clinical features: Chronic cholestasis,
congenital heart defects, ocular abnormalities (posterior
embryotoxon), vertebral defects (butterfly vertebra) and
characteristic facial phenotypes (pointed chin, broad forehead and
bulbous nose) (3–5). Renal and vascular abnormalities are
less commonly seen in a smaller percentage of cases (4). The phenotype of this disorder is
highly variable, ranging from no apparent clinical involvement to
severe disease leading to death. Therefore, the diagnosis of ALGS
is impaired by its highly variable expressivity.
Two members of the Notch signaling
pathway-JAG1 and NOTCH2-have been implicated in ALGS
(6–8). The Notch signaling pathway is
evolutionarily conserved, and is involved in both cell fate
determination and organogenesis (9). The JAG1 gene encodes the JAG1
protein belonging to the family of Notch ligands, and NOTCH2
encodes one of the four Notch receptors. Up to 94% of ALGS cases
are caused by mutations of the JAG1 gene (10). Only 1% cases are caused by
NOTCH2 mutations, in which renal malformations may be more
common, but heart, skeletal and facial feature anomalies may be
less frequent (11,12). More than 400 JAG1 mutations
have been identified to date, including whole gene deletions,
splice site alterations, and missense, nonsense and frameshift
mutations. Among JAG1 mutations, up to 70% have a premature
termination codon (PTC), leading to a truncated JAG1 protein
(13), or degradation by
nonsense-mediated mRNA decay (NMD) (14). Loss-of-function mutations have been
reported due to a truncated protein missing critical motifs
(15).
Haploinsufficiency is considered to be a pathogenic
mechanism of ALGS, since the same phenotypic features can be
observed in patients with complete gene deletion, missense
mutations, or protein-truncating mutations. Morrissette et
al (16), reported that mutant
JAG1 proteins R184J and L37S associated with functional
haploinsufficiency, resulting from abnormal glycosylation patterns.
Boyer-Di Ponio et al (15),
showed that the missense mutant protein C284F as well as truncated
proteins E1003X and Δ4 impaired activation of Notch signaling,
suggesting that a dominant-negative effect mechanism may also
explain the impaired biological function of some ALGS mutations.
Biomarker detection was widely used to monitor the development of
certain diseases (17–19). The finding that the developing
heart was more sensitive to a decreased dosage of JAG1 than the
developing liver implicates JAG1 as a plausible candidate biomarker
for cardiac severity in ALGS (20). Further studies are needed to
discover a more efficient and reliable detection method.
Here, we identified two protein-truncating mutations
in two twins and one unrelated patient with ALGS. Expression level
and transcriptional activation ability of these mutations were
studied to illuminate their pathogenic mechanism in ALGS.
Materials and methods
Subjects
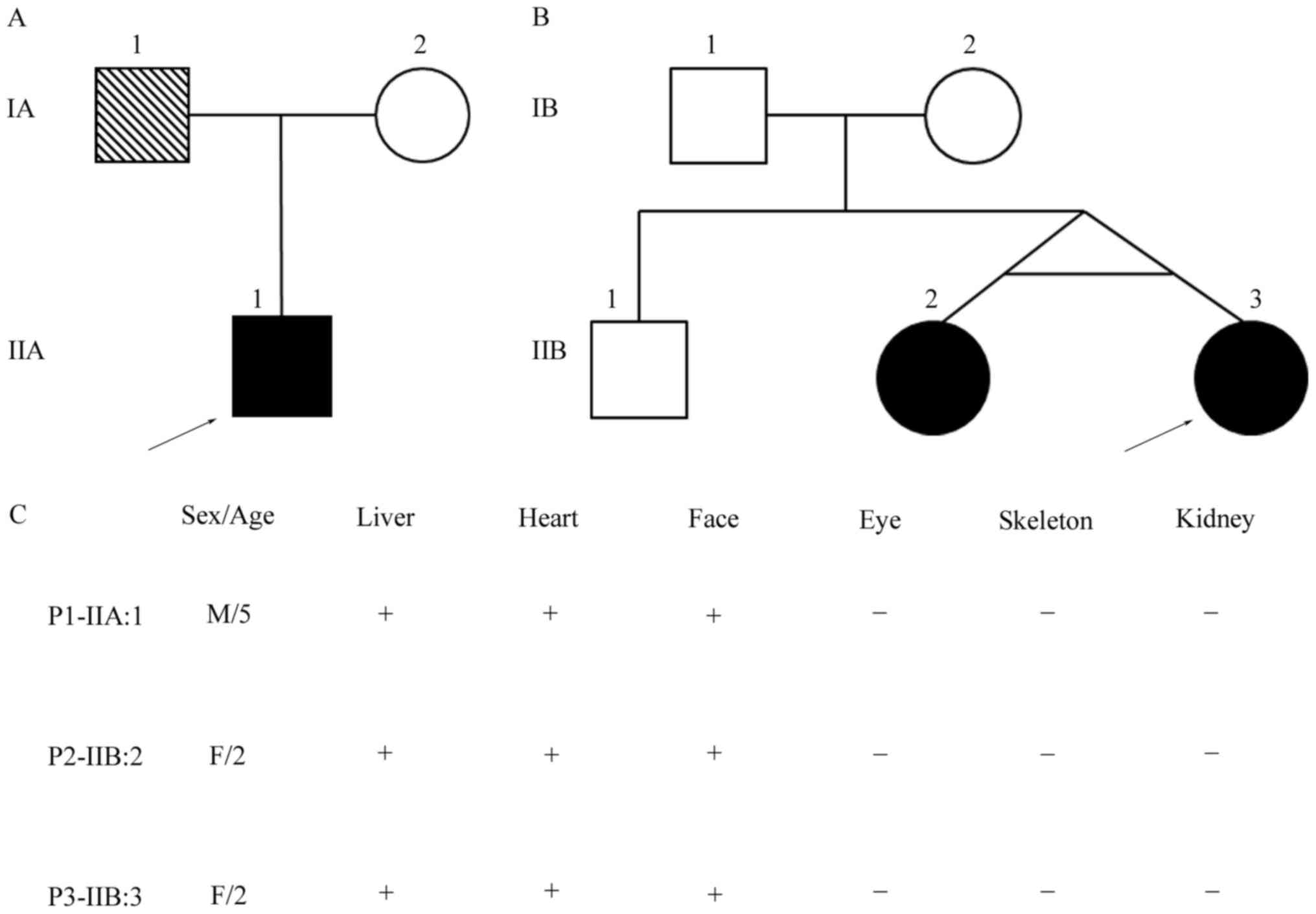
Eight individuals (three ALGS patients, their
siblings and parents) from two families were enrolled for clinical
and genetic studies (Fig. 1). All
of them were of Han ethnicity. The project was approved by the
Medical Ethics Committee of Xinhua Hospital (Shanghai, China).
