Introduction
Hepatitis B virus (HBV) infects ~350 million people
worldwide and is one of the most serious public health problems
(1). The virus belongs to the
Hepadnaviridae family and contains a 3.2-kb partially
double-stranded circular genome; HBV may be one of the smallest of
microbial pathogens affecting humans (2). HBV replication is highly dependent on
the accurate assembly of the capsid, which is also associated with
the covalently closed circular DNA (cccDNA) reservoir for
persistent infection (3,4). Following translation from full-length
pregenomic RNA (pgRNA), hepatitis B virus core (HBc) protein
interacts with pgRNA, reverse transcriptase, and host factors to
form icosahedral-shaped capsids and initiate viral replication
(5).
The HBV capsid is closely associated with genome
replication (3). A few small
molecules, including GLS4, Huntingtin-associated protein 1 and
AT130, disrupt capsid formation and inhibit viral replication
(6–9). These molecules alter the structure
and disrupt the function of capsids (7). In addition, capsids have been
considered to mediate the regulation of HBV replication (10). The secreted hepatitis B e-antigen,
which has a structure similar to that of HBc, may interact with HBc
monomers and form aberrant capsids that do not support the pgRNA
package (10).
HBc, which forms the icosahedral shell of capsids,
consists of 183 or 185 amino acid residues (aa), depending on
genotype (11). The primary
structure of the core protein can be divided into two domains,
namely, the N-terminal, which consists of 149 or 151 aa, depending
upon the genotype, and directs HBc self-assembly (12,13);
the C-terminal constitutes 34 aa and is rich in arginine residues,
essential for capsid formation. The deletion of the C-terminal
domain inhibits pgRNA encapsidation (14). Capsid assembly consists of two
steps, in which HBc monomers initially associate to yield a dimer
intermediate via an intradimer interface (15). Numerous dimers subsequently form an
intact capsid via a dimer-dimer interface. Whether higher-order HBc
oligomers exist remains controversial (16). Several studies have proposed that
aa 113–143 of HBc form the main dimer-dimer interface (17,18).
In the present study, the aim was to identify a
panel of residues located at the dimer-dimer interface based on the
HBc crystal structure, and to investigate their effect on capsid
function and viral replication. The results demonstrated that the
HBc dimer-dimer interface was required for capsid assembly and
viral replication. Targeting the dimer-dimer interface may be a
novel and powerful antiviral strategy.
Materials and methods
HBc crystal structure
The crystal structure of HBc was downloaded from the
PDB (1QGT, http://www.rcsb.org/pdb/home/home.do). The dimer-dimer
interface domains were visualized and mapped using Swiss-PdbViewer
v4.0 software (http://www.expasy.org/spdbv/).
Plasmids
The 1.2-length (3,215 bp) HBV adw genome (GenBank
accession no. AY518556; http://www.ncbi.nlm.nih.gov/genbank/) was obtained
from the pHBV1.2 plasmid and inserted into pUC18 vector (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) under the control of the
lac promoter, and was employed in the present study. Plasmid
pHBV1.2-core− was derived from pHBV1.2 by introducing a
stop codon (TAT→TAG) into the C gene at the Y38 position and
ligating to pUC18 vector by HindIII and PshAI
digestion, thereby preventing HBc production. In addition, the
plasmid 1–183 flag, which directs the expression of the HBc gene
under the control of the cytomegalovirus promoter, was generated
using XbaI and EcoRI enzymes, the pcDNA3.1 vector,
along with a FLAG tag (N-DYKDDDDK-C) fused to the C-terminus.
Site-directed mutagenesis
All HBc mutant plasmids (pHBc14-18M, pHBc120-135M,
pHBc23-39M and pHBc122-139M) were derived from the 1–183 flag
plasmid. The altered residues of each domain were presented in
Table I. Acidic amino acids were
mutated into basic amino acids and vice versa, and amino acids with
long side chains were mutated into glycine, to abolish the
hydrophobic effect of the four domains. All point mutations
generated within the core protein expression vector 1–183 flag were
similarly generated according to a previously described polymerase
chain reaction (PCR)-based method and the plasmid sequences were
analyzed with BioEdit version 7.0.5 (http://www.mbio.ncsu.edu/BioEdit/page2.html) (19). PCR reactions were performed using
Invitrogen™ PCR SuperMix (Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. In order to establish the 1–183
flag plasmid, a template pHBV1.2 plasmid, and 1–183 flag forward
(For) and 1–183 flag reverse (Rev) primers were used. The pHBC14-18
plasmid was established using 1–183 flag as a template, and aa14-18
For and 1–183 flag Rev primers. pHBc23-39M, pHBc120-135M and
pHBc122-139M plasmids were established via two cycles of PCR. For
example, for pHBc120-135M plasmids, aa120-135 For and 1–183 flag
Rev, and 1–183 flag For, and aa120-135 Rev primers were used for
the first round of PCR, and pHBV1.2 was used as a template; the two
products from the first round of PCR were subsequently mixed and
annealed at 72°C, and used as the template for the second round of
PCR, where 1–183 flag For and 1–183 flag Rev primers were used. All
PCR products were cloned into pcDNA3.1 vectors (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) using XbaI and
EcoRI enzymes for digestion and ligation, respectively. The
pHBV1.2-core− was generated using the aforementioned
method but instead using HindIII For and Y38 Rev, and Y38
For and PshAI Rev primers for the first round of PCR; the
two products from the first round of PCR were annealed and used as
a template for the second round of PCR, where HindIII For
and PshAI Rev primers were used. The PCR products were
introduced to pUC18 vector by HindIII and PshAI
digestion and ligation.
 | Table I.HBc mutants of the dimer-dimer
interface. |
Table I.
HBc mutants of the dimer-dimer
interface.
| HBc | Wild-type | Mutant |
|---|
| 14-18 | ELLSF | KLLSG |
| 120-135 |
VSFGVWIRTPPAYRPP |
GSFGGWIDTGPAARGG |
| 23-39 |
FFPSIRDLLDTASALYR |
GFGSIRKLLDTASAGYD |
| 122-139 |
FGVWIRTPPAYRPPNAPI |
GGVWIRTPPAYRPPNAPG |
Cell culture and transfection
Human hepatoma HepG2 cells (American Type Culture
Collection, Manassas, VA, USA) were cultured in Dulbecco's modified
Eagle's medium (DMEM)-F12 (Invitrogen; Thermo Fisher Scientific,
Inc.) containing 10% (v/v) fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C under 5% CO2. HepG2 cells were
co-transfected with HBc mutant plasmids and wild-type (WT) pHBV1.2.
1–183 flag and pcDNA3.1 were also transfected into HepG2 cells to
establish control groups. All transfections were conducted using
FuGENE HD transfection reagent (Promega Corporation, Madison, WI,
USA) according to the manufacturer's protocols. The day prior to
transfection, 5×105 cells were seeded into a 6-well
plate and cultured at 37°C. After 20 h, 6 µl FuGENE HD reagent and
2 µg HBc mutant plasmid or different amounts of HBc mutant plasmids
(0.2–3.2 µg) together with 1 µg pHBV1.2 were used for transfection.
