Introduction
Human natural killer (NK) cells are a subpopulation
of lymphocytes that are important in innate immunity, adaptive
immunity and reproduction (1). NK
cells are major effector cells of the innate immune system, being
important in tumor immunity, antiviral infection and the removal of
‘non-self’ cells (2–4). The T cell receptor is a specific
marker on the surface of all T cells which recognizes specific
antigens and exerts an immunological function. The difference
between T cells and NK cells is that the latter are not dependent
on the MHC-I molecules of tumor cells to initiate a toxic effect
and their activity is the result of the interaction of various
receptor molecules with corresponding ligands (5,6). The
interaction between the NK cell lectin-like receptor, NK group 2,
member D (NKG2D), and NKG2D ligands has been demonstrated to be
important in targeted cell killing by NK cells (5).
NKG2D is an activation receptor (7,8) and
is expressed on a variety of immune cells (5,9). NK
cells are heterogeneous cells and those expressing the NKG2D
receptor may be important in targeting and eliminating malignant
cells. There are two categories of NKG2D ligands: MHC class I
polypeptiderelated sequence (MIC)A and B, and UL16 binding protein
(ULBP)1–5 (10). NKG2D ligands are
expressed on the surface of tumor cells and MICs are induced cell
surface antigens (11,12). Previous studies have revealed that
numerous factors, including cell stress, heat shock, infection, DNA
damage and transformation, can upregulate the expression of MICs
(13–18).
Previous studies have demonstrated that specific
MHC-restricted cytotoxic T cells cannot function when MHC-I
molecules on tumor cells are lost or vary from the norm. In this
case, the NKG2D-NKG2D ligand signaling pathway is important in
antitumor immunity, with NKG2D as the main activating receptor of
NK cells that induces anti-tumor effects (19,20).
Following the interaction between NKG2D and corresponding induced
ligands on target cells, NKG2D binds to an adaptor protein and
transmits an activated signal to NK cells. In this way, NK cells
obtain the ability to lyse the target cells (21). In addition, CD8+ T cells
provide the necessary synergistic stimulating signals, and the
activation of the NKG2D pathway largely determines the intensity of
the cell's immune response against cancer.
At present, NK cell-based immunotherapy combined
with chemotherapy is widely used clinically; however, the mechanism
by which chemotherapy regulates the antitumor activity of NK cells,
particularly NKG2D-mediated cell death, remains to be fully
elucidated to the best of our knowledge. Although the coding
regions of NKG2D ligands are conservative, the homology of the 5′
non-translated region is low, which indicates that the regulation
of their expression may be specific to different stimuli or
lesions.
Therefore, the A549 cell line was used as the cell
model in the present study in order to investigate the regulatory
effect of a cancer treatment agent (the proteasome inhibitor MG132)
on NKG2D ligands and the susceptibility of A549 cells to NK lysis.
The possible regulatory mechanisms underlying the DNA damage
response pathway were also examined.
Materials and methods
Cell culture and reagents
The A549 (catalog no. 3111C0001CCC00002), NCI-H520
(catalog no. 3111C0001CCC000197) and NCI-H157 (catalog no.
3111C0001CCC000113) lung cancer cell lines were purchased from
Peking Union Culture Collection (Beijing, China). The PLA801D lung
cancer cell line (catalog no. CBP61005) was purchased from Nanjing
Cobioer Biotechnology Corporation (Nanjing, China). The cells were
maintained in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), supplemented with 100 IU/ml penicillin, 100
µg/ml streptomycin and 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), and incubated at 37°C with 5% CO2.
The NK92 cell line was purchased from the Peking Union Culture
Collection and cultured at 37°C with 5% CO2 in α-MEM
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
12.5% fetal bovine serum and 12.5% horse serum (Hyclone; GE
Healthcare, Inc., Logan, UT, USA). MG132, KU-55933, caffeine and
wortmannin were purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Mouse anti-human MICA (catalog no. sc-23870),
MICB (catalog no. sc-80527), ULBP1 (catalog no. sc-53131), ULBP2
(catalog no. sc-53135), ULBP3 (catalog no. sc-53132) and ULBP4
(catalog no. sc-53133) monoclonal antibodies (mAbs) were purchased
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). FITC-labeled
goat anti-mouse IgG (catalog no. ab6785) was purchased from Abcam
(Cambridge, MA, USA). Anti-Chk2 (catalog no. 2662) and -p-Chk2
(catalog no. 2661) mAbs were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA).
Treatment of the cells with the
inhibitors
A549 cells were treated with KU-55933 (10 µM) for 9
h or wortmannin (6 µM) or caffeine (10 mM) for 10 h, or MG132 for 8
h, or with KU-55933 for 9 h or wortmannin or caffeine for 10 h,
with the addition of MG132 during the final 8 h at 37°C. Then cells
were harvested at 300 × g at room temperature for 5 min.
Flow cytometry
The cells were harvested, washed three times with
PBS, and adjusted to a concentration of 5×105 cells/ml
with PBS. The cell suspensions (200 µl) were added to labeled tubes
and stained with mouse anti-human MICA, MICB, ULBP1, ULBP2, ULBP3
and ULBP4 antibodies (1:200) or an isotype antibody for 30 min at
4°C, followed by incubation with goat anti-mouse IgG in the dark.
Following incubation at 4°C for 30 min, the cells were washed three
times in PBS. The samples were examined using FACSCalibur flow
cytometry (BD Biosciences, Franklin Lakes, NJ, USA). Data were
analyzed with FlowJo version 7.6 software (FlowJo LLC, Ashland, OR,
USA).
Promoter analysis
The genomic DNA of the A549 cells was isolated and
the full-length MICB promoter was amplified using polymerase chain
reaction (PCR). The promoter fragments were cloned into the pGL3
luciferase reporter plasmid. The A549 cells were transfected with
0.79 µg MICB promoter plasmid and 0.01 µg SV40-Renilla plasmid
using Lipofectamine® 2000 and Plus reagents (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The cells (5×106) were cultured at 37°C for 24
h, following which they were treated with 10 µM MG132 at 37°C for 8
h and then lysed (Promega Corporation, Madison, WI, USA).
Luciferase and Renilla activity were measured as previously
described (22).