Written informed consents were provided by the participants or
their legal guardians. Peripheral venous blood samples were
collected after written informed consents were obtained. Control
population was 100 healthy children undergoing routine physical
examination, ethnically matched with patients. Genomic DNA was
isolated from peripheral lymphocytes using a QIAamp DNA Blood Mini
kit according to the manufacturer's instructions (Qiagen GmbH,
Hilden, Germany) and stored at minus 80°C.
Whole-exome sequencing and
analysis
SNPs tests showed that the two patients in family 2
were monozygotic twins, so genomic DNA sample of the proband
(P3-IIB:3 in family 2) was subjected to whole-exome sequencing. The
sample underwent whole-exome capture using the Sureselect Human All
Exon kit (Agilent Technologies, Inc., Santa Clara, CA, USA),
followed by sequencing on the Illummina HiSeq 2500 sequencer
(Illumina, Inc., San Diego, CA, USA) with an ~100× depth of
coverage. Sequence data were aligned and mapped to the human genome
reference (hg19). Variants (the single nucleotide variants and
insertions/deletions) were called by SAMTOOLS (samtools.sourceforge.net/) and compared with
common variant database, including dbSNP135 (ncbi.nlm.nih.gov/) and 1000 genomes project database
(www.1000genomes.org/).
Mutation validation by Polymerase
Chain Reaction (PCR) and Sanger sequencing
PCR and Sanger sequencing was used to detect
possible variants in family 1 and to confirm the candidate variant
in family 2. Parents or siblings were screened for the specific
variants in the affected exons by sequence analysis. DNA from 100
healthy controls were also screened. Primers that amplify 26 exons
(RefSeq NM_000214.2) and at least 100 bp of intron-exon boundaries
were designed (primer sequences available on request) by Primer
Premier v.5.0 (Premier Biosoft International, Palo Alto, CA, USA).
Standard PCR were performed with 100 ng genomic DNA using
MyTaq™ HS DNA Polymerase (Bioline Reagents Ltd., London,
UK) on an Applied Biosystems Veriti instrument (Applied Biosystems,
California, USA). PCR products were sequenced on an Applied
Biosystems 3730 sequencer (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and sequence traces were compared with the
reference sequence using the GenBank BLAST program (blast.ncbi.nlm.nih.gov/Blast.cgi).
Mutation nomenclature is based on the transcript NM_000214.2 and
NP_000205.1.
Plasmid construction
pCMV3-Flag-JAG1 vector containing human JAG1
cDNA was bought from Sino biological inc as a template, the FLAG
tag was at the N-terminus of the open reading frame (ORF) and the
signal peptide was at the N-terminus of the FLAG tag. The
c.1261delT (p.Cys421Valfs), c.1382_1383delAC (p.Asp461Glyfs) mutant
expression vectors were made using Quickchange Site-Directed
Mutagenesis kit (Agilent Technologies, Inc.). Complementary primers
were synthesized containing the desired mutation flanked by
unmodified sequences for the mutagenesis reaction. Forward primers
for these reactions were as follows, p.Cys421Valfs:
5′-GAATGTGAGGCCAAACCTGTGTAAACGCCAAATCC-3′; p.Asp461Glyfs:
5′-CAGTGTCAGAATGGCCTCCTGTCGG-3′. All plasmids used for transfection
were determined by NanoDrop 2000 Microvolume Spectrophotometers
(Thermo Fisher Scientific, Inc.), and according to our previous
studies (21,22), the concentration was controlled at
500 ng/µl.
Cell culture and transfection
NIH-3T3 cells (3,4,20)
(Cell Bank of the Chinese Academy of Science, Shanghai, China) were
cultured in Dulbecco's modified Eagle's Medium (HyClone; GE
Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum (MP Biomedicals, Santa Ana, California, USA),
penicillin (100 units/ml) and streptomycin (100 µg/ml) at 37°C in
5% CO2. Cells were transfected using Fugene HD
Transfection Reagent (Promega Corporation, Madison, WI, USA)
according to the manufacturer's instructions.
Reverse transcription-quantitative PCR
(RT-qPCR)
Cells were seeded in 12-well plate for 24 h, then
transfected with 1.1 µg pCMV3-C-FLAG vector, or with vector
expressing either wild-type JAG1 or JAG1 mutants
(p.Cys421Valfs, p.Asp461Glyfs). After 36 h, total RNA was extracted
by RNAiso (Takara Bio Inc., Otsu, Japan) following manufacturer's
instructions, and 1 µg of that RNA was reverse transcribed into
cDNA using Prime Script RT Master Mix (Takara Bio Inc.). The mRNA
level of JAG1 was quantified by RT-qPCR using SYBR Premix Ex
Taq (Takara Bio Inc.) on an Applied Biosystems 7500 system.
Reaction volume was 20 µl containing 100 ng reverse transcript
product, 0.2 µM primers (spanning exon-exon junction), 10 µl SYBR
Premix Ex Taq and 0.4 µl ROX Reference DyeII. PCR consisted of an
denaturation step at 95°C for 30 sec, followed by 40 cycles
amplification step (95°C for 5 sec, 60°C for 34 sec). The relative
quantitation of JAG1 mRNA was determined using
2−∆∆Cq method (23) and
normalized by an internal control GAPDH. Primers amplifying
cDNA of JAG1 and GAPDH were as follows,
JAG1-F: 5′-CAACCGCATCGTGCTGC-3′, JAG1-R:
5′-CGCCTCCACAAGCAACGTAT-3′; GAPDH-F:
5′-AGGTCGGTGTGAACGGATTTG-3′, GAPDH-R:
5′-TGTAGACCATGTAGTTGAGGTCA-3′.
Western blot analysis
Cells were grown in 12-well plates and transfected
with 1.1 µg empty vector, or with vector expressing either
wild-type JAG1 or mutant proteins. For protein from cell lysates,
cells were harvested 48 h later, lysed by RIPA lysis buffer
(Beyotime Institute of Biotechnology, Shanghai, China) and PMSF
(Beyotime Institute of Biotechnology). Supernatant was collected
after centrifugation (16,000 g, 20 min). Protein concentration was
determined using Enhanced BCA Protein Assay kit (Beyotime Institute
of Biotechnology). Then 20 µg of protein was loaded onto a 7.5%
SDS-polyacrylamide gel and subsequently transferred onto a
nitrocellulose membrane (EMD Millipore, Billerica, MA, USA),
blocked and incubated with rabbit anti-FLAG (1:1,000;
Sigma-Aldrich; Merck KGaA) or anti-GAPDH (1:10,000; Sigma-Aldrich;
Merck KGaA) overnight at 4°C. After washing three times in
TBS/Tween 20, the membrane was incubated with horseradish
peroxidase conjugated anti-rabbit secondary antibody (1:10,000;
Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for
1 h at room temperature. Proteins were revealed by Immobilon
Western Chemiluminescent HRP Substrate (EMD Millipore).