According to preliminary experiments using a pCMV-GFP plasmid, ~35%
of target HepG2 cells were successfully transfected. Five days
post-transfection, cells were harvested for further analysis.
Viral particle assays
The secreted virion and intracellular capsid were
assayed as previously described with some modifications (20). Briefly, after 5 days
post-transfection, 1 ml cell culture DMEM-F12 was harvested from
the 6-well plate and secreted HBV virion was precipitated by adding
dry PEG 8000 (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) to a
final concentration of 10% (w/v), and then incubated at 4°C for 1
h. The precipitate was collected by centrifugation (1,000 × g, 20
min at 4°C) and dissolved in 40 µl DMEM-F12. For the extraction of
the intracellular capsid, cells from each well were washed three
times with PBS and resuspended in 500 µl lysis buffer [150 mM NaCl,
50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.2% (v/v) Nonidet
P-40] on ice for 10 min. The supernatant of the cell lysate was
collected by centrifugation at 12,000 × g for 10 min at 4°C,
precipitated with PEG 8000 (final concentration 10%, w/v) for 1 h
at 4°C, and then dissolved in 60 µl DMEM-F12. A total of 10 µl
secreted virions and intracellular capsids were fractionated via
electrophoresis on a non-denaturing 1% agarose gel and transferred
onto a nitrocellulose membrane overnight by blotting with 20X SSC
buffer [3 M NaCl, 300 mM sodium citrate (pH 7)]. To detect the
secreted virions and intracellular capsids, the membrane was
blocked at room temperature for 1 h with Tris-buffered saline
supplemented with 5% non-fat dry milk and 0.2% Tween-20.
Subsequently, the virions were probed with monoclonal anti-envelope
antibodies (1:2,000; cat. no. 18-0023; Invitrogen; Thermo Fisher
Scientific, Inc.) or with a polyclonal antibody against core
antigen (1:1,000; cat. no. B0586; Dako; Agilent Technologies, Inc.,
Santa Clara, CA, USA) for 3 h at room temperature. Bound antibodies
were detected using horseradish peroxidase-labeled secondary
antibodies for 3 h at room temperature (goat anti-mouse; 1:5,000;
cat. no. RM3001 and goat anti-rabbit; 1:4,000; cat. no. RM3002;
both Beijing Ray Antibody Biotech, Beijing, China; http://www.rayantibody.com/). Proteins were visualized
using an ECL Plus chemiluminescence kit (Amersham Pharmacia
Biotech; GE Healthcare, Chicago, IL, USA). Densitometry was
performed using Quantity One® software version 4.6.3
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
SDS-PAGE and western blotting
Five days post-transfection, cells were treated with
lysis buffer. Protein concentration was determined using the
Pierce™ BCA Protein Assay kit (Thermo Fisher Scientific, Inc.) and
adjusted to 5 µg/µl with lysis buffer. A total of 50 µg/10 µl cell
lysate was separated by 12% SDS-PAGE and transferred onto a
polyvinylidene fluoride membrane. The membrane was blocked with 5%
non-fat milk in Tris-buffered saline for 0.5 h at room temperature,
and incubated with primary anti-flag (1:2,000; cat. no. PM020; MBL
International Co., Woburn, MA, USA), polyclonal rabbit anti-core
(1:1,000; cat. no. B0586; Dako; Agilent Technologies, Inc.) and
monoclonal mouse anti-GAPDH (1:3,000; cat. no. RM 2002; Beijing Ray
Antibody Biotech) antibodies for 2 h at room temperature. Bound
antibodies were detected with horseradish peroxidase-labeled
secondary antibodies for 3 h at room temperature (goat anti-rabbit;
1:4,000; cat. no. RM3002 and goat anti-mouse; 1:5,000; cat. no.
RM3001; both Beijing Ray Antibody Biotech) and visualized using an
ECL Plus chemiluminescence kit (Amersham Pharmacia Biotech; GE
Healthcare). The density of each band was analyzed using Quantity
One software version 4.6.3 (Bio-Rad Laboratories, Inc.).
Southern blotting
To detect capsid-associated HBV DNA, a digoxigenin
(DIG)-labeled double-stranded specific full-length HBV probe was
synthesized from pHBV1.2 with a PCR DIG Probe Synthesis Kit (Roche
Diagnostics, Indianapolis, IN, USA) according to the manufacturer's
protocols. Primers (P1,
5′-CCGGAAAGCTTGAGCTCTTCTTTTTCACCTCTGCCTAATCA-3′ and P2,
5′-CCGGAAAGCTTGAGCTCTTCAAAAAGTTGCATGGTGCTGG-3′) were used to
synthesize the HBV probe. Extraction of intracellular
capsid-associated HBV DNA and Southern blot analysis were conducted
as previously described (21)
using a DIG DNA Labeling and Detection Kit (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany). In brief, 15 µg of capsid-associated HBV
DNA per lane was electrophoresed on a 1% agarose gel. Nucleic acids
were then transferred onto a positively charged nylon membrane (GE
Healthcare). These blots were hybridized with a DIG-labeled
double-stranded specific full-length HBV probe overnight at room
temperature.
Quantification of HBV DNA and cccDNA
using quantitative (q)PCR
The nuclear cccDNA and virion HBV DNA in the culture
medium were quantified by qPCR on a LightCycler® 480
system (Roche Diagnostics, Indianapolis, IN, USA). Total DNA was
extracted using the QIAamp DNA Mini kit (Qiagen GmbH, Hilden,
Germany) according to the manufacturer's instructions. Isolated DNA
was treated with 10,000 U/ml T5 exonuclease (New England BioLabs,
Inc., Ipswich, MA, USA) for 30 min at 37°C in 10 µl reaction volume
followed by heat inactivation at 95°C for 5 min and 4-fold dilution
with distilled water. Two different primer and TaqMan probe sets
were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) for
the detection of HBV-DNA and HBV cccDNA in the culture medium. The
primers and probe design were according to a previous study
(22). The PCR primers for the
amplification of cccDNA were: For, 5′-GGGGCGCACCTCTCTTTA-3′ and
Rev, 5′-AGGCACAGCTTGGAGGC-3′. The TaqMan hydrolysis probe for the
detection of the amplified 367 bp DNA fragment was as follows:
5′-FAM-TTCTCATCTGCCGGACCGTG-BHQ-3′. The PCR primers for the
amplification of total DNA were: For, 5′-GCCAAAATTCGCAGTCC-3′ and
Rev, 5′-AAACTGAGCCAGGAGAAA-3′. The Taqman hydrolysis probe for the
detection of the amplified 376 bp DNA fragment was as follows:
5′-FAM-TTCCTCTTCATCCTGCTGCTATGCC-BHQ-3′. The amplification program
consisted of an initial denaturing step at 94°C for 10 min,
followed by 45 cycles of 95°C for 10 sec, 55°C for 20 sec and 72°C
for 20 sec.
