Introduction
Aniridia is characterized by the congenital
hypoplasia or absence of an iris and can be divided into hereditary
and sporadic forms (1). Hereditary
aniridia is commonly inherited in an autosomal-dominant manner
(1,2). Classic aniridia is usually
accompanied by a variety of ocular anomalies including keratopathy,
lens opacity (cataract), juvenile-onset glaucoma, foveal hypoplasia
and optic nerve hypoplasia. Aniridia can also occur as part of the
WAGR syndrome (including Wilms tumor, aniridia, genitourinary
anomalies and retardation) (3) or
Gillespie syndrome (including partial aniridia, ataxia, and
intellectual disability) (4).
Aniridia occurs in ~1 per 50,000–100,000 people, with a large range
of visual outcomes (5). The visual
acuity of the affected individuals largely depends on the extent of
the defects in other ocular components. Additionally, children with
aniridia frequently present with nystagmus, which leads to
decreased light sensitivity (6).
Tinted or photochromic lenses can be used to reduce light
sensitivity in patients with aniridia, and regular ocular
examinations should be performed to closely monitor intraocular
pressure and visual acuity (7).
Previous studies have demonstrated that both
isolated and syndromic aniridia are commonly associated with
mutations in the Paired Box 6 (PAX6) gene on chromosome
11p13 (8–10). The PAX6 gene, highly
conserved in both vertebrates and invertebrates, encodes a
transcription factor that is a master regulator during eye
development (11,12). The PAX6 protein contains a paired
domain (PD) at the N-terminus, a homeodomain (HD) linked by a
glycine-rich domain and a transactivation domain enriched with
proline-serine-threonine (PST) residues at the C terminus. The PD
domain consists of an N-terminal and a C-terminal subdomain, which
are important for DNA recognition (13,14).
The linker region between the PD and the HD provides a structural
base for DNA binding (15). The
PST domain, which is rich in proline, serine and threonine, has
transactivation function (16).
However, the role of PAX6 protein in ocular development is still
not fully understood and only a few target genes of PAX6 have been
identified (17). So far, >400
mutations in the PAX6 gene have been reported. Affected
patients with different mutations can present very distinct
clinical phenotypes (1). In the
present study, two PAX6 mutations were identified from two
patients in southern China with classic congenital aniridia and
cataract.
Materials and methods
Study subjects and clinical
examination
A total of two sporadic patients were diagnosed as
classic congenital aniridia and cataract at Zhongshan Ophthalmic
Center (Guangzhou, China). Patient 1 was a 4 year-old male and was
recruited in August 2015. Patient 2 was a 24 year-old male and was
recruited in March 2015. Visual acuity was examined using the Early
Treatment Diabetic Retinopathy Study chart (Precision Vision, La
Salle, IL, USA). Intraocular pressure (IOP) was measured by a
Goldmann applanation tonometer (Haag-Streit Diagnostics, Koeniz,
Switzerland). Anterior segment images were obtained using a BX 900
Slit Lamp (Haag-Streit Diagnostics). Optical coherence tomography
(OCT) scans (TOPCON Corporation, Tokyo, Japan) were performed to
assess the thickness and morphology of the posterior pole of the
retina. Anterior segment dimensions were measured by ultrasound
biomicroscopy (UBM; Suoer Electronic Ltd.; Model SW-3200L) and
Pentacam® HR version 70700 (OCULUS Optikgeräte GmbH,
Wetzlar, Germany). Complete physical examinations were performed to
exclude systemic diseases, such as diabetes, hypertension, Wilms
tumor and genitourinary anomalies.
Blood sample collection
Venous blood samples from the patients, their
unaffected parents, as well as 200 unrelated control subjects from
the same population (18–48 years old; male/female=108/92) were
collected. Genomic DNA was extracted from peripheral blood
leukocytes as described previously (18,19).
Briefly, 1 ml of blood sample was collected from each participant,
lysed in red blood cell lysis buffer (Sigma Aldrich; Merck KGaA,
Darmstadt, Germany) and centrifuged at a speed of 2,000 × g for 5
min at room temperature. Genomic DNA was extracted using a DNeasy
Blood & Tissue Kit following the manufacturer's protocol
(Qiagen GmbH, Hilden, Germany) (18,19).
Detection of the mutation
Exons of the PAX6 gene (PAX6) were amplified
by polymerase chain reaction (PCR) (20). The primer sequences used are listed
in Table I. All reagents in the
PCR reaction were obtained from a PCR amplification kit (Takara
Bio, Inc., Otsu, Japan). The thermocycling conditions used were as
follows: A single 5 min step at 94°C; followed by 40 cycles at 94°C
for 45 sec, 53–65°C for 45 sec and 72°C for 45 sec; and a final 10
min step at 72°C. The PCR products were sequenced from both
directions with an ABI3730 Automated Sequencer (Applied Biosystems;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). The sequencing
results were analyzed using Seqman (version 2.3; Technelysium Pty
Ltd., Brisbane, Queensland, Australia) and compared with the
reference sequences in the database at the National Center for
Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov). The variants identified in the
PAX6 gene were summarized using the Leiden Open source
Variation Database (LOVD; www.lovd.nl)
(21).
 | Table I.Summary of the primers and products
size used for the amplification of the PAX6 exons. |
Table I.