Densitometry of the bands was performed by Image Lab (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The relative intensity of
the wild-type band was set to 1 as a standard reference. Based on
this, relative intensities ± SEM were calculated for the two mutant
protein bands.
Luciferase assays
RBP-Jκ is an important transcriptional factor in the
Notch signaling pathway. When the JAG1 protein interacts with the
Notch receptor, the Notch intracellular domain (NICD) releases and
combines with RBP-Jκ, shifting RBP-Jκ from a transcriptional
repression state to a transcriptional activation state. Thus, we
used a RBP-Jκ-responsive reporter gene assay (SABiosciences; Qiagen
GmbH) to investigate the ability of mutant JAG1 proteins to
activate the Notch signaling pathway.
After being cultured in 48-well plates for 24 h,
cells transfected with pCMV3 control vector or with vector
expressing JAG1 (wild-type or mutant proteins) were used as ‘signal
sending cells’, while cells transfected with
pMyc-C-CMV5-NOTCH3 plasmid (kindly provided by Dr. M. Tada,
National Institute of Health Sciences, Japan) and RBP-Jκ-responsive
luciferase reporter plasmid were used as ‘signal receiving cells’.
The following day, cells were harvested by trypsin-EDTA and
counted, then signal sending cells and signal receiving cells were
co-cultured in a 24-well plates at the ratio of 1:1 for another 24
h as described previously (3).
Luciferase activities were measured using a Dual-Glo luciferase
assay system (Promega Corporation, Madison, WI, USA) according to
the manufacturer's instructions. Firefly luciferase activity levels
were normalized to that of Renilla. All assays were repeated three
times.
Extraction of protein from culture
medium
For protein from culture medium, 6 h after
transfection, the medium was replaced with serum-free medium and
cultured for 48 h. The medium was collected in an Amicon Ultra-4
centrifugal filter (50 kDa for wild-type and 10 kDa for 421fs and
461fs; EMD Millipore) and centrifuged at 3,500 g for 40 min. The
filtrate was collected to study JAG1 expression levels in
supernatant by western blotting. Densitometry of the bands was
performed as described above.
Statistical analysis
Data are presented as the mean ± standard error of
the mean of three independent experiments. Statistical differences
between groups were analyzed by one-way analysis of variance with
Tukey's post hoc test for multiple comparisons using GraphPad Prism
v.6.02 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Clinical manifestation
We performed clinical examinations on the two
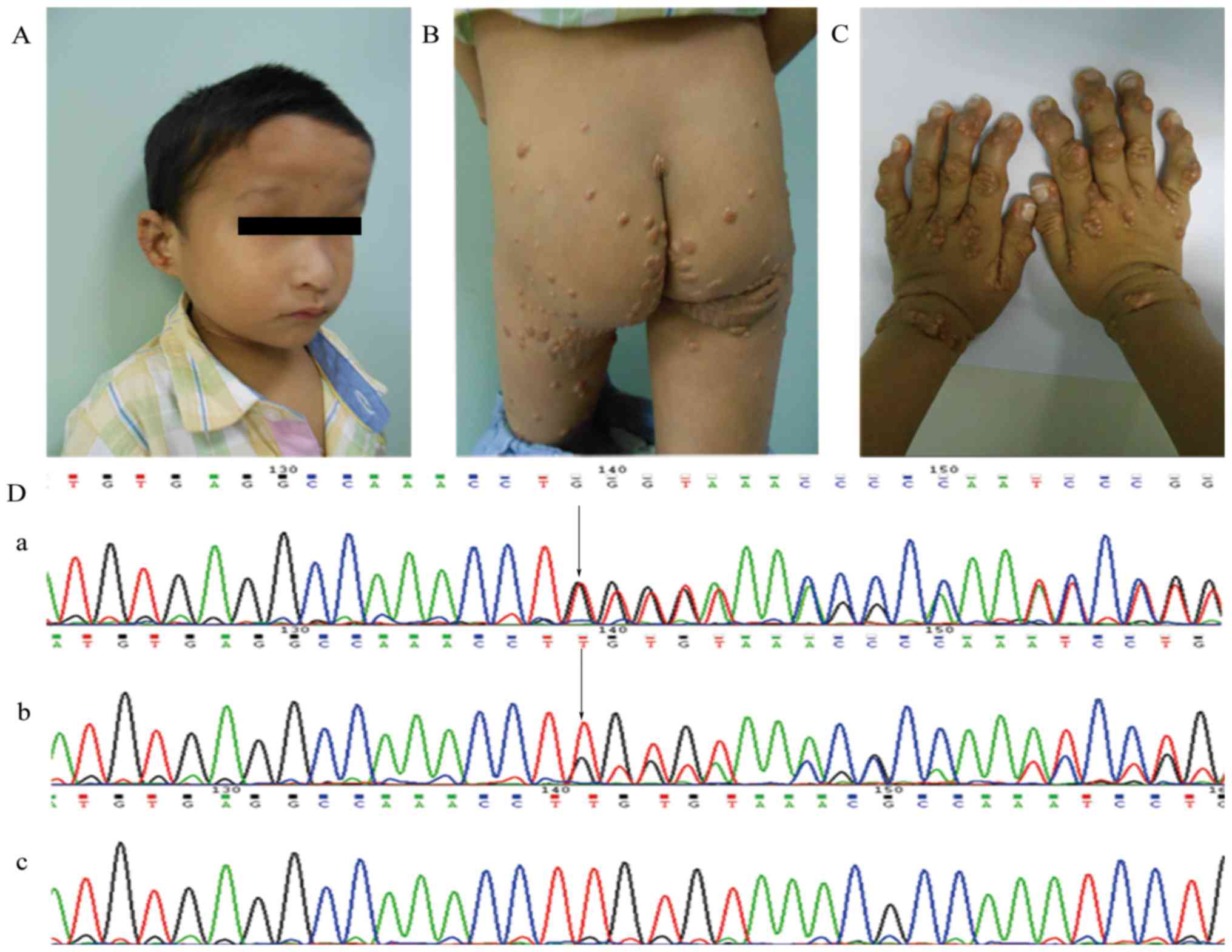
probands and their parents and siblings (Fig. 1). In family 1, the proband
(P1-IIA:1) was a five-year-old boy who had severe xanthomas
(Fig. 2B and C). Hepatic cell
damage and cholestasis were indicated by elevated biochemical
indexes (Table I). He had facial
abnormalities of a prominent forehead, deep-set eyes, a bulbous
nose and a pointed chin (Fig. 2A).
