Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) is a
serious disease that subsequently leads to a series of severe
complications, including cephaledema, vasospasm, hydrocephalus and
seizure (1). Previously, it was
suggested that delayed cerebral ischemia due to vasospasm was the
predominant cause of subacute complications of aSAH (2). However, an antagonist of vasospasm
failed to decrease patient mortality rates or to improve the
neurological outcomes (3).
Previously, early brain injury (EBI) has been increasingly
considered to be responsible for the unfavorable outcome of aSAH
(4). EBI is defined according to
the pathophysiological status of a patient typically occurring
within 72 h following an aneurysmal rupture, along with subsequent
complications, including brain edema, vasospastic ischemia and
delayed ischemic neurological deficits (5). In this phenomenon, neuronal apoptosis
has been suggested to be the key process leading to numerous
pathological events (6).
Therefore, inhibition of neuronal apoptosis in the early stages of
aSAH may be beneficial for the treatment of EBI.
Elucidation of the mechanism of aSAH-induced
apoptosis has been an area of investigation for several years.
Unlike cell apoptosis in a model of focal ischemia, aSAH-induced
apoptosis was partly attributed to the complex interaction of
injurious blood-derived factors (7). Essentially, this process comprises 4
primary signaling pathways: The death receptor/tumor protein p53
(p53) pathway; the caspase-dependent and -independent pathways; and
the mitochondrial pathway (5). The
death receptor/p53 signaling pathway, also referred to as the
extrinsic apoptotic pathway, is considered to be particularly
important in cerebral vasospasm following aSAH (8). The caspase-dependent cascade is
primarily associated with ischemia, whereas the caspase-independent
cascade is more closely associated with neurotoxin-induced
apoptosis (9). The mitochondrial
pathway, also termed the intrinsic pathway, is likely to function
as a signaling pathway downstream of p53 activation, which is
elicited by DNA damage (10). p53
appears to serve a central role as a cross-signaling molecule in
aSAH-induced apoptosis.
It has been well established that the c-Jun
N-terminal kinase (JNK)-associated signaling pathway serves a
particularly important role in cell apoptosis. The JNKs, as members
of the mitogen-activated protein kinase family, are encoded by
three genes (JNK1, JNK2 and JNK3) and may be activated by cellular
environmental stresses, inflammatory cytokines and growth factors
(11). Downstream factors of the
JNK signaling pathway comprise c-Jun, activating transcription
factor 2 (ATF-2), p53, ETS domain-containing protein Elk1, mothers
against decapentaplegic homolog 4, signal transducer and activator
of transcription 3 and nuclear factor of activated T cells 4
(12). In the case of apoptosis
induced by artificial DNA damage, JNK was demonstrated to be able
to activate p53 by phosphorylation in vivo (13). Furthermore, a subsequent study on
chemically-induced apoptosis in rat livers suggested that the JNK1
gene was associated with p53 activation (14). In the case of EBI, Dai et al
(15) demonstrated that sp600125,
a JNK-specific inhibitor, was able to ameliorate EBI by decreasing
levels of neuronal apoptosis in the brain tissue of rats. However,
the association between JNK1 and p53 in the EBI process remains
unclear. Therefore, a preliminary investigation of RNA interference
towards the JNK1 gene was performed in the present study to
investigate how JNK1 may interact with p53 in neuronal apoptosis
induced by EBI.
Materials and methods
Animal preparation and grouping
A total of 50 male Sprague-Dawley (SD) rats (Beijing
Vital River Laboratory Animal Technology Co., Ltd., Beijing, China)
weighing 300–350 g were used in the present study. The rats were
fed in a temperature- and humidity-controlled animal center with
ad libitum access to water and food. They were divided
randomly into three groups, termed the sham group (n=10), scramble
group (n=20) and siRNA group (n=20). Subsequently, 10 rats from the
scramble and the siRNA groups were monitored for survival analysis.
All procedures were approved by Ethics Committee of the Second
Affiliated Hospital of Harbin Medical University (Harbin, China)
and were conducted in accordance with the Guide for the Care and
Use of Laboratory Animals by the National Institutes of Health
(Bethesda, MD, USA) (16).
In vivo RNA interference (RNAi) prior
to reverse transcription quantitative polymerase (RT-qPCR)
verification
The rats were injected with JNK1 small interfering
RNA (siRNA) (Shanghai GeneChem Co., Ltd., Shanghai, China) via the
caudal vein, together with Entranster-in vivo RNA
transfection reagent (cat. no. 18668-11-1) or Entranster-in
vivo DNA transfection reagent (cat. no. 18668-11-2; all from
Engreen Biosystem, Ltd., Beijing, China) respectively, according to
the manufacturer's protocol, at a dosage of 3.0 mg/kg daily for 3
days. The sequences of the injected siRNAs were as follows: siRNA,
sense, 5′-AAGCCCAGTAATATAGTAGTA-3′, and antisense,
5′-ACGTGACACGTTCGGAGAATT-3′; scramble siRNA, sense,
5′-AATTCTCCGAACGTGTCACGT-3′, and antisense,
5′-ACGTGACACGTTCGGAGAATT-3′. After a 24 h period, the rats were
sacrificed for JNK1 transcript detection in hippocampal neuronal
tissue using RT-qPCR.