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
RNA was isolated using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol (23). RT
of 2 µg (20 µl) RNA into cDNA was performed using PrimeScript™
Reverse Transcriptase (Takara Biotechnology Co., Ltd., Dalian,
China). MICA, MICB, ULBP1 and ULBP2 PCR (cDNA 50 ng, 0.5 µl) was
performed with buffer TB Green Premix Ex Taq II (Takara
Biotechnology Co., Ltd.) under the following cycling conditions:
94°C for 40 sec, 61°C for 40 sec, 72°C for 50 sec, and extension at
72°C for 10 min for 40 cycles. The quantification of the NKG2D
ligands and β-actin was performed using specific primers and the
sequences were as follows: MICA, upstream,
5′-CGGGATCCTTTCTCACTGAGGTACAT-3′ and downstream
5′-CGGAATTCTGTCACGGTAATGTTGCC-3′; MICB, upstream
5′-CGGGATCCCACAGTCTTCGTTACAAC-3′ and downstream
5′-CGGAATTCCTATGTCACGGTGATGTTGC-3′; ULBP1, upstream
5′-CGGGATCCACACACTGTCTTTGCTAT-3′ and downstream
5′-CGGAATTCTCACAGCATTTGTTCCCAGTA-3′; ULBP2, upstream
5′-CGGGATCCGACCCTCACTCTCTTTGC-3′ and downstream
5′-CGGAATTCGAGGAGGAAGATCTGCC-3′; and β-actin, upstream
5′-ATCATGTTTGAGACCTTCAACA-3′ and downstream
5′-CATCTCTTGCTCGAAGTC-3′. The percentage change was calculated
using the following formula: 2−ΔΔCq (24).
Cytotoxicity assays
The cytotoxicity of the NK cells was measured using
a standard 51Cr-release assay (25). Briefly, the target tumor cells were
incubated for 1 h with 150 µCi 51Cr (PerkinElmer, Inc.,
Waltham, MA, USA) at 37°C in 5% CO2. The cells were then
washed three times with media and incubated for an additional 30
min. In order to detect the differential lysis effect of different
effector to target cell ratios, labeled target cells
(1×104 cells/well) were incubated with effector cells in
96-well plates in 10% FCS-RPMI-1640 at a total volume of 200 µl.
The plates were centrifuged at 300 × g at 37°C for 5 min following
incubation for 4 h. Aliquots (100 µl) of the supernatants from each
well were transferred to a new plate containing 100 µl/well of
Optiphase Supermix scintillation fluid. The NK cells were
pre-incubated at 37°C for 1 h with NKG2D antibodies (dilution
1:500) for antibody blocking experiments. Radioactivity was
measured using a gamma counter. The percentage of cytotoxicity was
calculated according to the following formula: 100× (experimental
release-spontaneous release)/(maximum release-spontaneous release).
Maximum release was determined by the addition of 100 µl 10% Triton
X-100 and spontaneous release was determined by incubating the
targets with 100 µl complete media.
Comet assay
The alkaline comet method of Singh et al
(26) was followed with minor
differences, and the application steps described. The cells were
harvested following treatment with 10 µM MG132 for 8 h. The slides
were pre-coated with 1% regular agarose. A low-melting-point
agarose (0.65%) suspension was added to the cell suspension at a
ratio of 4:1 and the suspension was immediately transferred onto
the slides. The cells on the slides were lysed with ice-cold
high-salt lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris pH 10,
1% Triton X-100 and 10% DMSO) in the dark at 4°C for 1 h for
disintegration of the cell and nuclear membranes. Following the
lysis phase, the slides were placed in an electrophoresis tank with
electrophoresis buffer (pH 10.0) and incubated in the dark and 4°C
for 30 min. Electrophoresis was then performed at 25 V for 20 min.
Subsequently, the slides were washed twice for 5 min with
neutralization buffer and allowed to air-dry until analysis. All
samples were evaluated within 24 h and were not subjected to
fixation. The samples were stained with 25 µg/ml propidium iodide
and then visualized under a fluorescence microscope (Leica
Microsystems GmbH, Wetzlar, Germany) and analyzed with CASPLab
version 1.2.2 software (University of Wroclaw, Wroclaw, Poland)
(27).
Western blotting
Tumor cells were collected, washed three times with
PBS and then lysed with RIPA buffer (Beyotime Institute of
Biotechnology, Shanghai, China) for 30 min at 4°C. Subsequently,
the suspension was centrifuged at 16,000 × g for 15 min at 4°C. The
concentration of proteins was detected using a BCA assay kit
(Sigma-Aldrich; Merck KGaA). Equal quantities (30 µg) of protein
were separated using SDS-PAGE (10% gels) and transferred onto
polyvinylidene fluoride membranes under 100 V for 1 h. The
membranes were blocked using 5% non-fat dry milk for 1 h at room
temperature. Subsequently, the membranes were blotted with an
appropriate primary antibody (Anti-Chk2, dilution 1:1,000, catalog
no. 2662 and p-Chk2, dilution 1:1,000, catalog no. 2661, both Cell
Signaling Technology, Inc.) overnight at 4°C. The membranes were
washed with TBST (TBS, pH 7.5, containing 0.05% Tween-20) and were
then incubated with horseradish peroxidase-conjugated secondary
antibodies (1:1,500) at room temperature for 1 h. The blots were
visualized using ECL reagents (Beyotime Institute of
Biotechnology).
Statistical analysis
All results presented are representative of a
minimum of three experiments. Statistical comparisons between
groups were made using Student's t-test or one-way analysis of
variance followed by NewmanKeul's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Statistical analysis was calculated using GraphPad, version 5.0
(GraphPad Software, Inc., La Jolla, CA, USA).