Echocardiography showed pulmonary branch stenosis and patent
foramen ovale. X-ray examination of the spine did not reveal
butterfly vertebrae or any other development anomalies in the
thoracic or lumbosacral segments. Ophthalmologic examination did
not reveal posterior embryotoxon. His parents were apparently
normal and not consanguineous. However serum biochemical tests of
the father illustrated mild hepatic cell damage with slightly
elevated serum levels of ALT 126, AST 48 and γ-GT 98 U/l.
 | Table I.Clinical features of the
probands. |
Table I.
Clinical features of the
probands.
| Clinical
indexes | Proband
(P1-IIA:1) | Proband
(P3-IIB:3) |
|---|
| Total bile acid
(1–10 µmol/l) | 284.3 | 313.0 |
| ALT (0–75 U/l) | 140 | 60 |
| AST (8–38 U/l) | 139 | 51 |
| Alkaline
phosphatase (34–114 U/l) | 829 | 814 |
| γ-GT (16–73
U/l) | 829 | 310 |
| Total bilirubin
(3.42–20.52 µmol/l) | 132.1 | 27.6 |
| Conjugated
bilirubin (0–6.8 µmol/l) | 59.5 | 18.4 |
| Total cholesterol
(3.36–6.46 mmol/l) | 21.53 | 8.73 |
| Triglyceride
(0.2–2.31 mmol/l) | 5.40 | 2.59 |
| LDL (2.07–3.1
mmol/l) | 7.14 | 4.01 |
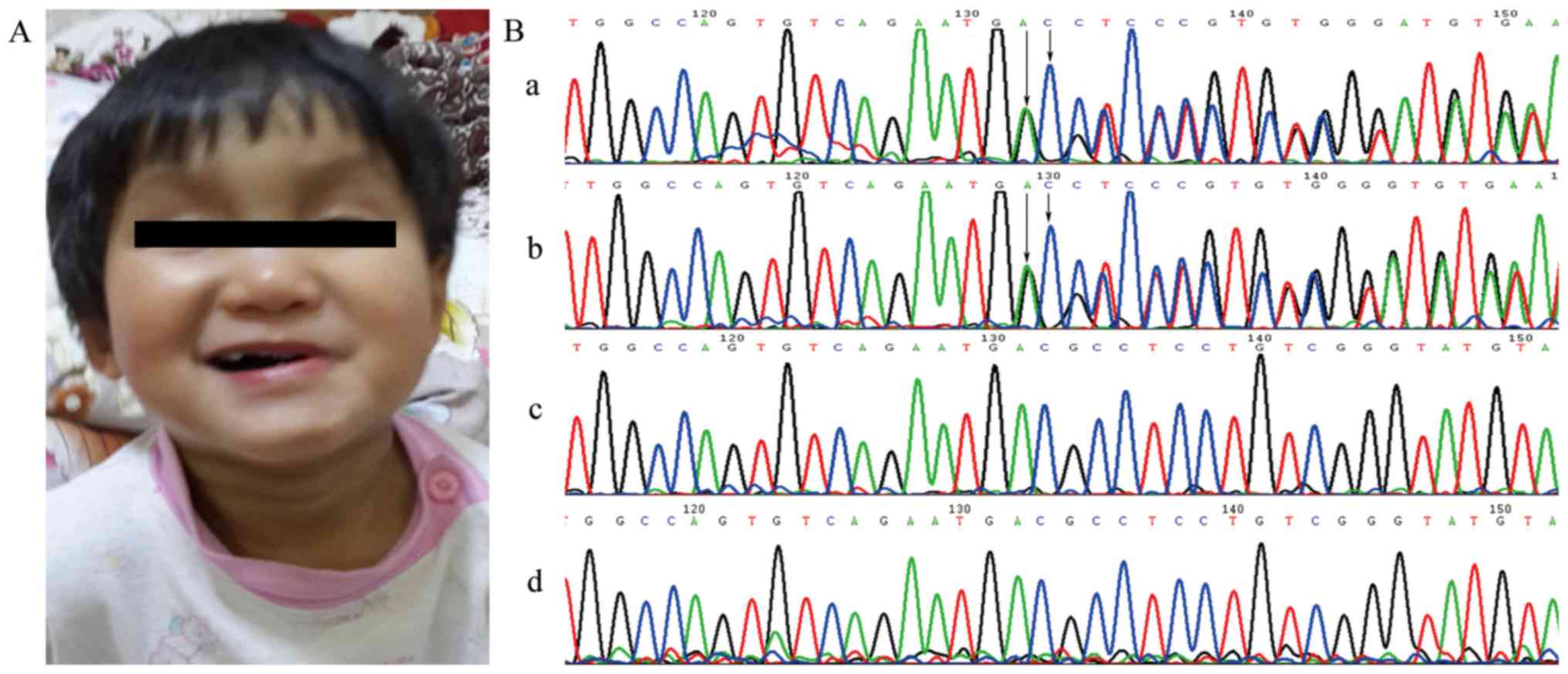
In family 2, the proband (P3-IIB:3) is one of
monozygotic twins. At 2 months of age, she had been referred to our
hospital for persistent jaundice. Echocardiography showed
supravalvular aortic stenosis and left pulmonary artery stenosis.
She had a dysmorphic face, with a broad forehead, a bulbous nose
and a pointed chin (Fig. 3A), but
no skeleton or ocular malformations. Biochemical tests revealed
elevated serum liver enzyme levels, and slightly elevated serum
bilirubin and lipid levels (Table
I). Her twin sister had face abnormalities, mild pulmonary
branch stenosis and hepatic damage. The other family members had
none of these features.
Whole-exome sequencing and
analysis
Whole-exome sequencing of P3-IIB:3 in family 2 was
at a mean depth of ~100×, covering more than 95% of exome regions.
After filtering the total 46 497 single nucleotide variants (SNVs)
and 3774 insertions/deletions (Indels), we located a frameshift
deletion on exon 11 in JAG1 (c.1382_1383delAC, p.Asp461Gly).
This mutation is presumed to result in a premature stop codon.
Direct Sanger sequencing
To validate the WES results from proband P3-IIB:3
and to detect mutations in proband P1-IIA:1, we performed PCR and
Sanger sequencing on the patients and extended family members.