The pHBV1.2 plasmid, which contained 1.2 copies of
the HBV genome, was extracted using the QIAGEN Plasmid Mini kit
(Qiagen GmbH), according to the manufacturer's instructions. The
plasmid concentration (mg/ml) was measured at 260 nm using a
spectrophotometer. The copy number of the plasmid was calculated
using the following formula: Copies/ml = C/M xA (where C is the
plasmid concentration, M is the molecular weight and A is the
Avogadro constant). Ten-fold serial dilutions of pHBV1.2 plasmid
stock (109−102 copies/ml) were used to
establish qPCR standard curves for detecting HBV cccDNA and HBV
total DNA. The primers and probe are the same as when the samples
were tested. Each experiment was performed in triplicate.
RNA extraction and northern
blotting
Total cellular RNA was extracted from transfected
cells with TRIzol reagent (Invitrogen; Thermo Fisher Scientific.,
Inc.). Total RNA (5 µg) per lane was separated on a 1% agarose gel
containing 2.2 M formaldehyde and transferred onto a positively
charged nylon membrane (GE Healthcare) with 20X SSC buffer. The
probe and hybridization procedure was conducted in accordance with
that for Southern blot analysis.
Conservation analysis of capsid
residues
A total of 9,386 HBc sequences were downloaded from
HBVdb (https://hbvdb.ibcp.fr/HBVdb/HBVdbIndex) (23) and submitted to Clustal Omega
(https://www.ebi.ac.uk/Tools/msa/clustalo/; version
1.2.4) for alignment (24).
Statistical analysis
SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA)
was employed to analyze the experimental data, which are presented
as the mean ± standard error of the mean. A Mann-Whitney U test was
performed to compare the levels of viruses between HBc mutant and
control groups. P<0.05 was considered to indicate a
statistically significant difference. All experiments were
performed in triplicate.
Results
Identification of HBc dimer-dimer
interface
The crystal structure of HBc has been reported
previously (18). HBc is mainly a
helical protein; the hydrophobic effect is the main force that
directs its assembly (Fig. 1A).
HBc monomers form a dimer via the intradimer interface to initiate
assembly, and then several dimers are assembled to form an
icosahedral capsid via the dimer-dimer interface (15). Based on its crystal structure, 16
aa located at the dimer-dimer interface were identified using
Swiss-PdbViewer v4.0: i) 14E, 18F, 120V, 124V, 127R, 129P, 132Y,
134P and 135P; and ii) 23F, 25P, 29D, 37L, 39R, 122F, and 139I
(Fig. 1B). These residues were
classified into four domains based on the HBc primary sequences and
spatial structure as follows: HBc14-18, HBc23-39, HBc120-135, and
HBc122-139 (Table I). To determine
the role of each of these domains in capsid assembly and function,
all 16 residues were mutated in accordance with the following
principles to abolish the hydrophobic effect of the four domains:
Acidic amino acids were mutated into basic amino acids and vice
versa, and amino acids with long side chains were mutated into
glycine. Of note, two mutants with similar primary structures,
HBc120-135 and HBc122-139, were generated in the present study as
the residues were located within different dimer units in the HBc
crystal structure. The two domains were presented in Fig. 1B as green and blue; the map of the
plasmids employed in the present study were presented in Fig. 1C.
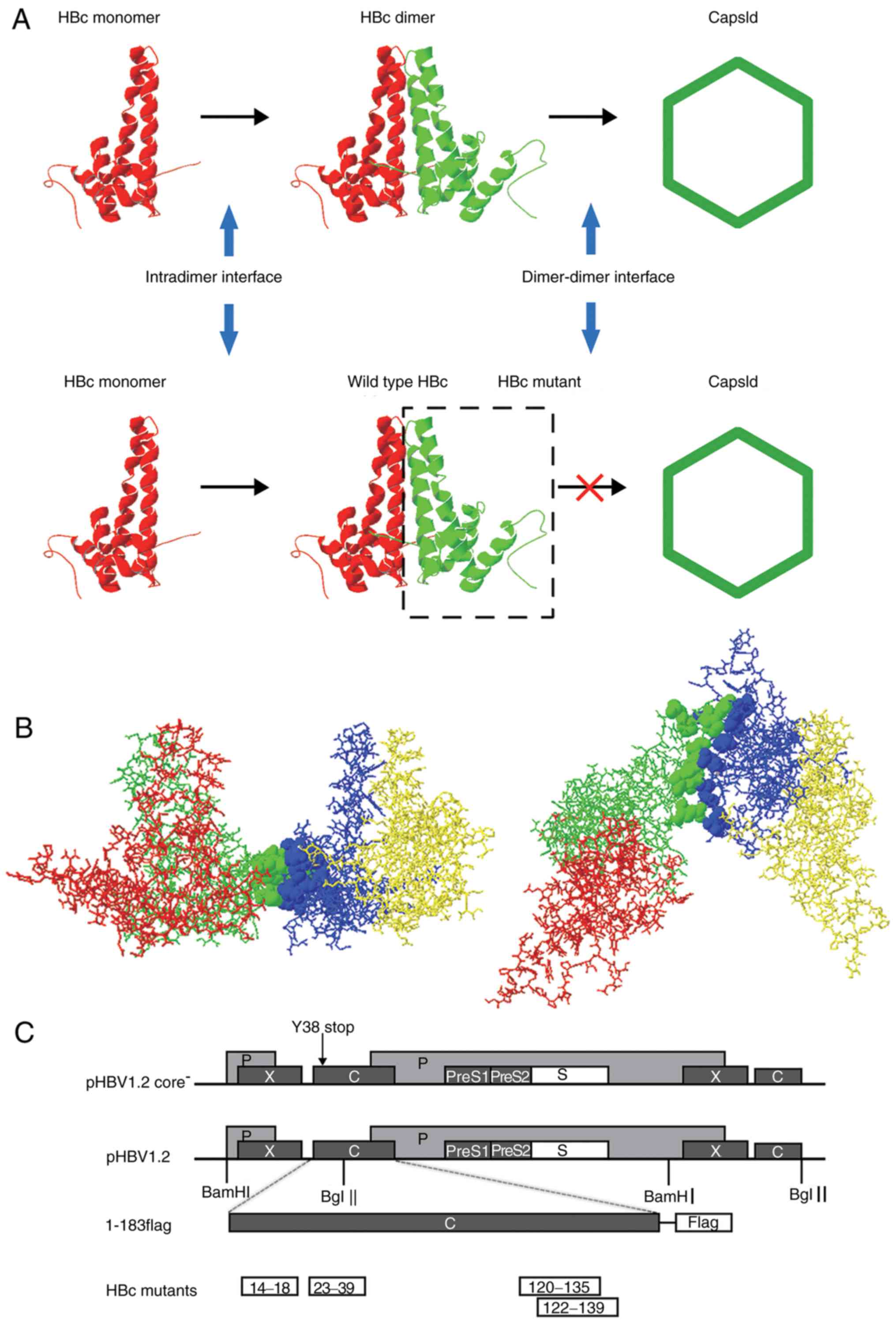 | Figure 1.HBV capsid assembly and dimer-dimer
interface. (A) Capsid assembly procedure. Upper panel, two
α-helical HBc monomers form a dimer via the intradimer interface,
and numerous dimers subsequently form an icosahedral capsid via the
dimer-dimer interface. Lower panel, HBc with mutations at the
dimer-dimer interface may interact with WT HBc monomer via the
intradimer interface to form an aberrant dimer, but disrupts the
capsid structure or function. (B) Orthogonal perspectives of HBc
(1QGT, Protein Data Bank) viewed normal to the local 2-fold axis
(right) and along the 2-fold axis from the outside of the HBc
(left); 4 HBc monomer units are indicated by four different colors
(red, green, blue and yellow). Residues at the dimer-dimer
interface are presented as ribbons, indicated in green (14E, 18F,
120V, 124V, 127R, 129P, 132Y, 134P and 135P) and blue (23F, 25P,
29D, 37L, 39R, 122F and 139I), respectively. (C) Map of plasmids.