Results
A restricted set of NKG2D ligands are
expressed in the A549 cell line
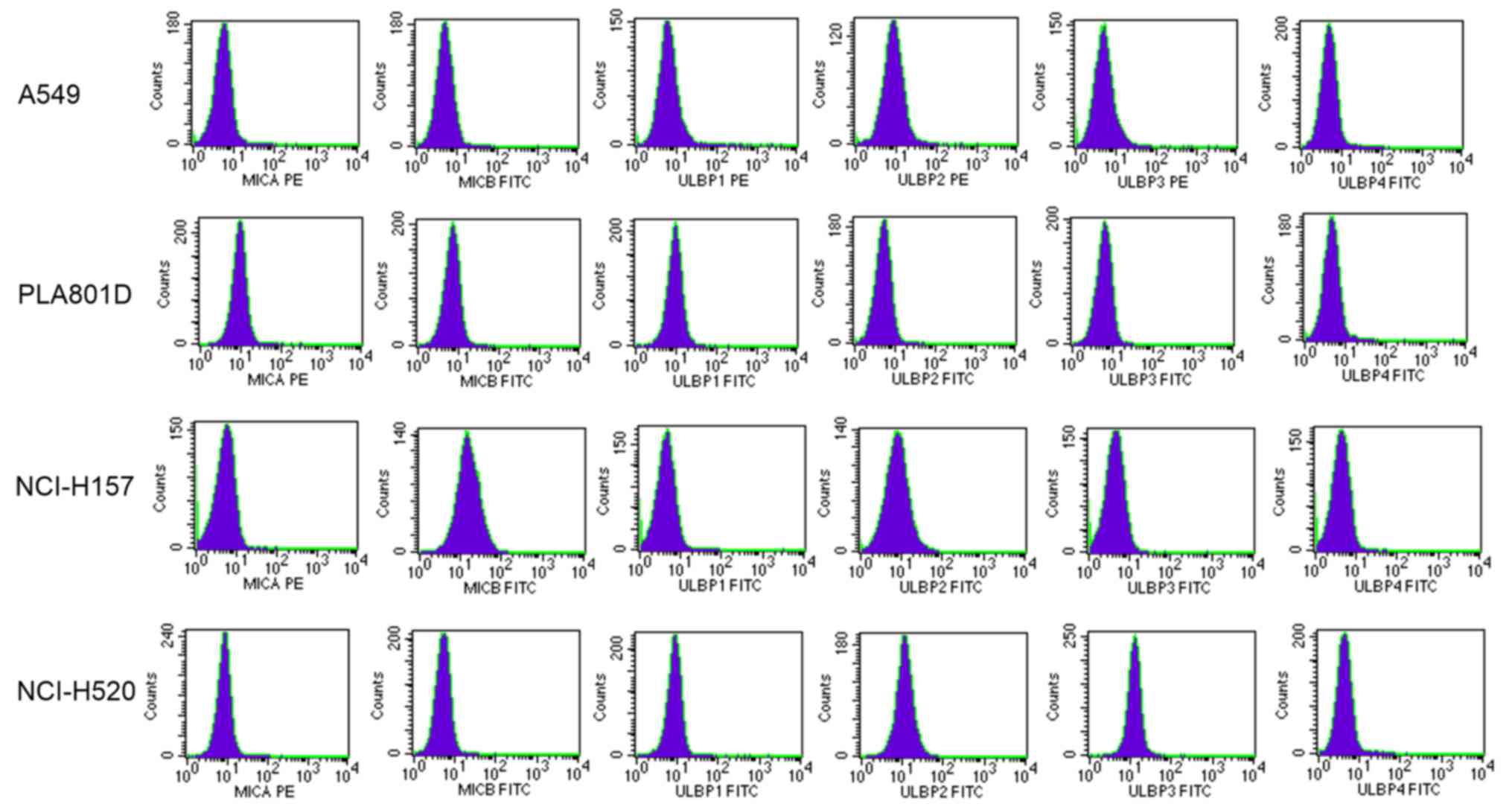
The expression levels of cell surface NKG2D ligands
were measured in the lung cancer cell lines using flow cytometry
(Fig. 1). The results demonstrated
that the expression levels of NKG2D ligands in PLA801D, NCI-H520,
NCI-H157 and A549 cells were different (Table I). In the A549 cells, MICA/B and
ULBP1 were weakly expressed, ULBP2 showed typical expression, and
neither ULBP3 nor ULBP4 were expressed (Table I).
 | Table I.Mean fluorescence intensity of NK
group 2, member D ligands on non-small cell lung cancer cells. |
Table I.
Mean fluorescence intensity of NK
group 2, member D ligands on non-small cell lung cancer cells.
| Cells | MICA | MICB | ULBP1 | ULBP2 | ULBP3 | ULBP4 |
|---|
| A549 | 1.34 | 1.27 | 1.22 | 1.80 | 0.95 | 1.03 |
| PLA801D | 1.77 | 1.49 | 2.11 | 1.14 | 1.47 | 1.04 |
| NCI-H157 | 1.03 | 3.83 | 1.10 | 1.92 | 1.10 | 1.04 |
| NCI-H520 | 1.76 | 1.14 | 2.04 | 2.79 | 2.99 | 1.06 |
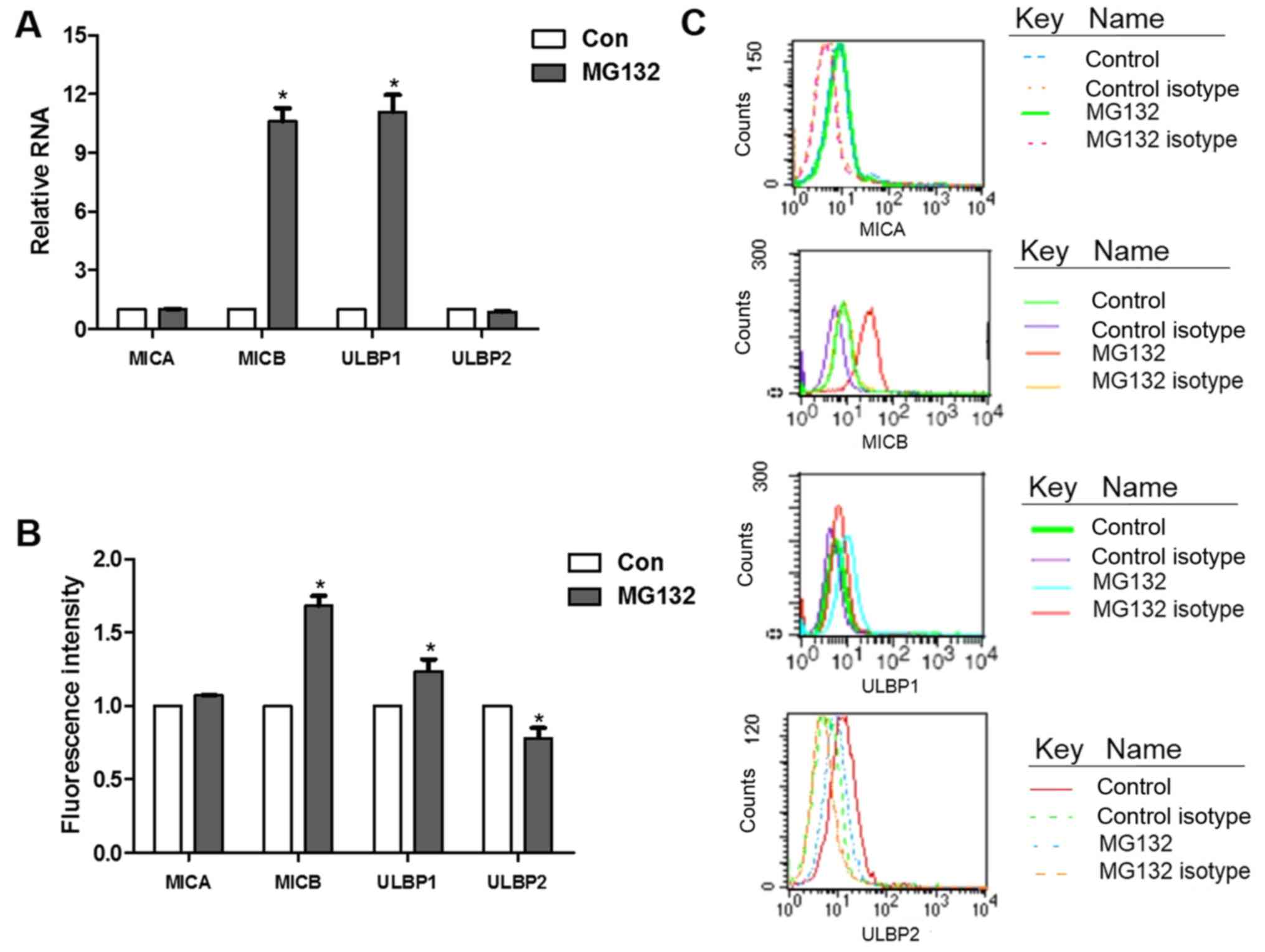
NKG2D ligand expression is selectively
induced by MG132
To investigate whether the cancer treatment agent
MG132 affects NKG2D ligands, the expression levels of NKG2D ligands
were measured following treatment with MG132 (22). Following treatment with MG132 for 8
h, the transcription levels of MICB and ULBP1 were upregulated by
10.62- and 11.09-fold, respectively (Fig. 2A), and the surface expression
levels of MICB and ULBP1 were increased by 68.18 and 23.65%,
respectively (Fig. 2B and C).