Summary of the primers and products
size used for the amplification of the PAX6 exons.
| Exon | Forward (5′-3′) | Reverse (5′-3′) | Product size
(bp) | Temperature (°C) |
|---|
| 4 |
TGCAGCTGCCCGAGGATTA |
GCACCCCGAGCCCGAAGTC | 144 | 65 |
| 5 |
TCCCTCTTCTTCCTCTTCACT |
GGGGTCCATAATTAGCATC | 301 | 58 |
| 5a,6 |
GCTCTCTACAGTAAGTTCTC |
AGGAGAGAGCATTGGGCTTA | 457 | 59 |
| 7 |
AATCCACCCACTGTCCCG |
CCAGCCACCTTCATACCG | 542 | 59 |
| 8 |
TCAGGTAACTAACATCGCA |
GTTGACTGTACTTGGAAGAA | 719 | 53 |
| 9,10,11 |
GAGGTGGGAACCAGTTTGATG |
CAAGCCAATCTCTGTAGTGCG | 890 | 55 |
| 12 |
GCTGTGTGATGTGTTCCTCA |
AAGAGAGATCGCCTCTGTG | 245 | 58 |
| 13 |
CATGTCTGTTTCTCAAAGGG |
CCATAGTCACTGACTGAATTAACAC | 202 | 59 |
Ethics
All experiments were carried out in accordance with
the guidelines approved by the ethics committee of Zhongshan
Ophthalmic Center at Sun Yat-sen University (Guangzhou, Guangdong,
China) and strictly followed the Declaration of Helsinki. Written