Sanger sequencing confirmed the two mutations: c.1261delT and
c.1382_1383delAC in exons 10 and 11 of JAG1, respectively
(Figs. 2D and 3B). The c.1261delT (p.Cys421Valfs)
frameshift mutation of JAG1 found in patient P1-IIA:1
(Fig. 2Da) was inherited from his
father IA:1 (Fig. 2Db) who had
mild hepatic cell damage, while his mother did not carry this
mutation (Fig. 2Dc). The other
frame-shift mutation c.1382_1383delAC (p.Asp461Glyfs), found in
P2-IIB:2 (Fig. 3Bb) and P3-IIB:3
(Fig. 3Ba) was de novo, for
it was not detected in their mother (Fig. 3Bc) or father (Fig. 3Bd). None of these mutations were
detected in the 100 healthy controls. Mutation c.1261delT has not
been reported in the Human Gene Mutation Database (www.hgmd.cf.ac.uk/) or the NHLBI Exome Sequencing
Project (evs.gs.washington.edu/). While c.1382_1383delAC had
been detected in a retrospective study (24), it had not been analyzed yet.
Mutations c.1261delT and c.1382_1383delAC resulted in the
conversion of the respective amino acid resides 421, a cysteine and
461, an aspartate to stop codons, leading to the loss of subsequent
domains.
Expression of JAG1 mutants
Relative quantitation of mRNA in control vector,
wild-type and two mutants was analyzed using RT-qPCR (Fig. 4A). Expression levels of
p.Cys421Valfs and p.Asp461Glyfs did not reveal any statistical
difference with that of wild-type (P>0.05), indicating
undisturbed mRNA transcription. We then performed western blotting
using expression vector with a Flag tag at the N-terminus of
JAG1 cDNA, thus truncated protein could be detected. As
expected, mutants p.Cys421Valfs and p.Asp461Glyfs both appeared as
truncated proteins (Fig. 4B, left
panel): p.Cys421Valfs was ~55 kDa and p.Asp461Glyfs was ~60 kDa,
almost 130 kDa smaller than the wild-type (~180 kDa). Densitometry
revealed that relative intensity of the wild-type was lower than
that of p.Cys421Valfs (P<0.001), but had no difference with that
of p.Asp461Glyfs (Fig. 4B, right
panel).
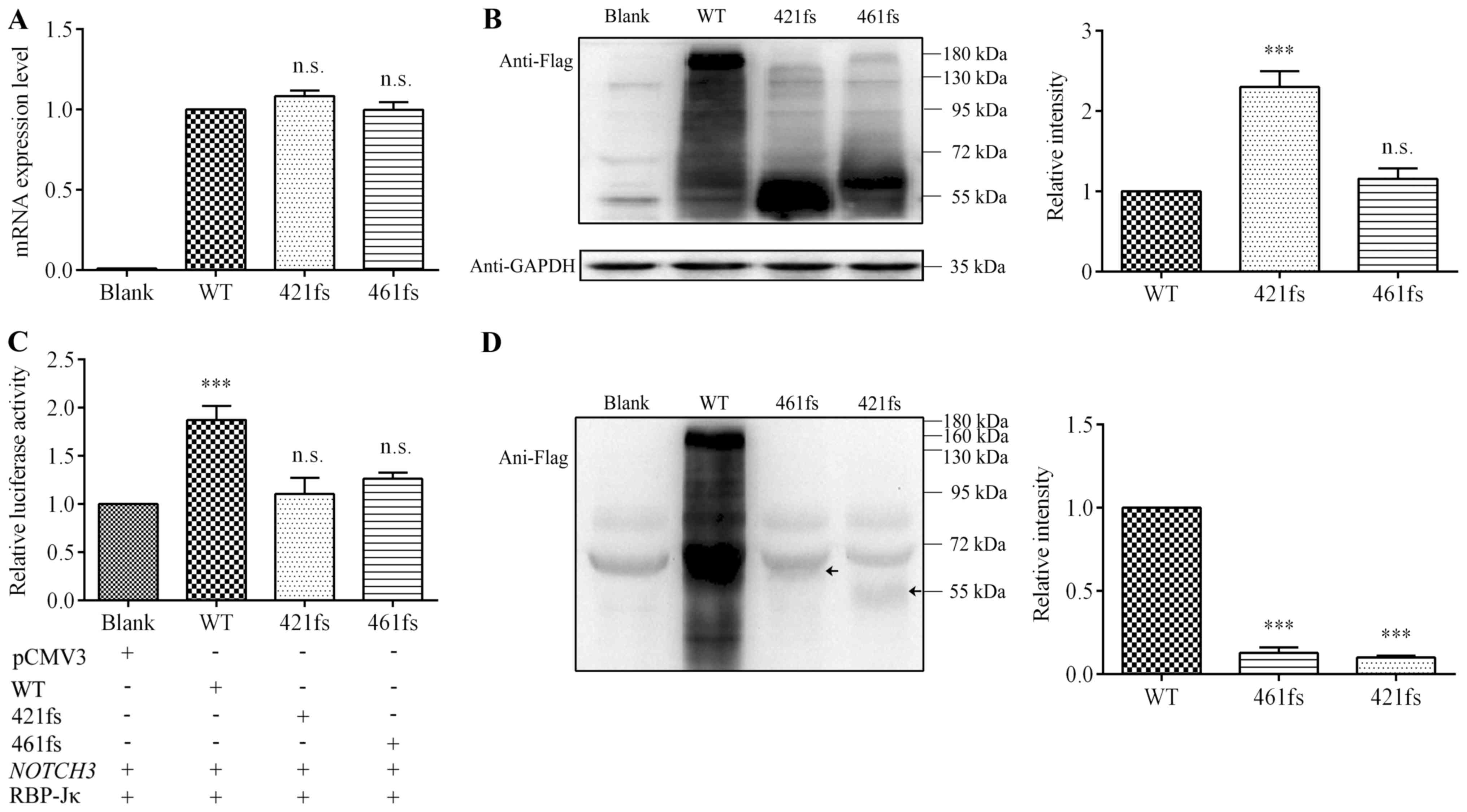 | Figure 4.Analysis of expression
characteristics and transcriptional activation ability of the
mutations. (A) NIH-3T3 cells transiently transfected with pCMV3
vector (blank), WT JAG1 (WT group), p.Cys421Valfs (421fs
group) and p.Asp461Glyfs (461fs group). Following 36 h, RNA was
isolated and reverse transcribed into cDNA. The mRNA expression
levels of both 421fs and 461fs exhibited no difference with that of
WT. (B) Flag-tagged WT, 421fs and 461fs mutant proteins were probed
by anti-Flag immunoglobulin G by western blotting. Left panel
reveals that the molecular weight of WT was ~180 kDa, while 421fs
(~55 kDa) and 461fs (~60 kDa) were ~130 kDa smaller than WT,
indicating impaired protein integrity. The right panel presents
densitometry analysis of the bands; WT had lower relative intensity
than 421fs, though there was no difference with 461fs. (C) When
compared with the control group (blank), WT had a statistically
significant increase in luciferase activity, suggesting activation
of Notch signaling. Conversely, the luciferase activity of 421fs
and 461fs was no different to that of blank, indicating impaired
transcriptional activation ability. (D) WT and mutant JAG1 proteins
were collected from the culture medium and analyzed by western
blotting. WT appeared at ~160 kDa, while 461fs and 421fs appeared
at ~60 and ~55 kDa, indicated by arrows, respectively (left panel).