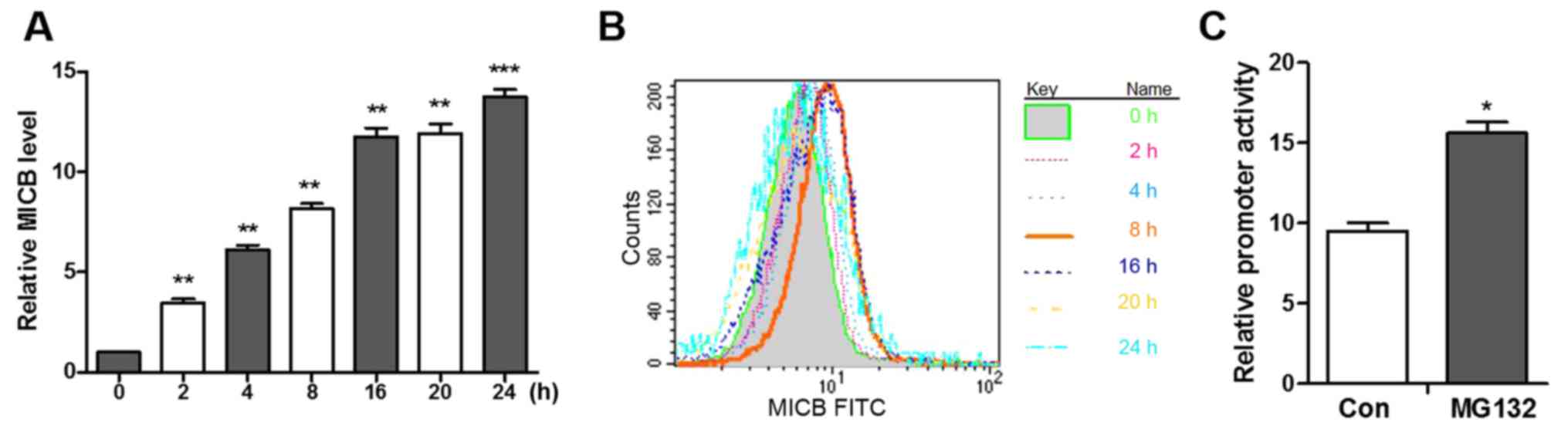
Among the ligands, the expression of MICB exhibited the most marked
change. Following treatment with MG132, the mRNA levels of MICB
increased linearly between 0 and 8 h and slowly increased after 8 h
(Fig. 3A). In addition, following
stimulation with MG132, MICB was not detectable between 2 and 4 h,
however, the expression of MICB rapidly increased between 4 and 8 h
and then slowly increased after 8 h (Fig. 3B), which indicated that the
MG132-induced expression of MICB is time-dependent.
MICB promoter is activated by a
proteasome inhibitor
As the proteasome inhibitor induced the
transcription of MICB, whether this change is initiated at the MICB
promoter was subsequently investigated. Promoter fragments of MICB,
from the MICB translation start site ATG to 480-bp upstream, were
cloned into the luciferase reporter vector pGL3 (28). The A549 cells transfected with the
pGL3-luciferase vector and incubated with MG132 for 8 h prior to
harvesting exhibited increased luciferase activity and a 1.77-fold
increase in promoter activity, as corrected for transfection
efficiency using the co-transfected pRL-SV40 (Renilla
luciferase) plasmid (Fig. 3C).
These results indicate that MG132 increases the transcription of
MICB and increases the activity of the MICB promoter.
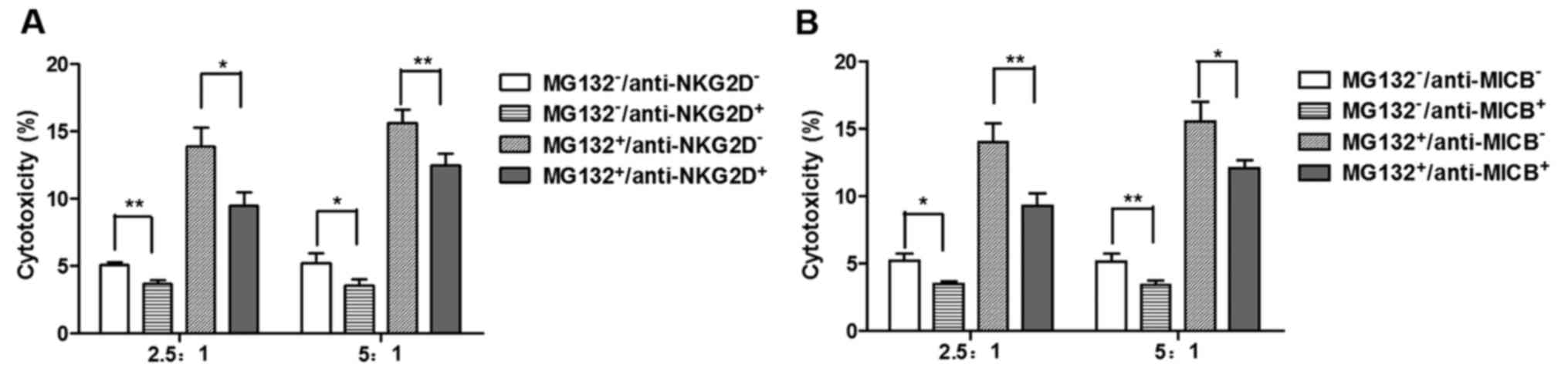
NKG2D-mediated tumor cell lysis is
enhanced by the upregulation of MICB
To determine the effect of increased levels of NKG2D
ligands in tumor cells, the cytotoxicity of NK cells against A549
cells pretreated with MG132 was measured. As shown in Fig. 4, MG132 significantly increased the
susceptibility of the A549 cell line to cytolysis by NK cells. When
the NKG2D-NKG2D ligand interactions were blocked with anti-NKG2D
antibody, the lysis of A549 cells treated with MG132 was markedly
reduced (Fig. 4A). The increased
lysis of the MG132-treated cells was partially blocked by the MICB
antibody (Fig. 4B). These results
indicate that the interaction between NKG2D and its ligands is
important in the NK-mediated lysis of the A549 cell line, and that
the increased susceptibility of MG132-treated cancer cells to the
cytotoxicity of NK cells may be mediated by upregulation of the
NKG2D ligand MICB.