informed consent was obtained from each participant. All
participants provided informed consent for the publication of their
clinical and genetic sequencing data in the present study.
Results
Clinical presentations
The sporadic patients were from the southern part of
China. Patient 1 was a 4-year-old male presented with aniridia,
congenital cataract and horizontal nystagmus. He had decreased
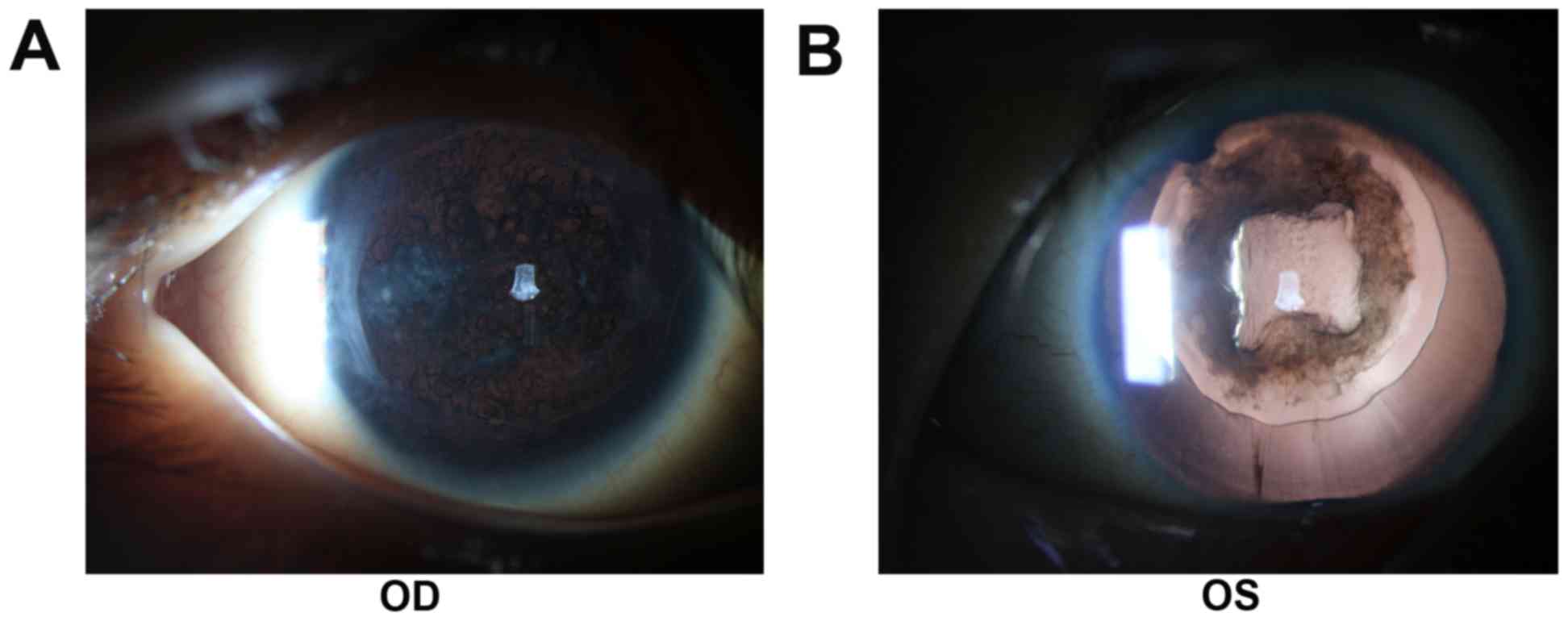
visual acuity from early childhood. Patient 1 had cataract surgery
two years ago and developed posterior capsular opacity
characterized by Elschnig pearls in the posterior capsule (Fig. 1). Applanation tonometry
demonstrated a normal IOP in both eyes. The corneas were
transparent and normal in size, with a width of 10.5 mm (oculus
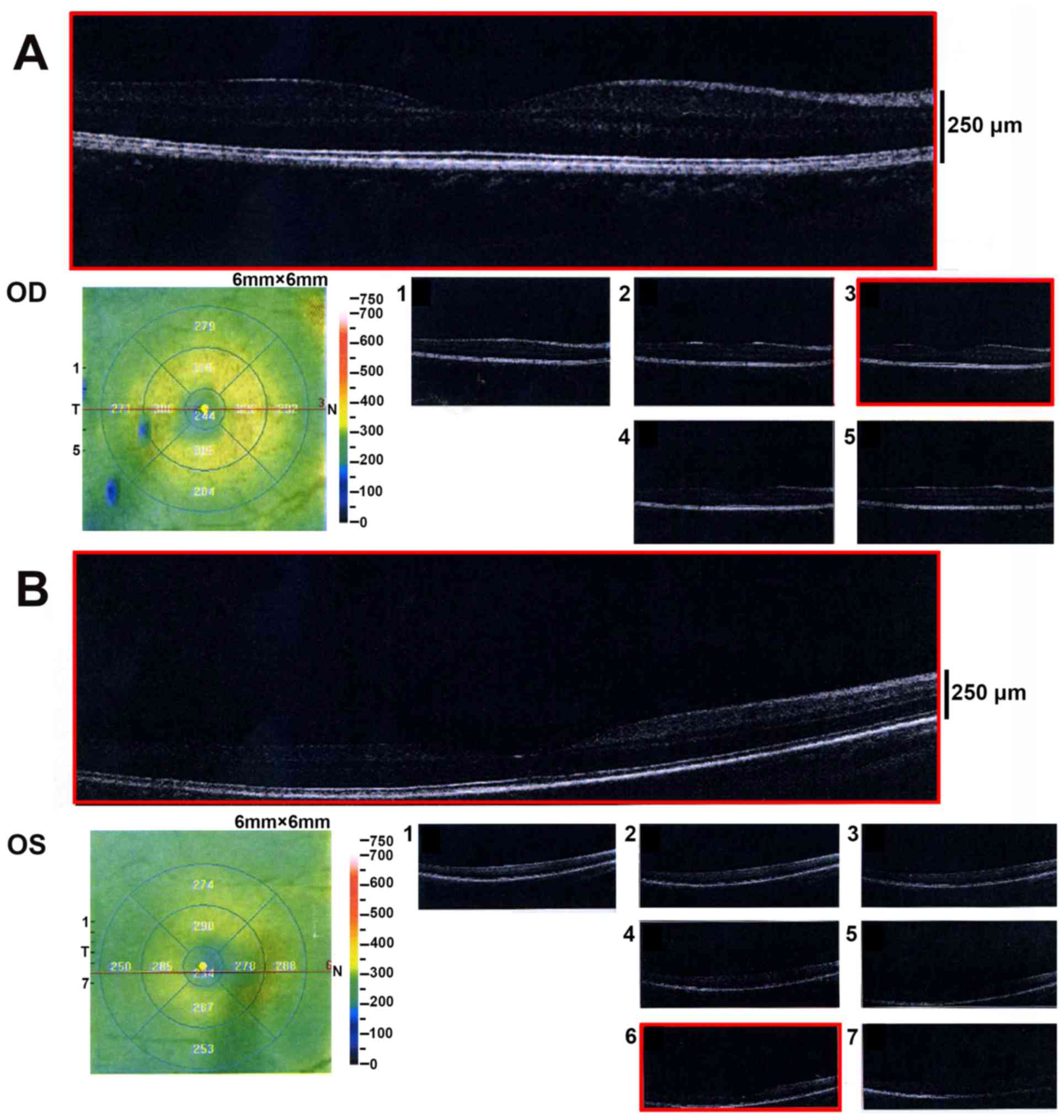
dexter; OD) and 10.5 mm (oculus sinister; OS). OCT revealed a
normal retina structure (Fig. 2).
He did not have other systemic diseases.