Relative intensities were calculated by comparing the intensity of
each band with that of the WT (right panel). Data are presented as
the mean ± standard error of the mean. ***P<0.001 vs. WT. n.s.,
not significant; WT, wild type; JAG1, jagged 1. |
Transcriptional activation ability of
JAG1 mutants
The RBP-Jκ-mediated the Notch signaling pathway
transcriptional activation was measured using a RBP-Jκ-responsive
luciferase reporter plasmid (Fig.
4C) 66. Wild-type JAG1 had a statistically significant increase
(almost two-fold, P<0.001) in luciferase activity compared to
the control vector, suggesting over-expression of wild-type JAG1
protein successfully activated the Notch signaling pathway. Both
p.Cys421Valfs and p.Asp461Glyfs had a lower activity than the
wild-type, nearly the same as the control vector (P>0.05),
indicating impaired transcriptional activation ability.
Protein expression level of JAG1
mutants in culture medium
After being transported to the plasma membrane, the
JAG1 protein undergoes proteolytic cleavage, and the soluble
extracellular fragment is secreted into the culture medium. In
western blotting analysis, the wild-type appeared at ~160 kDa,
while p.Cys421Valfs and p.Asp461Glyfs appeared at ~55 kDa and ~60
kDa, indicated by arrows, respectively (Fig. 4D, left panel). The relative
intensities of both p.Cys421Valfs and p.Asp461Glyfs were much lower
than that of the wild-type (Fig.
4D, right panel), suggesting less mutant JAG1 protein underwent
proteolytic cleavage, thus fewer soluble fragments were secreted
into the culture medium.
Discussion
Variable anomalies add intricacy to the differential
diagnosis of ALGS, while advances in molecular genetics contribute
to early diagnosis of the disease and improvement of survival
rates. Protein-truncating mutations account for up to 70% of
JAG1 mutations in ALGS patients. Nevertheless, no mutation
hotspots have been identified yet (13,24),
and the pathological mechanisms of these kinds of mutations have
been controversial. This study was aimed at analyzing the
biological functions of two JAG1 protein-truncating
mutations detected in Chinese ALGS patients, which may allow for
the possibility of future genetic counseling.
JAG1 is a single-transmembrane protein composed of
five main structures, an intracellular region, a transmembrane
domain and an extracellular section, which includes a signal
peptide, an N-terminal region, and a DSL domain, followed by 16
epidermal growth factor-like (EGF-like) repeats and a CR region
(25). The signal peptide
associates with correct target of the JAG1 protein in the cell
membrane, and the DSL domain is necessary in binding JAG1 to the
Notch receptors (26). EGF-like
repeats are evolutionarily highly conserved and have a vital role
in maintaining conformation and stability of the protein, as well
as facilitating the interaction between the ligand and the receptor
(25). Mutations p.Cys421Valfs and
p.Asp461Glyfs were located in the sixth and seventh EGF-like
repeats, respectively. These frameshift deletions resulted in the
conversion of the respective amino acid resides 421, a cysteine and
461, an aspartate to stop codons, leading to the loss of subsequent
domains. The loss of the intracellular domain may affect adhesion
between cells, inhibiting cell motility (26). Even when the signal peptide and the
DSL domain remained intact, assays found that Serrate DSL mutants
retained the ability to bind to the Notch receptor despite the loss
of transactivation function, suggesting that binding alone does not
ensure activation of the Notch signaling pathway (27). Therefore, it is highly suspected
that these two mutations are the cause of the nonfunctional JAG1
protein.
The mRNA expression levels of p.Cys421Valfs and
p.Asp461Glyfs were similar to that of the wild-type, suggesting
that these mutations had no effect on normal transcription
processes. Mutations with PTC are typical targets of NMD, which
prevents deleterious proteins from being produced (28,29).
However, in some cases mRNAs may escape NMD for one or more rounds
of translation, because of the inefficient recruitment of the
NMD-activating complex (30,31).
Besides, Researches showed that in mammalian cells, the induction
of NMD requires a long 3′UTR or the presence of an exon-junction
complex downstream of a PTC (32,33).
Thus, PTC in eukaryotic over-expression plasmid with intronless
JAG1 CDS will not subjected to NMD. To assess NMD, it is
more convincing to test mRNA levels using patient-derived
lymphoblasts and/or tissues (14),
but unfortunately, due to social reasons, we could not obtain
enough blood samples or tissues, so we studied the mRNA expression
level of mutant JAG1 in transient transfected cell as previous
researches did (34). We used
western blotting to affirm the abnormality of the mutant JAG1
protein by detecting the Flag tag that was attached to the
N-terminus of the JAG1 ORF. Since thep.Cys421Valfs and
p.Asp461Glyfs mutations express truncated JAG1 proteins, both of
them had smaller molecular weights than the wild-type. Mutant
protein p.Cys421Valfs had higher expression level than the
wild-type, possibly due to posttranslational modification (16,20).
The ability of JAG1 mutants to activate Notch
signaling was measured by the reporter gene assay. The truncated
mutants had lower luciferase activity than the wild-type, almost at
the same level as the control vector, demonstrating a loss of
function in the transcriptional activation process. After being
transported to the plasma membrane, JAG1 can interact with the
NOTCH receptor and initiate downstream transcription.
Membrane-bound JAG1 protein undergoes proteolytic cleavage,
secreting a soluble extracellular fragment into the culture medium.