MG132 induces DNA damage in A549
cells
Previous studies have demonstrated that genotoxic
agents that activate the DNA damage response pathway are
responsible for the upregulation of NKG2D ligand expression in
numerous tumor cell lines (13,22,23).
Several of the chemotherapeutic drugs used clinically have the
ability to induce the activation of ATM. Therefore, it was
hypothesized that the MG132-induced upregulation of MICB in A549
cells may be dependent on activation of the DNA damage response
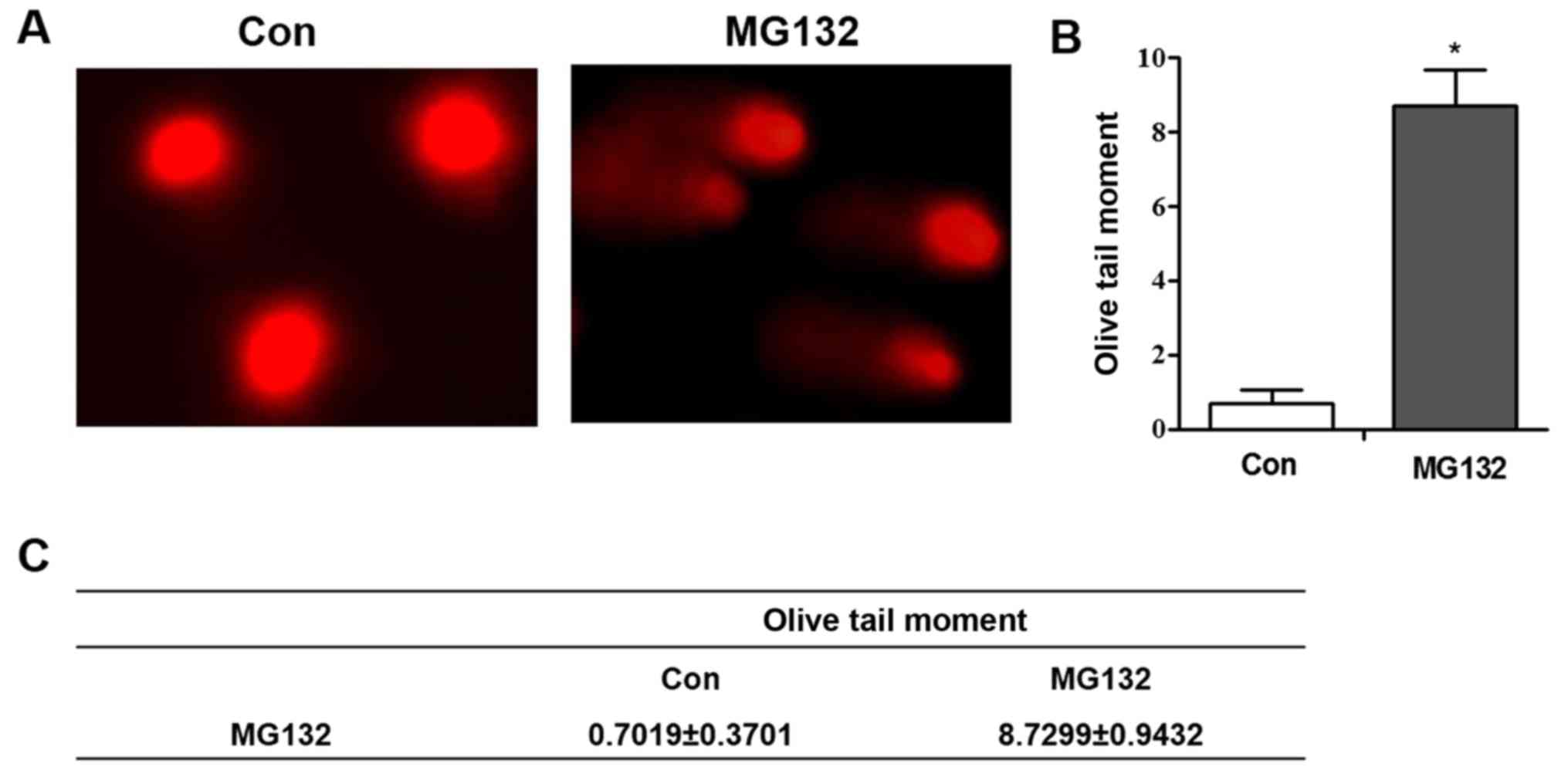
pathway. Following MG132 treatment, the results produced a ‘comet
tail’ in the comet assay, which indicates DNA strand breakage
(Fig. 5A-C).