For the expression of the HBV genome, 1.2 units of the HBV genome
was inserted at the 3′ end of the lac promoter in pUC18 to
construct the pHBV1.2 plasmid. The open reading frames of the viral
C, P, E and X genes are indicated by dark grey boxes. The C gene of
pHBV1.2core− contained a point mutation of a stop codon
at codon 38 (arrow). For the expression of WT and mutant HBc, the
pcDNA3.1 vector was used. A FLAG tag was fused to the C-terminus of
HBc. Each HBc mutant is indicated by a white box. HBV, hepatitis B
virus; HBc, HBV core protein; WT, wild-type. |
The HBV sequence alignment results demonstrated that
residues 14E and 18F are relatively conserved, whereas 29D and 132Y
are highly conserved, and 25P and 139I are completely conserved
across all 9,386 sequences (Fig.
2).
Capsid-like structure formed by HBc
mutants
To determine whether all HepG2 cells were
successfully transfected with HBc mutants, a FLAG tag was fused to
the C-terminus of WT HBc (1–183 flag) and all mutant plasmids
(Fig. 1C). After 5 days
post-transfection, an anti-FLAG antibody was used in western
blotting to detect its expression. A 21 kDa band was observed in
all mutant and 1–183 flag-transfected groups, indicating that all
HBc mutants exhibited normal expression (Fig. 3A).
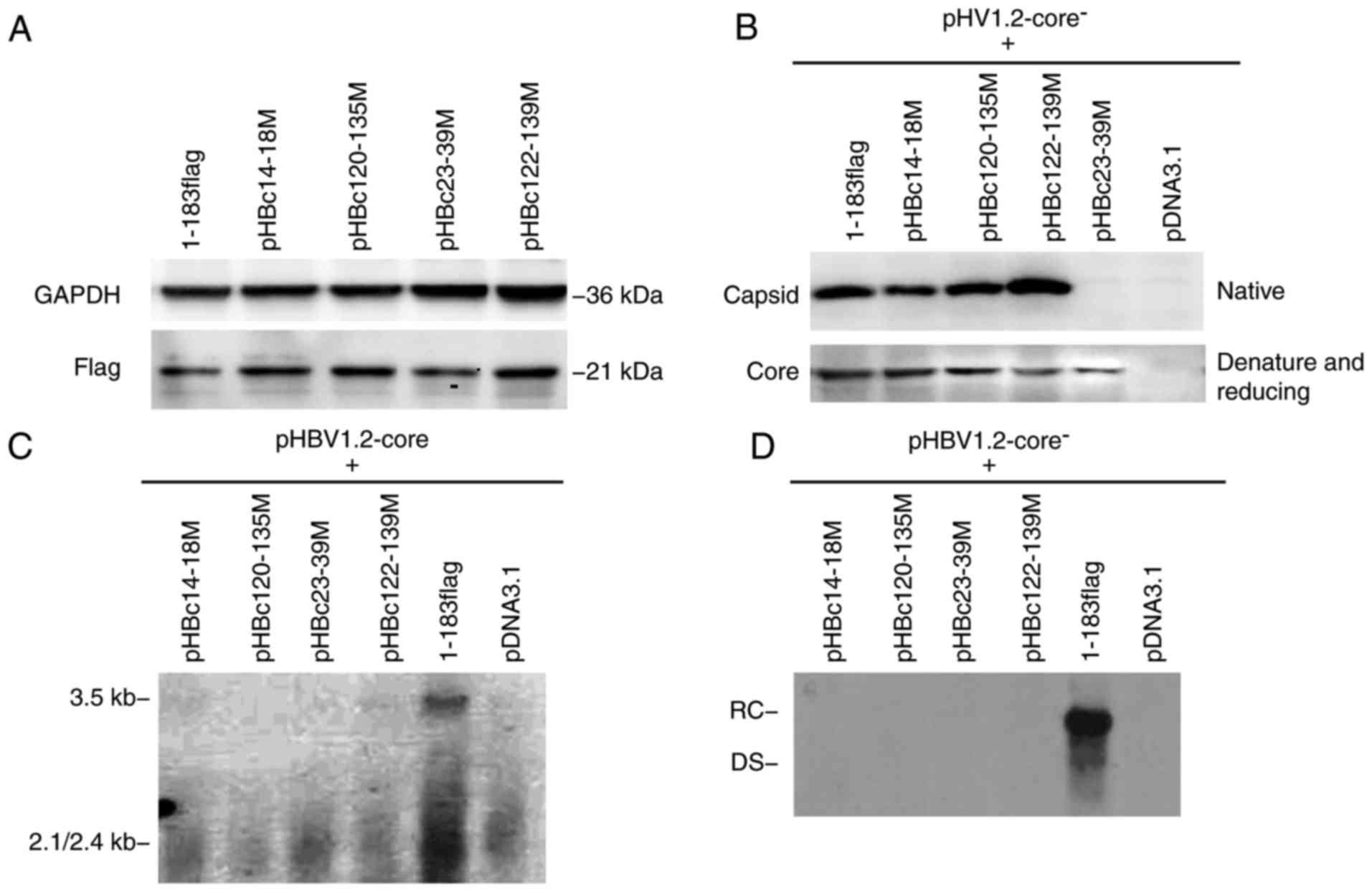 | Figure 3.Capsid formed by HBc with dimer-dimer
mutations. (A) HepG2 cells were transfected with 2 µg HBc mutant or
1–183 flag (control) using FuGENE HD transfection reagent according
to the manufacturer's instructions. After 5 days post-transfection,
cells lysates were isolated, resolved with SDS-PAGE and probed with
anti-GAPDH or -flag antibody. (B) HepG2 cells were co-transfected
with an equal amount of HBc mutant and pHBV1.2core−; 5
days post-transfection, cell lysates were analyzed on a native
agarose gel (for capsid detection) or subjected to SDS-PAGE (for
core protein detection), and then detected with a polyclonal
antibody against the core antigen. The major epitope of the
antibody is at the tip of the spike on the capsid surface (amino
acid residues 78–83), which were unaltered in mutants. Under native
conditions, the spike epitope emerging from the capsid should be
exposed and accessible to the anti-HBc antibody. (C) HepG2 cells
were co-transfected with equal amounts of HBc mutant and
pHBV1.2core−. After 5 days post-transfection, the
capsid-associated 3.5 kb pregenomic RNA was detected with a
specific HBV DNA probe. The positive and negative controls were
1–183 flag and pcDNA3.1, respectively. (D) After 5 days post
co-transfection, capsid-associated HBV DNA was purified and
subjected to Southern blot analysis with a specific HBV DNA probe.