If the protein was not transported correctly to the plasma
membrane, proteolytic cleavage might not occur, and the soluble
extracellular fragment might not be secreted into the culture
medium. Western blotting using wild-type and mutant JAG1 proteins
from cell culture medium suggested that fewer soluble extracellular
fragments were generated by either mutant protein than by the
wild-type. To detect a possible cleavage process of the JAG1
protein, an anti-Jag1 antibody directly against the C-terminus
should be used to identify the membrane associated and
intracellular fragments of JAG1 (15). However, both p.Cys421Valfs and
p.Asp461Glyfs were truncated without the transmembrane and
intracellular domains, so it is difficult to analyze their
proteolytic cleavage process directly. Tada et al (3) found that P163L and R184H localized in
the endoplasmic reticulum, so their soluble forms were not detected
in the culture medium, which resulted in a failure to activate
RBP-Jκ. Additionaly, trucated JAG1 protein E1003 exhibited
decreased soluble protein levels with damaged transcriptional
activity (15). This evidence
suggests that abnormal localization of JAG1 may affect its
regulation of down-stream target elements. Further studies of
p.Cys421Valfs and p.Asp461Glyfs are still needed to elucidate the
association between their subcellular localization and functional
abnormality.
Lack or absence of Notch signaling affects
development of those organs mainly involved in ALGS, such as liver,
heart and face. A study by Sparks et al (35) found a dose-dependent relationship
between impaired Notch signaling and diminished peripheral
intrahepatic bile ducts (IHBD) derived from bipotential hepatoblast
progenitor cells, suggesting that Notch signaling is a critical
requirement for the formation of IHBD. Notch signaling is also
closely implicated in cardiogenesis, as Notch mutant mouse models
demonstrate various cardiac abnormalities, from septal defects to
heart looping defects (36). Mice
with attenuated JAG1 had midfacial hypoplasia phenotypes similar to
ALGS, unveiling the necessity of Notch signaling in midfacial
cranial neural crest cells during development (37). Consequently, we believe that the
frameshift mutations are deleterious and might contribute to ALGS
phenotypes.
This study is the first to analyze the biological
function of two protein-truncating JAG1 mutations detected
in Chinese ALGS patients. Similarities among the three ALGS
patients are frameshift mutations in the EGF domain lead to
truncated protein with function loss that are associated with
manifestations involving liver and heart. There are also some
differences. In family 1, follow-up with P1-IIA:1 after treatment
with UDCA (ursodeoxycholic acid) indicated decreased bilirubin
levels, but hepatic enzymes elevated significantly, and eventually
the patient received a liver transplantation. His liver function
has improved, and his xanthomas reduced. His father carried the
same mutation, but had only sub-clinical findings: Slightly
elevated liver enzymes that could result from profound reasons
apart from ALGS. A cohort study showed that the frequency of
serious clinical findings in relatives is much lower compared to
the probands in their families (38). It still remains to be explored why
the same mutation can lead to variable manifestations, from full
exhibition of ALGS features to no detectable findings at all. In
family 2, the monozygotic twins carried a de novo mutation.
This is a primary report of Chinese monozygotic twins with ALGS.
The twins had similar face abnormalities, pulmonary branch stenosis
and hepatic damage, but they did not finish their follow up, so we
were unable to evaluate their present conditions. Nevertheless,
future genetic counseling of the two families will still benefit
from these findings.
This is the first study to analyze the biological
function of two protein-truncating JAG1 mutations detected
in three Chinese ALGS patients. According to the results, mutations
c.1261delT and c.1382_1383delAC produced truncated JAG1 proteins
with impaired function in activating the Notch signaling pathway.
These findings suggested that the two mutations might associate
with ALGS manifestations in these patients, enriched the spectrum
of JAG1 mutation known in ALGS patients and provided
necessary information for genetic counseling of families with ALGS
patients.
Acknowledgements
The authors would like to thank Dr. Minoru Tada
(Division of Biological Chemistry and Biologicals, National
Institute of Health Sciences, Tokyo, Japan) for generously offering
the plasmids used in the present study.
Funding
The present study was supported by grants from the
National Basic Research Program of China (grant no. 2010CB529501),
the National Natural Science Foundation of China (grant nos.
81300068/H0201 and 81270233/H0204) and the Major Key Project for
Fundamental Research from Shanghai Science and Technology Committee
(grant no. 13JC1401705).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
EZ and YX performed the experiments and drafted the
manuscript. SC and YuY the acquired data, performed statistical
analysis, and analyzed and interpreted the data. YgY provided
follow-up information of the patients, and analyzed and interpreted
the data in the revised manuscript. KS conceived and designed the
research, and revised the manuscript for important intellectual
content.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of Xinhua Hospital (Shanghai, China). Written informed
consents were provided by the participants or their legal
guardians.
Patient consent for publication
Written informed consent was obtained from all
patients or their legal guardians for the publication of any
identifiable data or images included in this article.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Loomes KM, Underkoffler LA, Morabito J,
Gottlieb S, Piccoli DA, Spinner NB, Baldwin HS and Oakey RJ: The
expression of Jagged1 in the developing mammalian heart correlates
with cardiovascular disease in Alagille syndrome. Hum Mol Genet.
8:2443–2449. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Giannakudis J, Röpke A, Kujat A,
Krajewska-Walasek M, Hughes H, Fryns JP, Bankier A, Amor D,
Schlicker M and Hansmann I: Parental mosaicism of JAG1 mutations in
families with Alagille syndrome. Eur J Hum Genet. 9:209–216. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tada M, Itoh S, Ishii-Watabe A, Suzuki T
and Kawasaki N: Functional analysis of the Notch ligand Jagged1
missense mutant proteins underlying Alagille syndrome. FEBS J.
279:2096–2107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bauer RC, Laney AO, Smith R, Gerfen J,
Morrissette JJ, Woyciechowski S, Garbarini J, Loomes KM, Krantz ID,
Urban Z, et al: Jagged1 (JAG1) mutations in patients with tetralogy
of Fallot or pulmonic stenosis. Hum Mutat. 31:594–601. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Krantz ID, Piccoli DA and Spinner NB:
Clinical and molecular genetics of Alagille syndrome. Curr Opin
Pediatr. 11:558–564. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li L, Krantz ID, Deng Y, Genin A, Banta
AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, et al: Alagille
syndrome is caused by mutations in human Jagged1, which encodes a
ligand for Notch1. Nat Genet. 16:243–251. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oda T, Elkahloun AG, Pike BL, Okajima K,
Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS
and Chandrasekharappa SC: Mutations in the human Jagged1 gene are
responsible for Alagille syndrome. Nat Genet. 16:235–242. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McDaniell R, Warthen DM, Sanchez-Lara PA,
Pai A, Krantz ID, Piccoli DA and Spinner NB: NOTCH2 mutations cause
Alagille syndrome, a heterogeneous disorder of the notch signaling
pathway. Am J Hum Genet. 79:169–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Penton AL, Leonard LD and Spinner NB:
Notch signaling in human development and disease. Semin Cell Dev
Biol. 23:450–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leonard LD, Chao G, Baker A, Loomes K and
Spinner NB: Clinical utility gene card for: Alagille Syndrome
(ALGS). Eur J Hum Genet. 22:2014.(doi: 10.1038/ejhg.2013).