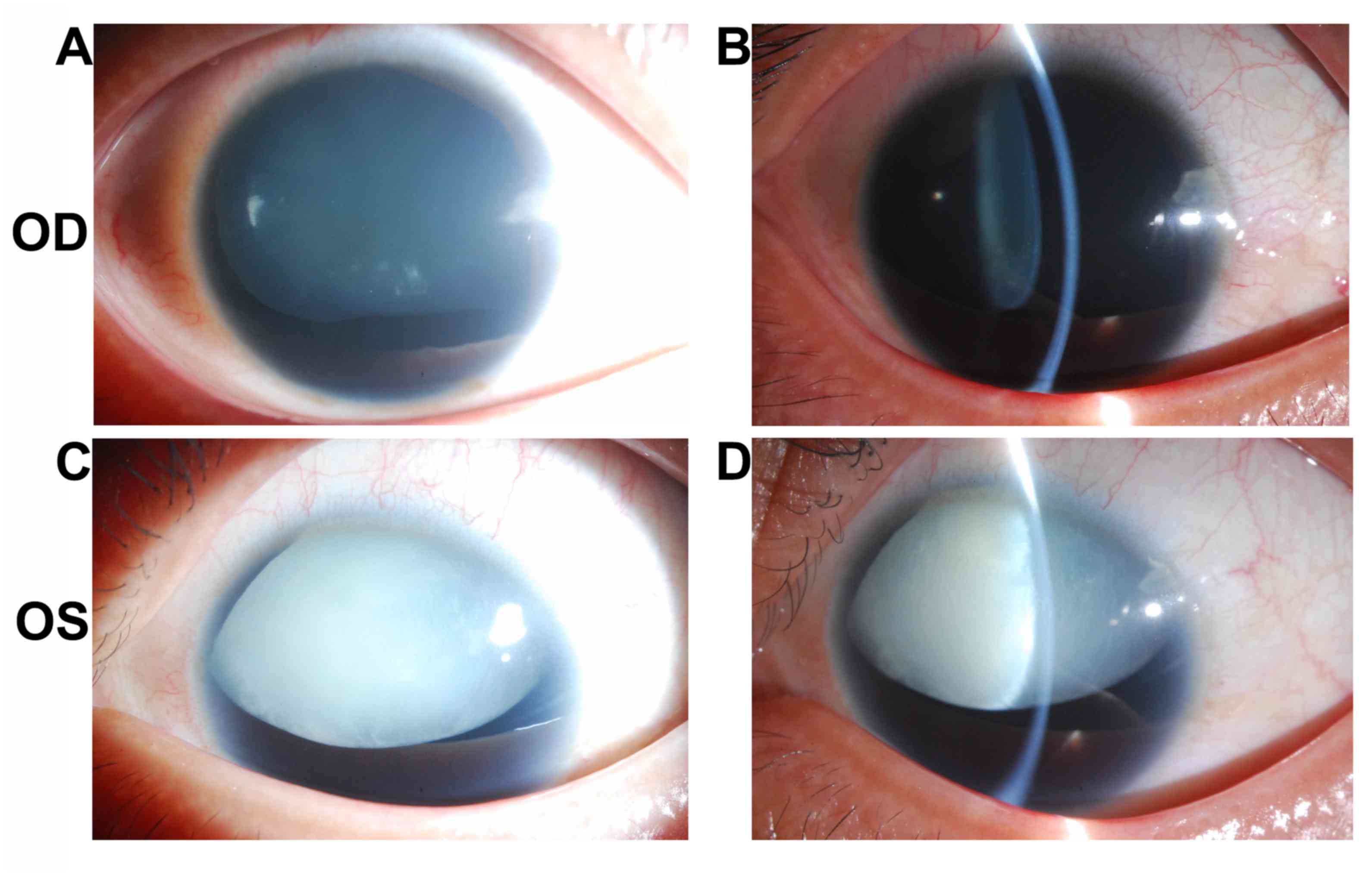
Patient 2 was a 24-year-old male with impaired
vision and eye pain. The visual acuity (LogMAR) was 0.5 (OD) and
0.7 (OS), which could not be corrected. The width of the cornea was
11.5 mm (OD) and 11.5 mm (OS). The intraocular pressure was 22.9
mmHg (OD) and 26.6 mmHg (OS). The axial length of the eyeballs was
26.45 mm (OD) and 26.51 mm (OS). Anterior segment imaging exhibited
aniridia and cataract. The degree of lens opacity in the left eye
was more severe compared with in the right eye. Both eyes exhibited
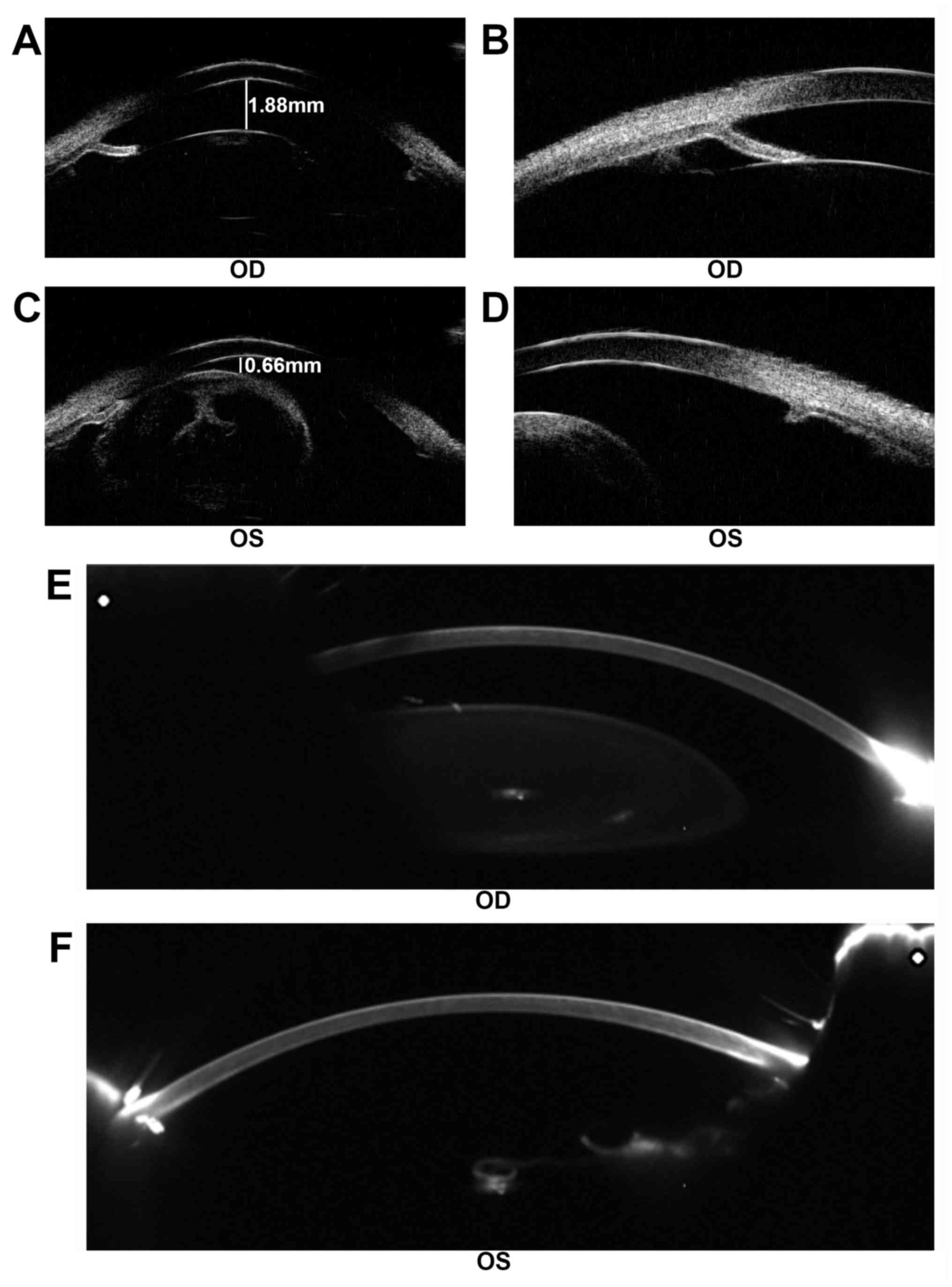
lens subluxation (Fig. 3). UBM
demonstrated a shallow anterior chamber with uneven anterior
chamber depth (ACD). The central ACD (CACD) was 1.88 mm (OD) and
0.66 mm (OS). The anterior chamber angles in both eyes were closed
(Fig. 4A-D). Following cataract
surgery of the left eye, Pentacam examination demonstrated that the
CACD (OS) raised to 4.17 mm (Fig. 4E
and F). The IOP of the left eye decreased to 14.6 mmHg