Chk2 is activated by MG132 in A549
cells
Numerous types of cancer cell, including A549 cells,
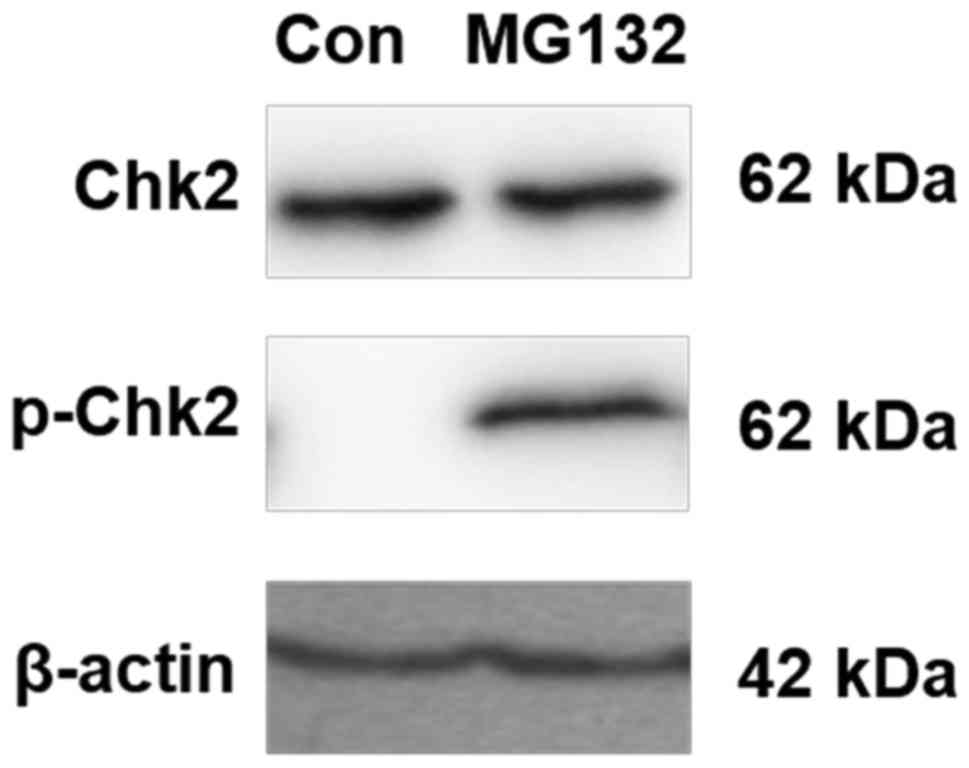
exhibit defective DNA repair mechanisms. Chk2 autophosphorylation
at Thr68 is a key early signaling event in the DNA damage response
cascade (22,29). Therefore, whether Chk2 was
functionally activated in MG132-treated A549 cells was investigated
in the present study. The A549 cells were treated with 10 µM MG132
for 8 h and lysed, following which the phosphorylation of Chk2 at
Thr68 was measured using western blotting. The results demonstrated
that the phosphorylation of Chk2 at Thr68 was induced by 10 µM
MG132 (Fig. 6). Although other
aspects of the DNA damage response pathway have not been excluded,
these results indicate that the autophosphorylation of Chk2 is
involved in the increased expression of MICB induced by MG132.
MG132-induced expression of MICB is
eliminated following treatment with KU-55933 (ATM kinase
inhibitor), wortmannin [phosphoinositide 3 (PI3) kinase inhibitor]
and caffeine (ATM/R inhibitor)
Gasser et al (30) demonstrated that the expression of
NKG2D ligands is induced by ATM/ATM-Rad3-related (ATR) signaling in
the DNA damage response pathway and that induction is prevented by
ATM/ATR inhibitors, including caffeine. Therefore, whether the
ATM/ATR inhibitors KU-55933, wortmannin and caffeine can prevent
drug-induced MICB transcription was investigated in the present
study. Treatment with KU-55933, wortmannin and caffeine inhibited
the MG132-induced upregulation of MICB (Fig. 7A). Consistent with the RT-qPCR
results, the flow cytometry revealed a similar trend (Fig. 7B). These results indicate that the
ATM/ATR signaling pathway is a possible mechanism by which MG132
induces the expression of MICB.
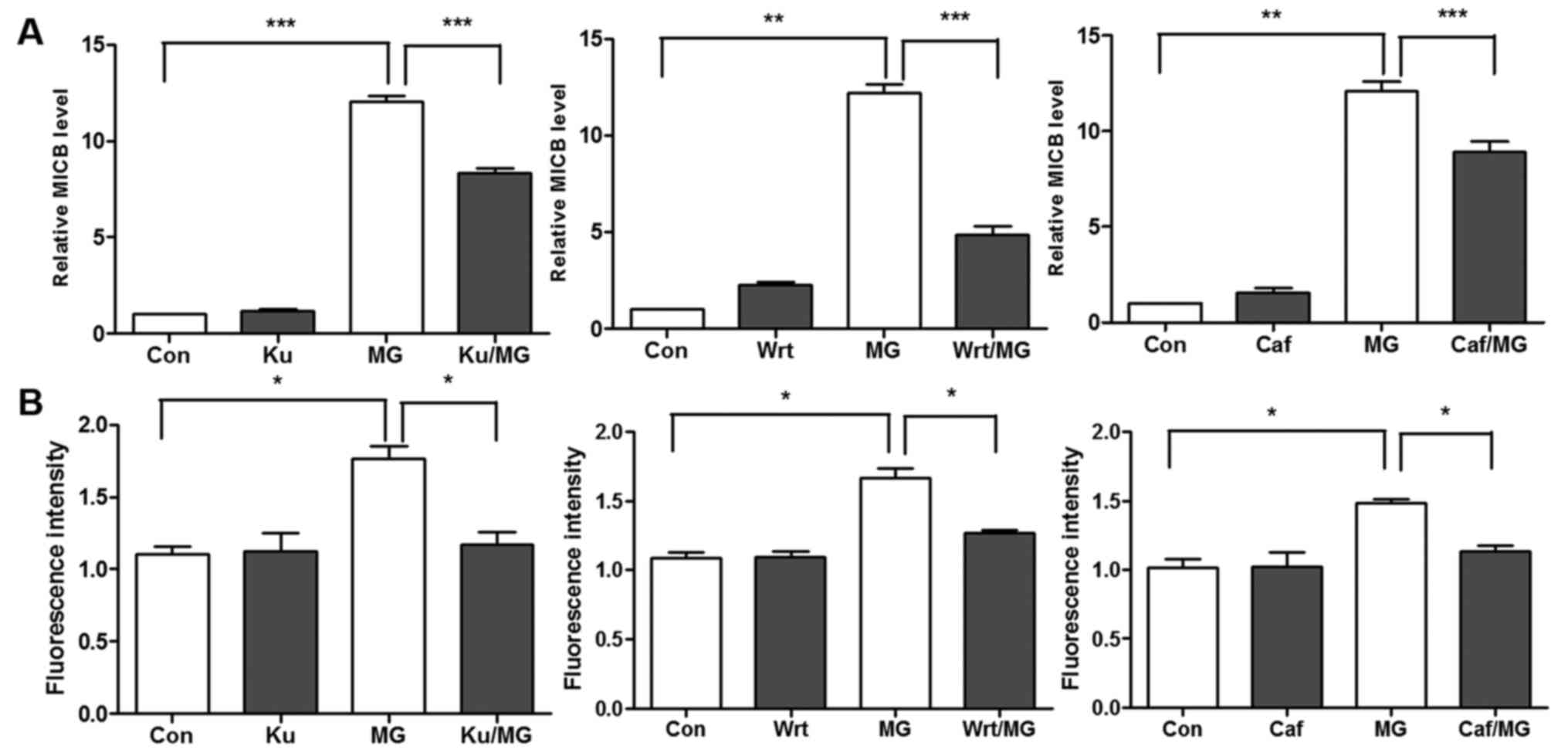 | Figure 7.ATM kinase inhibitor (KU-55933),
phosphoinositide 3 kinase inhibitor (wortmannin) and
ATM/ATM-Rad3-related inhibitor (caffeine) prevent MG132-induced
upregulation of MICB. A549 cells were treated with KU-55933 (10 µM)
for 9 h or wortmannin (6 µM) or caffeine (10 mM) for 10 h, or MG132
for 8 h, or with KU-55933 for 9 h or wortmannin or caffeine for 10
h, with the addition of MG132 during the final 8 h. The expression
of MICB was measured and was normalized to that of control
DMSO-treated cells (Con). (A) RNA levels and (B) flow cytometry.