HBV, hepatitis B virus; HBc, HBV core protein; DS, double-stranded
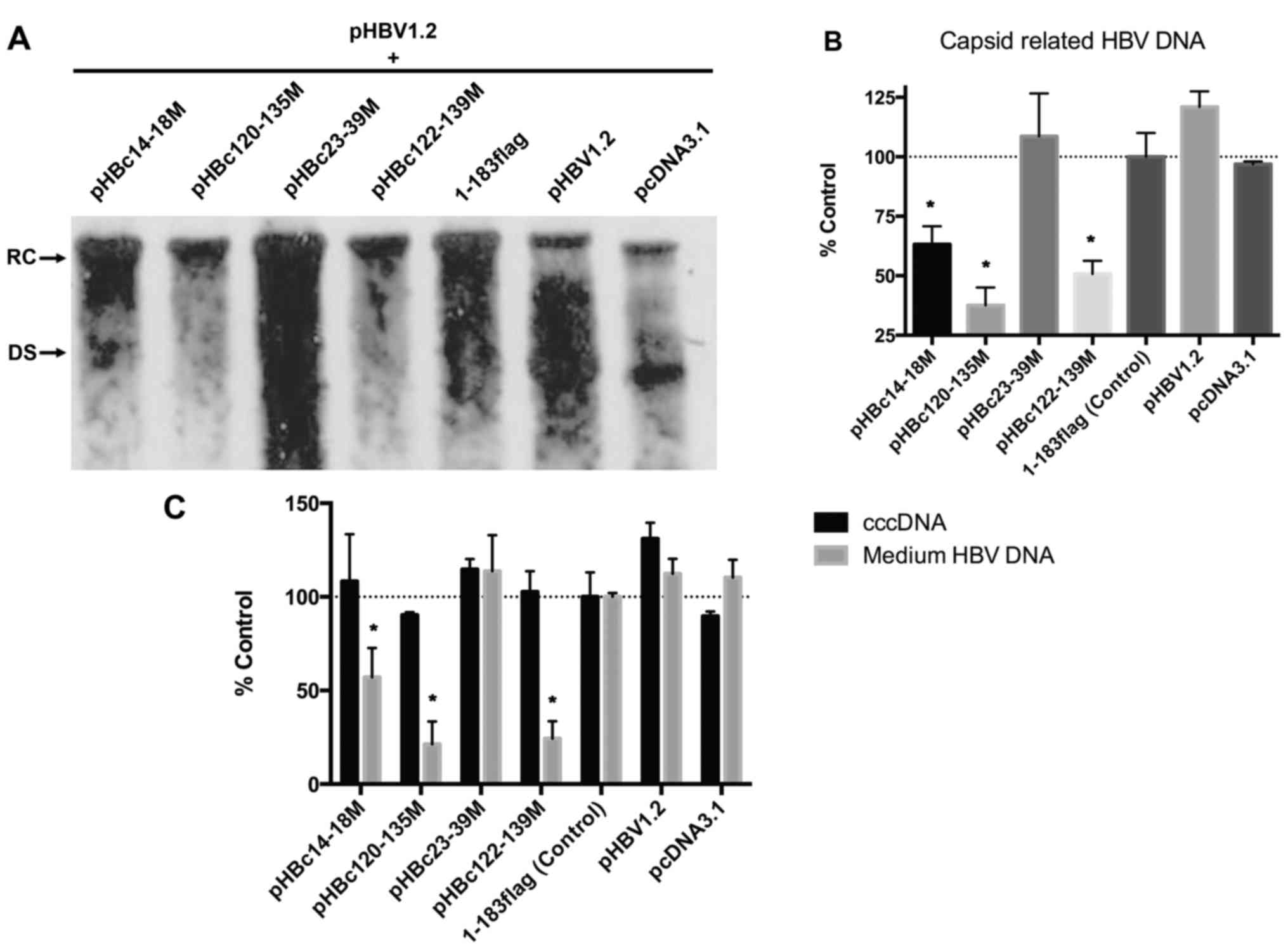
DNA; RC, relaxed circular DNA. |
To determine whether mutations of the dimer-dimer
interface affect capsid formation, HepG2 cells were co-transfected
with each HBc mutant and pHBV1.2-core−. A total of 5
days post-transfection, the intracellular capsid was isolated and
analyzed on an agarose gel (native condition) or subjected to
SDS-PAGE (denaturing and reducing conditions), and then detected
with an anti-HBc antibody (Fig.
3B). The major epitope of the antibody is at the tip of the
spike on the capsid surface (aa 78–83), which appeared to remain
unaltered in the mutants. In the capsid structure, the spike
epitope is exposed and accessible to the anti-HBc antibody
(18). Following SDS-PAGE, 1–183
flag and all mutants were detected with the anti-core antibody,
which indicated that the plasmids were successfully generated;
however, under native conditions, pHBc23-39M was not detected via
an anti-HBc antibody, suggesting that the spike epitope was not
properly exposed, thereby preventing the formation of capsid-like
structures. Conversely, the three mutants, pHBc14-18M, pHBc120-135M
and pHBc122-139M, assembled a capsid-like structure that could be
detected with the corresponding antibody under native conditions
(Fig. 3B).
As some of the mutants assembled capsid-like
structures, whether these supported normal capsid function was
investigated in the present study. To address this issue, each HBc
mutant was respectively co-transfected into cells with
pHBV1.2-core−, and the capsid-associated 3.5 kb pgRNA
and HBV DNA were isolated 5 days post-transfection and detected
with a specific HBV probe via northern and Southern blotting. None
of the HBc mutants supported the assembly of the pgRNA package or
viral replication (Fig. 3C and D).
These findings indicated that these residues at the dimer-dimer
interface are critical for capsid structure and function. Mutations
within these regions may completely disrupt HBV replication.