postoperatively.
Mutation screening
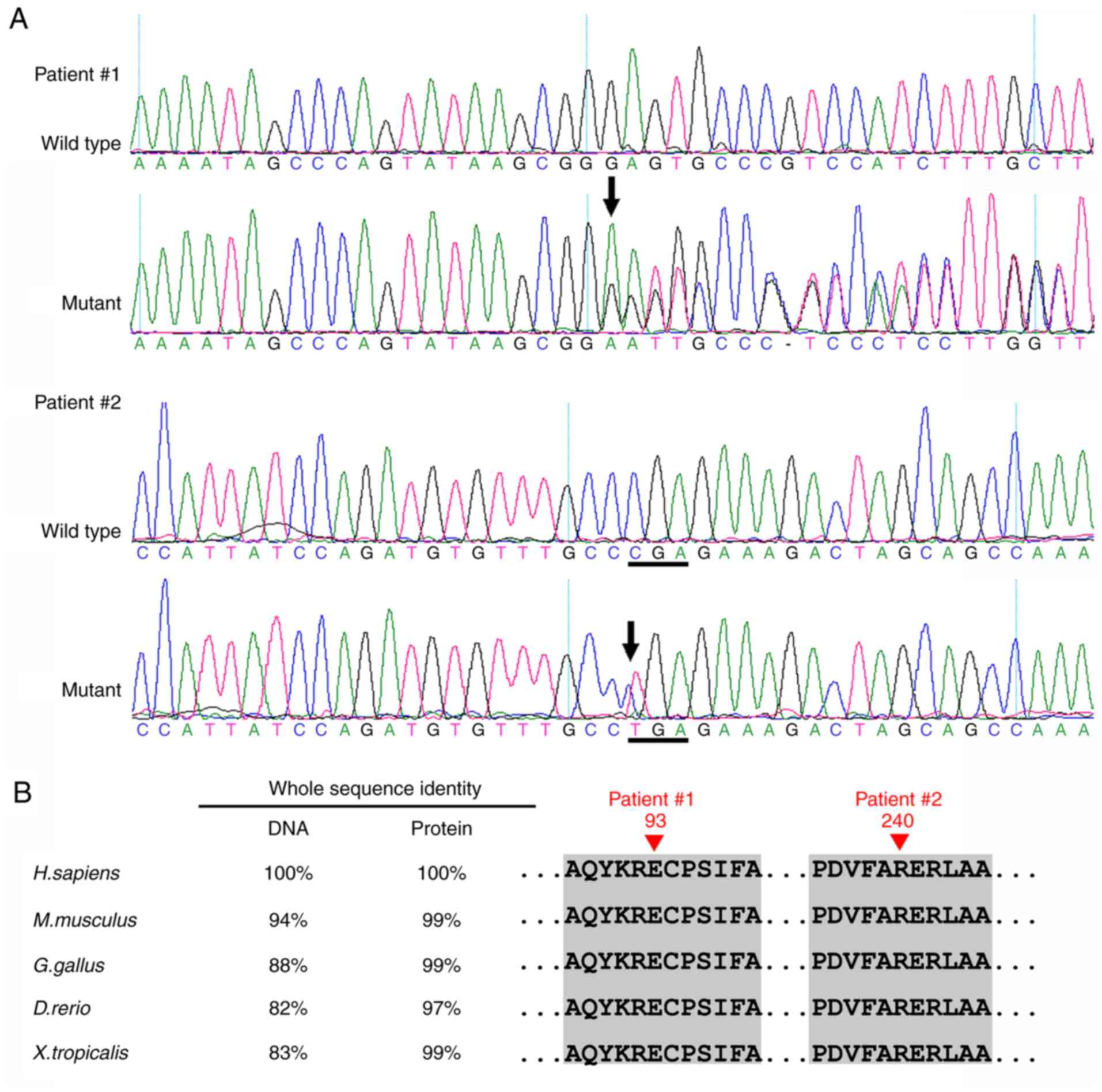
In Patient 1, a novel heterozygous mutation
c.277delG (p.Glu93SerfsX31) in exon 6 of the PAX6 gene was
identified (Fig. 5). This
frameshift mutation is predicted to produce a premature stop codon,
and thus is likely to produce a truncated protein. In Patient 2, a
recurrent nonsense mutation c.718C>T (p.Arg240X) in exon 9 was
detected (Fig. 5A). Multiple
sequencing alignment indicated that the residues at positions 93
and 240 of the PAX6 protein are highly conserved across species
(Fig. 5B). These mutations were
not detected in their unaffected parents or the 200 unrelated
control subjects from the same population. The variants identified
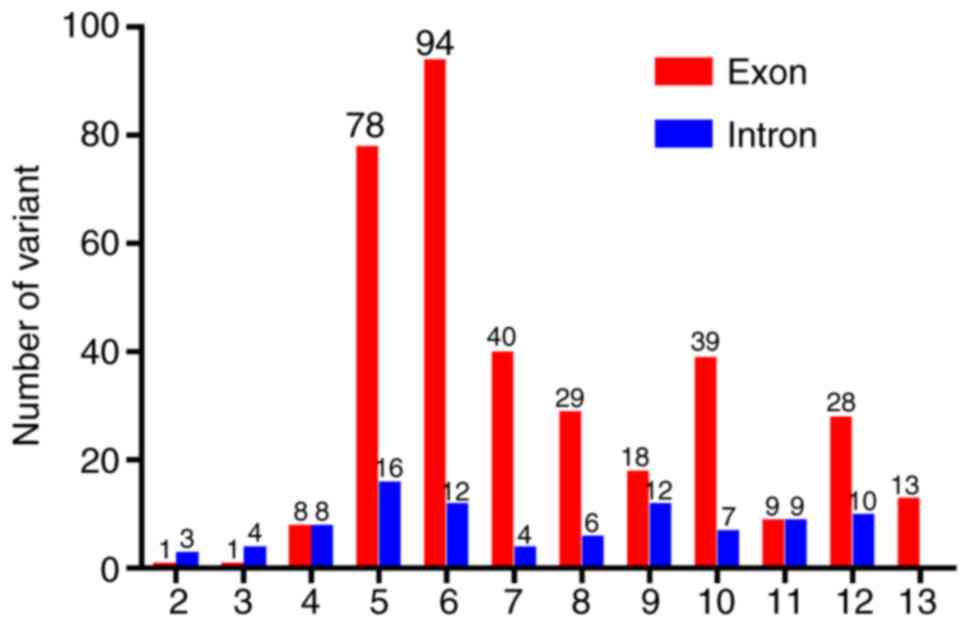
in the PAX6 gene are summarized in Fig. 6 (data LOVD; www.lovd.nl).
Discussion
Mutations in the exons 6 and 9 of PAX6 are
commonly identified in patients with congenital aniridia (22,23).
In the present study, two mutations in two Chinese patients with
classic aniridia and cataract were identified. In Patient 1, the
mutation c.277delG (p.Glu93SerfsX31) in exon 6 is a novel one; two
other mutations (c.277G>A and c.277G>T) have previously been
reported at this site (24,25).
In Patient 2, the mutation c.718C>T (p.Arg240X) in exon 9 has
been consistently reported according to LOVD. Another mutation
c.718delC at this site has been reported recently (26). These two mutations are predicted to
result in a premature stop codon and produce a truncated PAX6
protein. The C-terminal region of PAX is enriched in PST residues
that are tightly regulated by phosphorylation and serves an
essential role in transactivation of downstream targets, such as
MAF bZIP transcription factor and GATA binding protein 3 (16). Therefore, the two mutations
identified in the present study may result in impaired PAX6
function and subsequent abnormal eye development, but this requires
further investigation.