Multiple comparisons were performed with one-way analysis of
variance. *P<0.05, **P<0.01 and ***P<0.001. MIC, MHC class
I polypeptiderelated sequence; Ku, KU-55933; Wrt, wortmannin; Caf,
caffeine; MG, MG132; Con, control. |
Discussion
In experimental animals and patients with cancer,
the expression of tumor NKG2D ligands is associated with tumor
eradication and survival rate (22). The expression levels of NKG2D
ligands are increased in tumor cells compared with those in the
surrounding normal tissue (21),
which can be induced further by cancer treatment agents (30,31).
Therefore, effective cancer treatments may directly damage tumor
cells and induce the expression of NKG2D ligands, causing NK cell
attack. In the present study, the expression levels of NKG2D
ligands in A549 cells and other lung cancer cell lines, including
PLA801D, NCI-H520 and NCI-H157, were detected. The results
demonstrated that different lung cancer cell lines express
different NKG2D ligands and have different levels of sensitivity to
NKG2D-mediated cell death by NK cells. The regulation of NKG2D
ligands in human A549 cells, a tumor system that remains to be
fully elucidated, was investigated. The regulation of NKG2D ligands
by the proteasome inhibitor MG132 and the susceptibility of A549
cells to NK lysis were also investigated. The possible regulatory
mechanisms involved were examined, focusing on the role of the DNA
damage response pathway.
Proteasome inhibitors have attracted interest as a
novel type of antitumor drug. MG132 is an aldosterone inhibitor
that reversibly inhibits proteasome chymotrypsin-like activity,
inhibiting its degradation of protein substrates, inhibiting tumor
cell growth and inducing apoptosis. MG132 is a potential
therapeutic and preventive agent for cancer cachexia (32,33).
Butler et al (22) reported
that proteasome inhibitors that regulate NKG2D ligands are
multifaceted, multilevel and can selectively upregulate expression
of the ULBP1 promoter, and mRNA and cell surface proteins in head
and neck squamous cell carcinoma. As the role of MG132 is
complicated, the regulation of MICB may also be multilinked and
multilevel. Therefore, the effects of MG132 on the promoter
activity, mRNA expression and cell surface expression of MICB were
investigated in the present study. Whether MG132 can cause DNA
damage and activate key molecules in the DNA damage response
pathway were also examined. The results demonstrated that MG132
upregulated the activity of the MICB promoter, mRNA expression and
cell surface protein expression. Additionally, MG132 induced DNA
single-strand breaks and activated Chk2 molecules. KU-55933 (ATM
kinase inhibitor), wortmannin (PI3 kinase inhibitor) and caffeine
(ATM/R inhibitor) inhibited the expression of MICB that was induced
by MG132. These results indicate that DNA damage induced by MG132
activates protein kinase molecules, including ATM and Chk2, and
those of other DNA damage pathways, which may affect the expression
of MICB on the cell surface.
Based on analysis with Gene Runner software and
experimental data (28), it was
hypothesized that the core region of the MICB promoter includes
elements for transcription regulation, including TATA-like elements
and HSE. These elements may regulate the transcription of MICB, and
this requires further investigation. Winter et al (34) reported that, following DNA damage,
ATM molecules can phosphorylate nuclear E3 ubiquitin ligase Siah-1
and inhibit Siah-1-mediated homeodomain-interacting protein 2
(HIPK2) ubiquitination and degradation. HIPK2 activates
phosphorylated p53 and promotes apoptosis. Therefore, the role of
MG132 is associated not only with the DNA damage response but also
with the ubiquitination and degradation of signaling molecules.
However, the detailed mechanism requires further investigation.
In conclusion, the present study demonstrates that
MG132 selectively upregulates the surface expression of MICB in
A549 cells, and increases the NKG2D-mediated cytotoxicity of NK
cells. The regulatory effect of MG132 is associated with the
activation of Chk2, an event associated with DNA damage. The
combination of MG132 with NK cell immunotherapy may have a
synergistic effect that improves the therapeutic effect of lung
cancer treatment.
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science
Foundation of China (grant no. 81503391), the China Youth
Foundation (grant no. 31500137) and the China Postdoctoral Science
Foundation (grant no. 2015M582847).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
ZNC, FL and ZLW conceived and designed the
experiments. DL, XWD and BY performed the experiments and drafted
the manuscript. ML and JHY were involved in the data analysis. HL
and THX assisted with the experiments. All authors reviewed and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATM
|
ataxia telangiectasia mutated
|
|
ATR
|
ATM-Rad3-related
|
|
mAb
|
monoclonal antibody
|
|
MICA
|
MHC class I polypeptiderelated
sequence A
|
|
MICB
|
MHC class I polypeptiderelated
sequence B
|
|
NK
|
natural killer
|
|
NKG2D
|
NK group 2, member D
|
|
NSCLC
|
non-small cell lung cancer
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
ULBP
|
UL16 binding protein
|
References
|
1
|
Bellora F, Castriconi R, Dondero A,
Carrega P, Mantovani A, Ferlazzo G, Moretta A and Bottino C: Human
NK cells and NK receptors. Immunol Lett. 161:168–173. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gasser S and Raulet DH: Activation and
self-tolerance of natural killer cells. Immunol Rev. 214:130–142.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bae DS, Hwang YK and Lee JK: Importance of
NKG2D-NKG2D ligands interaction for cytolytic activity of natural
killer cell. Cell Immunol. 276:122–127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kruse PH, Matta J, Ugolini S and Vivier E:
Natural cytotoxicity receptors and their ligands. Immunol Cell
Biol. 92:221–229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yin X, Lu X, Zhang X, Min Z, Xiao R, Mao Z
and Zhang Q: Role of NKG2D in cytokine-induced killer cells against
lung cancer. Oncol Lett. 13:3139–3143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mayes K, Elsayed Z, Alhazmi A, Waters M,
Alkhatib SG, Roberts M, Song C, Peterson K, Chan V, Ailaney N, et
al: BPTF inhibits NK cell activity and the abundance of natural
cytotoxicity receptor co-ligand. Oncotarget. 8:64344–64357. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Borchers MT, Harris NL, Wesselkamper SC,
Vitucci M and Cosman D: NKG2D ligands are expressed on stressed
human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol.