Effect of HBc mutants on WT viral
replication
As all the mutations were situated within the
dimer-dimer interface, the mutants should remain to possess intact
intradimer interfaces. The present study hypothesized that these
interact with the WT HBc monomer to form aberrant dimers, which in
turn may affect WT virus capsid assembly and function (Fig. 1A, lower panel). To validate this
hypothesis, HepG2 cells were co-transfected with an equal amount of
HBc mutant plasmid and WT pHBV1.2; after 5 days post-transfection,
capsid-associated HBV DNA was extracted and identified by Southern
blot analysis (Fig. 4A). The
results demonstrated that pHBc14-18M significantly inhibited WT
viral replication as presented by a 36.8% reduction in the
expression levels of capsid-associated HBV DNA compared with in the
1–183 flag control; pHBc120-135M and pHBc122-139M significantly
inhibited HBV DNA production by 62.4 and 49.3%, respectively,
compared with the control (Fig.
4B). However, pHBc23-39M did not exhibit an inhibitory effect
on WT viral replication. The present study proposed that pHBc23-39M
could not form a capsid by itself, indicating that mutations within
this domain may also inhibit HBc monomer interactions. Thus,
pHBc23-39M may not interact with WT HBc to affect capsid function
and viral replication.
In addition, whether the mutants affect cccDNA and
virion-associated HBV DNA in the medium was investigated in the
present study. After 5 days post-transfection, the nuclear cccDNA
and HBV DNA in the medium were isolated and quantitated using qPCR.
The results revealed that HBc mutants exhibited no significant
effect on the levels of cccDNA; however, the number of viruses in
the medium of the pHBc14-18M, pHBc120-135M, and pHBc122-139M groups
was significantly decreased compared with in the 1–183 flag
control, which coincided with alterations in capsid-associated HBV
DNA (Fig. 4C).
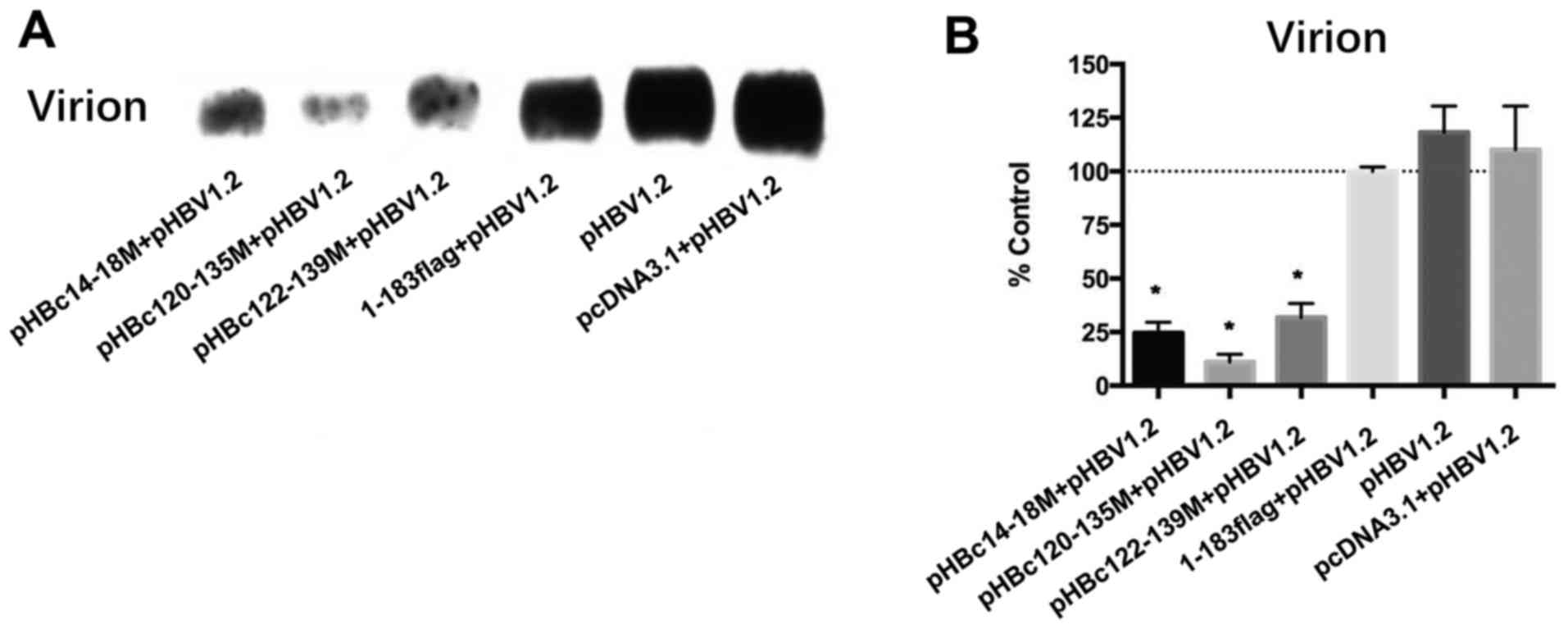
Effect of HBc mutants on WT virion
production
In addition, how the HBc mutants affect WT virion
production was investigated. After 5 days post co-transfection,
virions in the media were collected and detected using an
anti-envelope antibody. Since it was confirmed that pHBc23-39M
could not form capsids by itself or inhibit WT HBV replication, its
effect on HBV virion production was not analyzed. The results
revealed that pHBc14-18M, pHBc120-135M and pHBc122-139M
significantly inhibited WT virion production by >50% compared
with in the 1–183 flag control (Fig.
5; P<0.05). pHBc14-18M significantly decreased
capsid-associated HBV DNA by 36.8% compared with the 1–183 flag
control (Fig. 4B; P<0.05),
indicating that this domain may also participate in the late events
of the HBV life cycle and virion formation.