Congenital aniridia is commonly accompanied by
cataract (in 50–85% of patients), reflecting an abnormal
development of the anterior segment (27,28).
Cataracts are rarely presented in infancy, but visually significant
lens opacity can develop in the teenage or early adulthood stage
(29,30). These patients frequently suffer
from severe glare and decreased visual acuity. A number of
strategies have been developed to manage the disabling effects of
aniridia, including colored contact lens, eyelid surgery, corneal
tattooing and implantation of artificial iris (31). For Patient 1, implantation of an
artificial iris-lens diaphragm may effectively correct aphakia and
reduce glare.
Congenital aniridia is also frequently associated
with glaucoma (in 30–50% of patients) (27,32).
Similar to cataract, glaucoma usually develops in late childhood or
adulthood, but can also be manifested early in infancy with an
enlarged corneal diameter and corneal edema (30). Therefore, IOP needs to be routinely
checked in patients with congenital aniridia. Notably, these
patients may have increased central corneal thickness, which may
lead to a significant overestimation of IOP (33). Therefore, careful and regular
examinations of the optic disc morphology by fundoscopy or OCT are
required to detect early optic nerve damage.
In conclusion, two mutations in the PAX6 gene
in two sporadic Chinese patients with classic congenital aniridia
and cataract were identified. The c.277delG (p.Glu93SerfsX31)
mutation in exon 6 is a novel one, while the c.718C>T
(p.Arg240X) mutation in exon 9 is a recurrent one. These findings
expand the mutation spectrum of PAX6 and may be valuable for
future genetic counseling, and prenatal diagnosis in families with
aniridia, but larger studies are needed in order to confirm the
above findings.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81500709, 81570862
and 81670872), Guangzhou Science and Technology Project (grant no.
2014Y2-00064), and the State Scholarship Fund from the China
Scholarship Council.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XH, YL, CC, YZ, TL, CJ, XL and LL analyzed and
interpreted the patient data. HG, BL, CL, YH, QW and HL examined
the patients and performed polymerase chain reaction and gene
sequence analysis. YL, CC and YZ interpreted the sequencing data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments were carried out in accordance with
the guidelines approved by the ethics committee of Zhongshan
Ophthalmic Center at Sun Yat-sen University (Guangzhou, Guangdong,
China) and strictly followed the Declaration of Helsinki. Written
informed consent was obtained from each subject.
Patient consent for publication
All participants provided informed consent for the
publication of their clinical and genetic sequencing data in the
present study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hanson IM, Seawright A, Hardman K, Hodgson
S, Zaletayev D, Fekete G and van Heyningen V: PAX6 mutations in
aniridia. Hum Mol Genet. 2:915–920. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen JH, Lin W, Sun G, Huang C, Huang Y,
Chen H, Pang CP and Zhang M: A novel PAX6 deletion in a Chinese
family with congenital aniridia. Mol Vis. 18:989–995.
2012.PubMed/NCBI
|
|
3
|
Fischbach BV, Trout KL, Lewis J, Luis CA
and Sika M: WAGR syndrome: A clinical review of 54 cases.
Pediatrics. 116:984–988. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarsfield JK: The syndrome of congenital
cerebellar ataxia, aniridia and mental retardation. Dev Med Child
Neurol. 13:508–511. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hingorani M and Moore A:
AniridiaGeneReviews® [Internet]. Adam MP, Ardinger HH,
Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A:
University of Washington; Seattle, WA: pp. 1993–2018
|
|
6
|
Calvão-Pires P, Santos-Silva R,
Falcão-Reis F and Rocha-Sousa A: Congenital aniridia: Clinic,
genetics, therapeutics, and prognosis. Int Sch Res Notices.
2014:3053502014.PubMed/NCBI
|
|
7
|
Lee H, Khan R and O'Keefe M: Aniridia:
Current pathology and management. Acta Ophthalmol. 86:708–715.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yokoi T, Nishina S, Fukami M, Ogata T,
Hosono K, Hotta Y and Azuma N: Genotype-phenotype correlation of
PAX6 gene mutations in aniridia. Hum Genome Var. 3:150522016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jordan T, Hanson I, Zaletayev D, Hodgson
S, Prosser J, Seawright A, Hastie N and van Heyningen V: The human
PAX6 gene is mutated in two patients with aniridia. Nat Genet.
1:328–332. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Axton R, Hanson I, Danes S, Sellar G, van
Heyningen V and Prosser J: The incidence of PAX6 mutation in
patients with simple aniridia: An evaluation of mutation detection
in 12 cases. J Med Genet. 34:279–286. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ton CC, Hirvonen H, Miwa H, Weil MM,
Monaghan P, Jordan T, van Heyningen V, Hastie ND, Meijers-Heijboer
H, Drechsler M, et al: Positional cloning and characterization of a
paired box- and homeobox-containing gene from the aniridia region.