291:L222–L231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burgess SJ, Maasho K, Masilamani M,
Narayanan S, Borrego F and Coligan JE: The NKG2D receptor:
Immunobiology and clinical implications. Immunol Res. 40:18–34.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coudert JD and Held W: The role of the
NKG2D receptor for tumor immunity. Semin Cancer Biol. 16:333–343.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Champsaur M and Lanier LL: Effect of NKG2D
ligand expression on host immune responses. Immunol Rev.
235:267–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stern-Ginossar N, Elefant N, Zimmermann A,
Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichardi M,
Hahn G, et al: Host immune system gene targeting by a viral miRNA.
Science. 317:376–381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bauer S, Groh V, Wu J, Steinle A, Phillips
JH, Lanier LL and Spies T: Activation of NK cells and T cells by
NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fionda C, Soriani A, Malgarini G, Iannitto
ML, Santoni A and Cippitelli M: Heat shock protein-90 inhibitors
increase MHC class I-related chain A and B ligand expression on
multiple myeloma cells and their ability to trigger NK cell
degranulation. J Immunol. 183:4385–4394. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haque M, Ueda K, Nakano K, Hirata Y,
Parravicini C, Corbellino M and Yamanishi K: Major
histocompatibility complex class I molecules are down-regulated at
the cell surface by the K5 protein encoded by Kaposi's
sarcoma-associated herpesvirus/human herpesvirus-8. J Gen Virol.
82:1175–1180. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Friese MA, Wischhusen J, Wick W, Weiler M,
Eisele G, Steinle A and Weller M: RNA interference targeting
transforming growth factor-beta enhances NKG2D-mediated antiglioma
immune response, inhibits glioma cell migration and invasiveness,
and abrogates tumorigenicity in vivo. Cancer Res.
64:7596–7603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nachmani D, Stern-Ginossar N, Sarid R and
Mandelboim O: Diverse herpesvirus microRNAs target the
stress-induced immune ligand MICB to escape recognition by natural
killer cells. Cell Host Microbe. 5:376–385. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lanier LL: NKG2D receptor and its ligands
in host defense. Cancer Immunol Res. 3:575–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stern-Ginossar N, Gur C, Biton M, Horwitz
E, Elboim M, Stanietsky N, Mandelboim M and Mandelboim O: Human
microRNAs regulate stress-induced immune responses mediated by the
receptor NKG2D. Nat Immunol. 9:1065–1073. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morisaki T, Onishi H and Katano M: Cancer
immunotherapy using NKG2D and DNAM-1 systems. Anticancer Res.
32:2241–2247. 2012.PubMed/NCBI
|
|
20
|
Tallerico R, Todaro M, Di FS, Maccalli C,
Garofalo C, Sottile R, Palmieri C, Tirinato L, Pangigadde PN and La
Rocca R: Human NK cells selective targeting of colon
cancer-initiating cells: A role for natural cytotoxicity receptors
and MHC class I molecules. J Immunol. 190:2381–2390. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu J, Song Y, Bakker AB, Bauer S, Spies T,
Lanier LL and Phillips JH: An activating immunoreceptor complex
formed by NKG2D and DAP10. Science. 285:730–732. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Butler JE, Moore MB, Presnell SR, Chan HW,
Chalupny NJ and Lutz CT: Proteasome regulation of ULBP1
transcription. J Immunol. 182:6600–6609. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang C, Wang Y, Zhou Z, Zhang J and Tian
Z: Sodium butyrate upregulates expression of NKG2D ligand MICA/B in
HeLa and HepG2 cell lines and increases their susceptibility to NK
lysis. Cancer Immunol Immunother. 58:1275–1285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maniar A, Zhang X, Lin W, Gastman BR,
Pauza CD, Strome SE and Chapoval AI: Human gammadelta T lymphocytes
induce robust NK cell-mediated antitumor cytotoxicity through CD137
engagement. Blood. 116:1726–1733. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Singh NP, Mccoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang KF, Ren H, Cao J, Zeng GL, Xie J,
Chen M, Wang L and He CX: Decreased dicer expression elicits DNA
damage and up-regulation of MICA and MICB. J Cell Biol.
182:233–239. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Venkataraman GM, Suciu D, Groh V, Boss JM
and Spies T: Promoter region architecture and transcriptional
regulation of the genes for the MHC class I-Related chain a and b
ligands of NKG2D. J Immunol. 178:961–969. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bartek J, Bartkova J and Lukas J: DNA
damage signalling guards against activated oncogenes and tumour
progression. Oncogene. 26:7773–7779. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gasser S, Orsulic S, Brown EJ and Raulet
DH: The DNA damage pathway regulates innate immune system ligands
of the NKG2D receptor. Nature. 436:1186–1190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ljunggren HG and Malmberg KJ: Prospects
for the use of NK cells in immunotherapy of human cancer. Nat Rev
Immunol. 7:329–339. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu J, Shen W, Tang Y, Zhou J, Li M, Zhu
W, Yang H, Wu J, Zhang S and Cao J: Proteasome inhibitor MG132
enhances the antigrowth and antimetastasis effects of radiation in
human nonsmall cell lung cancer cells. Tumour Biol. 35:7531–7539.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang L, Tang H, Kou Y, Li R, Zheng Y,
Wang Q, Zhou X and Jin L: MG132-mediated inhibition of the
ubiquitin-proteasome pathway ameliorates cancer cachexia. J Cancer
Res Clin Oncol. 139:1105–1115. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Winter M, Sombroek D, Dauth I,
Moehlenbrink J, Scheuermann K, Crone J and Hofmann TG: Control of
HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases
ATM and ATR. Nat Cell Biol. 10:812–824. 2008. View Article : Google Scholar : PubMed/NCBI
|