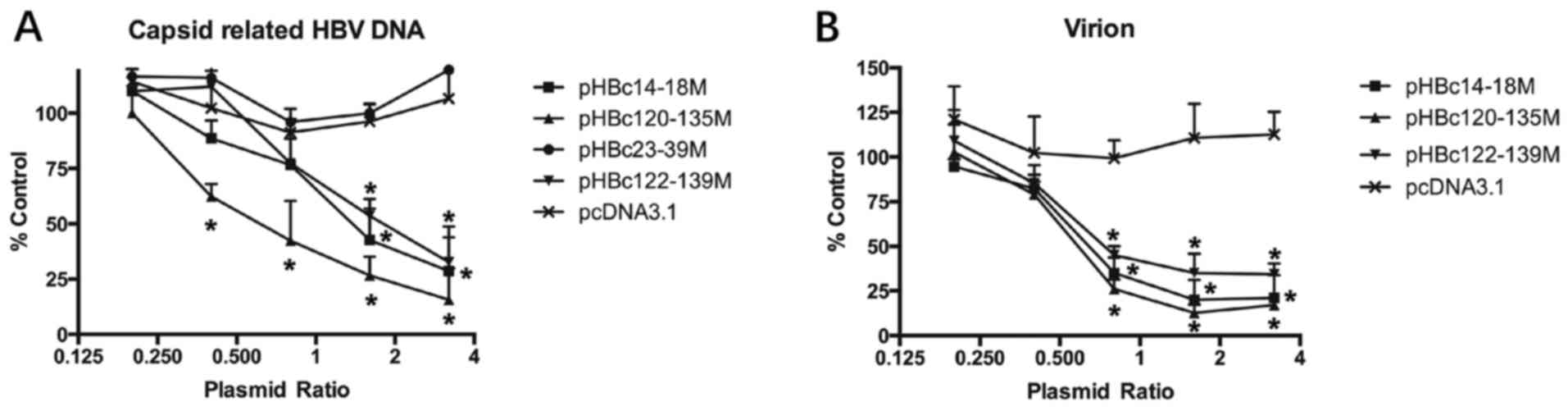
HBc mutants inhibit WT replication and
virion production in a dose-dependent manner
To further confirm the effect of HBc dimer-dimer
mutations on WT replication and virion production, different
amounts of HBc mutant plasmids (0.2–3.2 µg) together with 1 µg
pHBV1.2 were transfected into HepG2 cells. After 5 days
post-transfection, the production of HBV DNA and virions in the
culture medium was detected as aforementioned (Fig. 6). HBc mutants, including
pHBc14-18M, pHBc120-135M and pHBc122-139M significantly inhibited
HBV DNA and virion production in a dose-dependent manner (Fig. 6; P<0.05). pHBc120-135 exhibited
the most potent antiviral effect, as it inhibited WT replication
and virion production by 84 and 83.1%, when co-transfected with a
plasmid ratio of 3.2:1 and 1.6:1 µg, respectively.
Discussion
The aim of the present study was to identify the
role of the HBc dimer-dimer interface in capsid assembly and
function. HBV consists of an outer lipidic envelope protein shell
and inner icosahedral capsid, which is crucial for pgRNA packing,
genome replication, and viral envelopment. A previous study
demonstrated that capsid assembly is strongly associated with the
presence of supplementary cccDNA (25). Dysregulation of capsid assembly
could be a powerful tool for inhibiting viral replication. A
variety of agents that inhibit capsid assembly, including
intracellular single-chain antibody (26), peptide aptamers (27), N-nonyl-deoxy-galactonojirimycin
(28), and bis-ANS (29) demonstrated potent antiviral effects
in vitro and in vivo. In the present study, it was
reported that some domains of the dimer-dimer interface are may be
essential for capsid function and virus viability.
The successful identification of the HBc crystal
structure provides insight to improve understanding of its assembly
and function. In the present study, four domains of the HBc
dimer-dimer interface were identified based on its crystal
structure; two of the domains (HBc14-18 and HBc23-39) were located
at the N-terminus of HBc, whereas the two other domains (HBc120-135
and HBc122-139) encompassed aa 113–143, which have been proposed to
comprise the key dimer-dimer interaction site (18,19).
The present study demonstrated that mutations involving HBc23-39
completely disable capsid assembly, and pHBc23-39 could not
interact with WT HBc to interfere with its replication. These
findings indicated that HBc23-39 may be associated with the
intradimer interface, which is critical for HBc monomer
interactions. In addition, P25 and D29 of HBc23-39 are absolutely
and highly conserved, respectively across 9,386 HBc sequences. The
mutation of these two residues may account for failure in capsid
assembly. Conversely, pHBc14-18M formed a capsid-like structure,
but it did not support pgRNA packaging or HBV DNA replication in
the present study.
The S17 and F18 mutations, which were introduced in
pHBc14-18M, allow capsid assembly but inhibit mature virion
formation (20,21). Similarly, HBc120-135 and HBc122-139
formed capsid-like structures, but did not support replication. The
V124 mutation only permitted the assembly of non-capsid polymers as
observed by electron microscopy (30). Collectively, these findings
suggested that the HBc dimer-dimer interface serves pivotal roles
in maintaining normal capsid structure and function. Interestingly,
HBc23-39 may be a potential antiviral target as minor alterations
of its residues efficiently inhibited capsid formation.
As the approved nucleotide analogs could not
eradicate nuclear cccDNA, the main source of persistent infection,
further investigation and evaluation of novel antiviral agents is
warranted. HBc is recruited to the cccDNA minichromosome in
vivo and maintains the permissive epigenetic state in the
critical region of cccDNA (4,31).
The present study demonstrated that pHBc14-18M, pHBc120-135M and
pHBc122-139M do not support viral replication. Furthermore, these
mutants interacted with WT HBc, and inhibited viral replication and
virion production. Therefore, these domains may be utilized as
anti-HBc targets; once bound to small molecules, these domains are
prevented from participating in capsid assembly and may also
interfere with WT capsid formation and function (7).
In the present study, pHBc14-18M significantly
inhibited HBV replication by 36.8%, but effectively inhibited
virion formation by 75.1%. F18 of pHBc14-18M was reported to also
inhibit particle envelopment (19)
which may explain the observed differences in the effects on viral
replication and virion production.
pHBc120-135M exhibited the most efficient antiviral
effect. Y132 of pHBc120-135 is a highly conserved residue of HBc
and is almost fully buried in the capsid crystal structure
(18). Whether Y132 or other
residues, or their combined synergistic effect is responsible for
the potent antiviral effect of pHBc120-135M requires further study;
the generation of a series of single point mutants may further
elucidate these associations. In the present study pHBc122-139M
contains only two mutated residues, F122 and I139; however,
effective inhibition of WT viral replication was observed,
indicating the importance of these two residues in capsid function.
A single mutation of F122 has been reported to inhibit pgRNA
packaging (20).
The present study also determined whether
dimer-dimer mutations affect the quantity of nuclear cccDNA. No
significant alterations in the quantity of cccDNA were observed
after 5 days post-co-transfection. The results of the present study
revealed that HBc dimer-dimer mutants inhibited WT replication in a
dose-dependent manner, thereby excluding the possibility of
nonspecific interaction between mutants and WT HBc.
Our study demonstrated that the HBc dimer-dimer
interface is required for capsid assembly and viral replication.
Targeting the dimer-dimer interface may be a novel and powerful
antiviral strategy. The findings of the present study provide an
improved understanding of the HBV life cycle and may contribute to
the development of novel antiviral treatments.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Major Program
of National Natural Science Foundation of China (grant no.
81500287), the Natural Science Foundation of Guangdong (grant no.
2016A030313357), the Science and technology Planning Project of
Guangdong (grant nos. 2014A020212575 and 2016A020215215).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author on reasonable request.
Authors' contributions
KD and YZ conceived and designed the study. CLZ,
YMF, ZXX, YZ and KD performed the experiments. CLZ, YZ and KD wrote
the manuscript. All authors read and approved the manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board of the Third Hospital of Sun Yat-Sen University (Guangzhou,
China). Informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HBV
|
hepatitis B virus
|
|
HBc
|
hepatitis B virus core protein
|
|
pgRNA
|
pre-genomic RNA
|
|
cccDNA
|
covalently closed circular DNA
|
|
aa
|
amino acid
|
|
WT
|
wild-type
|
References
|
1
|
Dienstag JL: Hepatitis B virus infection.