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Turnpenny PD and Ellard S: Alagille
syndrome: Pathogenesis, diagnosis and management. Eur J Hum Genet.
20:251–257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kamath BM, Bauer RC, Loomes KM, Chao G,
Gerfen J, Hutchinson A, Hardikar W, Hirschfield G, Jara P, Krantz
ID, et al: NOTCH2 mutations in Alagille syndrome. J Med Genet.
49:138–144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Warthen DM, Moore EC, Kamath BM,
Morrissette JJ, Sanchez-Lara PA, Piccoli DA, Krantz ID and Spinner
NB: Jagged1 (JAG1) mutations in Alagille syndrome: Increasing the
mutation detection rate. Hum Mutat. 27:436–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Boyer J, Crosnier C, Driancourt C, Raynaud
N, Gonzales M, Hadchouel M and Meunier-Rotival M: Expression of
mutant JAGGED1 alleles in patients with Alagille syndrome. Hum
Genet. 116:445–453. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boyer-Di Ponio J, Wright-Crosnier C,
Groyer-Picard MT, Driancourt C, Beau I, Hadchouel M and
Meunier-Rotival M: Biological function of mutant forms of JAGGED1
proteins in Alagille syndrome: Inhibitory effect on Notch
signaling. Hum Mol Genet. 16:2683–2692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morrissette JD, Colliton RP and Spinner
NB: Defective intracellular transport and processing of JAG1
missense mutations in Alagille syndrome. Hum Mol Genet. 10:405–413.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu C, Yang C, Lu L, Wang W, Tan W, Leung
CH and Ma DL: Luminescent iridium(iii) complexes as COX-2-specific
imaging agents in cancer cells. Chem Commun (Camb). 53:2822–2825.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lincoln R, Greene LE, Zhang W, Louisia S
and Cosa G: Mitochondria alkylation and cellular trafficking mapped
with a lipophilic BODIPY-acrolein fluorogenic probe. J Am Chem Soc.
139:16273–16281. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin S, Gao W, Tian Z, Yang C, Lu L, Mergny
JL, Leung CH and Ma DL: Luminescence switch-on detection of protein
tyrosine kinase-7 using a G-quadruplex-selective probe. Chem Sci.
6:4284–4290. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu F, Morrissette JJ and Spinner NB:
Conditional JAG1 mutation shows the developing heart is more
sensitive than developing liver to JAG1 dosage. Am J Hum Genet.
72:1065–1070. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang E, Hong N, Chen S, Fu Q, Li F, Yu Y
and Sun K: Targeted sequencing identifies novel GATA6 variants in a
large cohort of patients with conotruncal heart defects. Gene.
641:341–348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pu T, Liu Y, Xu R, Li F, Chen S and Sun K:
Identification of ZFPM2 mutations in sporadic conotruncal heart
defect patients. Mol Genet Genomics. 293:217–223. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li L, Dong J, Wang X, Guo H, Wang H, Zhao
J, Qiu Y, Abuduxikuer K and Wang J: JAG1 mutation spectrum and
origin in chinese children with clinical features of alagille
syndrome. PLoS One. 10:e01303552015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grochowski CM, Loomes KM and Spinner NB:
Jagged1 (JAG1): Structure, expression, and disease associations.
Gene. 576:381–384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chillakuri CR, Sheppard D, Lea SM and
Handford PA: Notch receptor-ligand binding and activation: Insights
from molecular studies. Semin Cell Dev Biol. 23:421–428. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cordle J, Johnson S, Tay JZ, Roversi P,
Wilkin MB, de Madrid BH, Shimizu H, Jensen S, Whiteman P, Jin B, et
al: A conserved face of the Jagged/Serrate DSL domain is involved
in Notch trans-activation and cis-inhibition. Nat Struct Mol Biol.
15:849–857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lykke-Andersen S and Jensen TH:
Nonsense-mediated mRNA decay: An intricate machinery that shapes
transcriptomes. Nat Rev Mol Cell Biol. 16:665–677. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He F and Jacobson A: Nonsense-mediated
mRNA decay: Degradation of defective transcripts is only part of
the story. Annu Rev Genet. 49:339–366. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mocquet V, Durand S and Jalinot P: How
retroviruses escape the nonsense-mediated mRNA decay. AIDS Res Hum
Retroviruses. 31:948–958. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Neu-Yilik G, Amthor B, Gehring NH, Bahri
S, Paidassi H, Hentze MW and Kulozik AE: Mechanism of escape from
nonsense-mediated mRNA decay of human beta-globin transcripts with
nonsense mutations in the first exon. RNA. 17:843–854. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ferraiuolo MA, Lee CS, Ler LW, Hsu JL,
Costa-Mattioli M, Luo MJ, Reed R and Sonenberg N: A nuclear
translation-like factor eIF4AIII is recruited to the mRNA during
splicing and functions in nonsense-mediated decay. Proc Natl Acad
Sci USA. 101:4118–4123. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Neu-Yilik G, Gehring NH, Thermann R, Frede
U, Hentze MW and Kulozik AE: Splicing and 3′end formation in the
definition of nonsense-mediated decay-competent human beta-globin
mRNPs. EMBO J. 20:532–540. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan ZR, Kobayashi N and Kohsaka T: Human
Jagged 1 mutants cause liver defect in Alagille syndrome by
overexpression of hepatocyte growth factor. J Mol Biol.
356:559–568. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sparks EE, Huppert KA, Brown MA,
Washington MK and Huppert SS: Notch signaling regulates formation
of the three-dimensional architecture of intrahepatic bile ducts in
mice. Hepatology. 51:1391–1400. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
High FA and Epstein JA: The multifaceted
role of Notch in cardiac development and disease. Nat Rev Genet.
9:49–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Humphreys R, Zheng W, Prince LS, Qu X,
Brown C, Loomes K, Huppert SS, Baldwin S and Goudy S: Cranial
neural crest ablation of Jagged1 recapitulates the craniofacial
phenotype of Alagille syndrome patients. Hum Mol Genet.
21:1374–1383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kamath BM, Bason L, Piccoli DA, Krantz ID
and Spinner NB: Consequences of JAG1 mutations. J Med Genet.
40:891–895. 2003. View Article : Google Scholar : PubMed/NCBI
|