Cell. 67:1059–1074. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Halder G, Callaerts P and Gehring WJ:
Induction of ectopic eyes by targeted expression of the eyeless
gene in Drosophila. Science. 267:1788–1792. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Czerny T, Schaffner G and Busslinger M:
DNA sequence recognition by Pax proteins: Bipartite structure of
the paired domain and its binding site. Genes Dev. 7:2048–2061.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mishra R, Gorlov IP, Chao LY, Singh S and
Saunders GF: PAX6, paired domain influences sequence recognition by
the homeodomain. J Biol Chem. 277:49488–49494. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu HE, Rould MA, Xu W, Epstein JA, Maas RL
and Pabo CO: Crystal structure of the human Pax6 paired domain-DNA
complex reveals specific roles for the linker region and
carboxy-terminal subdomain in DNA binding. Genes Dev. 13:1263–1275.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singh S, Chao LY, Mishra R, Davies J and
Saunders GF: Missense mutation at the C-terminus of PAX6 negatively
modulates homeodomain function. Hum Mol Genet. 10:911–918. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coutinho P, Pavlou S, Bhatia S, Chalmers
KJ, Kleinjan DA and van Heyningen V: Discovery and assessment of
conserved Pax6 target genes and enhancers. Genome Res.
21:1349–1359. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin Y, Li T, Ma C, Gao H, Chen C, Zhu Y,
Liu B, Lian Y, Huang Y, Li H, et al: Genetic variations in
Bestrophin-1 and associated clinical findings in two Chinese
patients with juvenile-onset and adult-onset best vitelliform
macular dystrophy. Mol Med Rep. 17:225–233. 2018.PubMed/NCBI
|
|
19
|
Huang X, Lin Y, Chen C, Zhu Y, Gao H, Li
T, Liu B, Lyu C, Huang Y, Wu Q, et al: Targeted next-generation
sequencing identifies two novel COL2A1 gene mutations in Stickler
syndrome with bilateral retinal detachment. Int J Mol Med.
42:1819–1826. 2018.PubMed/NCBI
|
|
20
|
Lin Y, Liu X, Yu S, Luo L, Liang X, Wang
Z, Chen C, Zhu Y, Ye S, Yan H and Liu Y: PAX6 analysis of two
sporadic patients from southern China with classic aniridia. Mol
Vis. 18:2190–2194. 2012.PubMed/NCBI
|
|
21
|
Fokkema IF, Taschner PE, Schaafsma GC,
Celli J, Laros JF and bden Dunnen JT: LOVD v. 2.0: the next
generation in gene variant databases. Hum Mutat. 32:557–563. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Azuma N, Hotta Y, Tanaka H and Yamada M:
Missense mutations in the PAX6 gene in aniridia. Invest Ophthalmol
Vis Sci. 39:2524–2528. 1998.PubMed/NCBI
|
|
23
|
Dharmaraj N, Reddy A, Kiran V, Mandal A,
Panicker S and Chakrabarti S: PAX6 gene mutations and
genotype-phenotype correlations in sporadic cases of aniridia from
India. Ophthalmic Genet. 24:161–165. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Villarroel CE, Villanueva-Mendoza C,
Orozco L, Alcántara-Ortigoza MA, Jiménez DF, Ordaz JC and
González-del Angel A: Molecular analysis of the PAX6 gene in
Mexican patients with congenital aniridia: Report of four novel
mutations. Mol Vis. 14:1650–1658. 2008.PubMed/NCBI
|
|
25
|
Park SH, Kim MS, Chae H, Kim Y and Kim M:
Molecular analysis of the PAX6 gene for congenital aniridia in the
Korean population: Identification of four novel mutations. Mol Vis.
18:488–494. 2012.PubMed/NCBI
|
|
26
|
Pérez-Solórzano S, Chacón-Camacho OF,
Astiazarán MC, Ledesma-Gil G and Zenteno JC: PAX6 allelic
heterogeneity in Mexican congenital aniridia patients: Expanding
the mutational spectrum with Seven novel pathogenic variants. Clin
Exp Ophthalmol. 45:875–883. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Angmo D, Jha B and Panda A: Congenital
aniridia. J Curr Glaucoma Pract. 5:01–13. 2011.
|
|
28
|
Neuhann IM and Neuhann TF: Cataract
surgery and aniridia. Curr Opin Ophthalmol. 21:60–64. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hashmani S, Haider SI, Khatri B and Rind
GK: Management of Aniridia with Ectopia Lentis. Pak J Ophthalmol.
31:48–50. 2015.
|
|
30
|
Parekh M, Poli B, Ferrari S, Teofili C and
Ponzin D: Aniridia. Springer; New York, NY: 2015, View Article : Google Scholar
|
|
31
|
Adeoti CO, Afolabi AA, Ashaye AO and
Adeoye AO: Bilateral sporadic aniridia: Review of management. Clin
Ophthalmol. 4:1085–1089. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brémond-Gignac D: Glaucoma in aniridia. J
Fr Ophtalmol. 30:196–199. 2007.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park SH, Park YG, Lee MY and Kim MS:
Clinical features of Korean patients with congenital aniridia.
Korean J Ophthalmol. 24:291–296. 2010. View Article : Google Scholar : PubMed/NCBI
|