N Engl J Med. 359:1486–1500. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seeger C and Mason WS: Molecular biology
of hepatitis B virus infection. Virology. 479-480:672–686. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rabe B, Vlachou A, Panté N, Helenius A and
Kann M: Nuclear import of hepatitis B virus capsids and release of
the viral genome. Proc Natl Acad Sci USA. 100:9849–9854. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guo YH, Li YN, Zhao JR, Zhang J and Yan Z:
HBc binds to the CpG islands of HBV cccDNA and promotes an
epigenetic permissive state. Epigenetics. 6:720–726. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seeger C and Mason WS: Hepatitis B virus
biology. Microbiol Mol Biol Rev. 64:51–68. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu G, Liu B, Zhang Y, Li J, Arzumanyan A,
Clayton MM, Schinazi RF, Wang Z, Goldmann S, Ren Q, et al:
Preclinical characterization of GLS4, an inhibitor of hepatitis B
virus core particle assembly. Antimicrob Agents Chemother.
57:5344–5354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bourne CR, Finn MG and Zlotnick A: Global
structural changes in hepatitis B virus capsids induced by the
assembly effector HAP1. J Virol. 80:11055–11061. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stray SJ, Bourne CR, Punna S, Lewis WG,
Finn MG and Zlotnick A: A heteroaryldihydropyrimidine activates and
can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci
USA. 102:8138–8143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Delaney WE IV, Edwards R, Colledge D, Shaw
T, Furman P, Painter G and Locarnini S: Phenylpropenamide
derivatives AT-61 and AT-130 inhibit replication of wild-type and
lamivudine-resistant strains of hepatitis B virus in vitro.
Antimicrob Agents Chemother. 46:3057–3060. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scaglioni PP, Melegari M and Wands JR:
Posttranscriptional regulation of hepatitis B virus replication by
the precore protein. J Virol. 71:345–353. 1997.PubMed/NCBI
|
|
11
|
Chain BM and Myers R: Variability and
conservation in hepatitis B virus core protein. BMC Microbiol.
5:332005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Birnbaum F and Nassal M: Hepatitis B virus
nucleocapsid assembly: Primary structure requirements in the core
protein. J Virol. 64:3319–3330. 1990.PubMed/NCBI
|
|
13
|
Zlotnick A, Cheng N, Conway JF, Booy FP,
Steven AC, Stahl SJ and Wingfield PT: Dimorphism of hepatitis B
virus capsids is strongly influenced by the C-terminus of the
capsid protein. Biochemistry. 35:7412–7421. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gallina A, Bonelli F, Zentilin L, Rindi G,
Muttini M and Milanesi G: A recombinant hepatitis B core antigen
polypeptide with the protamine-like domain deleted self-assembles
into capsid particles but fails to bind nucleic acids. J Virol.
63:4645–4652. 1989.PubMed/NCBI
|
|
15
|
Zhou S and Standring DN: Hepatitis B virus
capsid particles are assembled from core-protein dimer precursors.
Proc Natl Acad Sci USA. 89:10046–10050. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lingappa JR, Martin RL, Wong ML, Ganem D,
Welch WJ and Lingappa VR: A eukaryotic cytosolic chaperonin is
associated with a high molecular weight intermediate in the
assembly of hepatitis B virus capsid, a multimeric particle. J Cell
Biol. 125:99–111. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
König S, Beterams G and Nassal M: Mapping
of homologous interaction sites in the hepatitis B virus core
protein. J Virol. 72:4997–5005. 1998.PubMed/NCBI
|
|
18
|
Wynne SA, Crowther RA and Leslie AG: The
crystal structure of the human hepatitis B virus capsid. Mol Cell.
3:771–780. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ponsel D and Bruss V: Mapping of amino
acid side chains on the surface of hepatitis B virus capsids
required for envelopment and virion formation. J Virol. 77:416–422.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pairan A and Bruss V: Functional surfaces
of the hepatitis B virus capsid. J Virol. 83:11616–11623. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Günther S, Sommer G, Von Breunig F,
Iwanska A, Kalinina T, Sterneck M and Will H: Amplification of
full-length hepatitis B virus genomes from samples from patients
with low levels of viremia: Frequency and functional consequences
of PCR-introduced mutations. J Clin Microbiol. 36:531–538.
1998.PubMed/NCBI
|
|
22
|
Singh M, Dicaire A, Wakil AE, Luscombe C
and Sacks SL: Quantitation of hepatitis B virus (HBV) covalently
closed circular DNA (cccDNA) in the liver of HBV-infected patients
by LightCycler real-time PCR. J Virol Methods. 118:159–167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hayer J, Jadeau F, Deléage G, Kay A,
Zoulim F and Combet C: HBVdb: A knowledge database for hepatitis B
virus. Nucleic Acids Res. 41:D566–D570. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sievers F, Wilm A, Dineen D, Gibson TJ,
Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al:
Fast, scalable generation of high-quality protein multiple sequence
alignments using clustal omega. Mol Syst Biol. 7:5392011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bock CT, Schwinn S, Locarnini S, Fyfe J,
Manns MP, Trautwein C and Zentgraf H: Structural organization of
the hepatitis B virus minichromosome. J Mol Biol. 307:183–196.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamamoto M, Hayashi N, Takehara T, Ueda K,
Mita E, Tatsumi T, Sasaki Y, Kasahara A and Hori M: Intracellular
single-chain antibody against hepatitis B virus core protein
inhibits the replication of hepatitis B virus in cultured cells.
Hepatology. 30:300–307. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Butz K, Denk C, Fitscher B,
Crnkovic-Mertens I, Ullmann A, Schröder CH and Hoppe-Seyler F:
Peptide aptamers targeting the hepatitis B virus core protein: A
new class of molecules with antiviral activity. Oncogene.
20:6579–6586. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mehta A, Conyers B, Tyrrell DL, Walters
KA, Tipples GA, Dwek RA and Block TM: Structure-activity
relationship of a new class of anti-hepatitis B virus agents.
Antimicrob Agents Chemother. 46:4004–4008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zlotnick A, Ceres P, Singh S and Johnson
JM: A small molecule inhibits and misdirects assembly of hepatitis
B virus capsids. J Virol. 76:4848–4854. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tan Z, Pionek K, Unchwaniwala N, Maguire
ML, Loeb DD and Zlotnick A: The interface between hepatitis B virus
capsid proteins affects self-assembly, pregenomic RNA packaging,
and reverse transcription. J Virol. 89:3275–3284. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pollicino T, Belloni L, Raffa G, Pediconi
N, Squadrito G, Raimondo G and Levrero M: Hepatitis B virus
replication is regulated by the acetylation status of hepatitis B
virus cccDNA-bound H3 and H4 histones. Gastroenterology.
130:823–837. 2006. View Article : Google Scholar : PubMed/NCBI
|