Establishment of the rat SAH model and
preparation of brain samples
The experimental rat SAH model was established by
autologous arterial blood injection into the pre-chiasmatic
cistern, as described previously (17), but with certain modifications.
Briefly, the rats were anesthetized with 10% chloral hydrate (300
mg/kg body weight) intraperitoneally (IP). They were subsequently
fixed in a stereotaxic instrument. A microinjector was placed in
the sagittal plane with a tilt angle of 45°. The entry point was 5
mm posterior to the external occipital protuberance in the midline.
A longitudinal incision of 1.5 cm was made along the midline, with
the entry point in the center. The atlantooccipital membrane was
then exposed by removing part of the occipital bone surrounding the
foramen magnum. The tip of the microinjector was lowered to
penetrate the atlantooccipital membrane, and then inserted 2–3 mm
into the pre-chiasmatic cistern. A total of 300 µl non-heparinized
fresh autologous arterial blood was aseptically injected into the
pre-chiasmatic cistern slowly for 3 min. To maintain fluid balance,
all rats were injected with 2 ml 0.9% NaCl subcutaneously. The rats
were then returned to their cages, and free access to food and
water was provided following their recovery from anesthesia. In
this model, the inferior basal temporal lobe of the SAH group was
stained with blood. The rats in the sham group underwent the
identical procedure with the exception of the blood injection, and
these rats were sacrificed 24 h following the operation. All the
rats in the present study were euthanized by cervical dislocation
following anesthesia with 10% chloral hydrate (300 mg/kg body
weight) IP. Brain samples were collected following intracardial
perfusion with 4% paraformaldehyde at 4°C. The samples were
subsequently immersed into 4% paraformaldehyde at 4°C for at least
48 h prior to subsequent use in the histological analyses.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick-end labeling
(TUNEL) assay
TUNEL assays were performed using a TUNEL detection
kit, following the manufacturer's protocol (cat. no. WLA029;
Wanleibio Co., Ltd., Shenyang, China). In brief, previously fixed
hippocampus tissue was dehydrated in graded ethyl alcohol (70, 80,
90 and 100%), embedded in paraffin, sliced into 5 µm sections, and
rehydrated (100, 95, 85 and 75%). The slides were subsequently
incubated with 50 µl TUNEL reaction mixture for 1 h at 37°C in the
dark. Slides were then developed with 3,3′-diaminobenzidine (DAB)
and counterstained with 0.2% hematoxylin for 3 min at room
temperature, and finally mounted in Neutral balsam mounting medium
(cat. no. g8590; Shanghai Haoran Biological Technology Co., Ltd.,
Shanghai, China). The apoptotic cells were identified as those with
hyperchromatic nuclei, and counted under a light microscope by an
investigator blinded to the grouping. The extent of brain damage
was evaluated by determining the average percentage of apoptotic
cells in each section counted at magnification, ×400. A total of
three sections from each animal were used for quantification. The
final average percentages of apoptotic cells of the sections were
recorded for analysis.
RT-qPCR
Tissues were stored prior to RT-qPCR analysis at
−80°C. Total RNA was extracted using TRIzol® reagent
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). RT was
performed using M-MLV Reverse Transcriptase (cat. no. 28025021;
Invitrogen; Thermo Fisher Scientific, Inc.). Briefly, 1 µl oligo
(dT)15 primer (cat. no. C1101-20; Promega Corporation,
Madison, WI, USA), 1 µl random primers (cat. no. C1181; Promega
Corporation), 2 µl of 2.5 mM dNTP (cat. no. U1511; Promega
Corporation) were mixed with ddH2O in a final reaction
volume of 14.5 µl, and heated to 70°C for 5 min and then cooled on
ice for 2 min. Subsequently, 4 µl 5X First Strand Buffer [250
mmol/l Tris-Cl (pH 8.3), 375 mmol/l KCl, 15 mmol/l
MgCl2], 0.5 µl RNasin® ribonuclease
inhibitors (cat. no. N2111; Promega Corporation) and 1 µl M-MLV
were added. The temperature protocol was as follows: 25°C for 10
min, 42°C for 50 min and 95°C for 5 min. The transcriptional levels
of genes were subsequently assessed using a SYBR Green PCR Master
mix kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
sequences of the primers used are summarized in Table I. β-actin was used as an internal
control. The thermocycling conditions were as follows: 94°C for 10
min, then 41 cycles of 94°C for 10 sec, 60°C for 20 sec and 72°C
for 30 sec, followed by 72°C for 2.5 min, 40°C for 5.5 min, and a
melting temperature gradient from 60 to 94°C (1 sec for every 1.0°C
rise; 25°C for 1 min). RT-qPCR products were assessed using an
Exicycler™ 96 fluorescence quantitative instrument
(Bioneer Corporation, Daejeon, Korea). Data obtained for each of
the experimental groups were recorded as fold change values
relative to the control group (the mean 2−ΔΔCq ±
standard error of the mean) (18).
 | Table I.Sequences of primers in reverse
transcription quantitative polymerase chain reaction. |
Table I.
Sequences of primers in reverse
transcription quantitative polymerase chain reaction.
| Genes | Sequences | Primer, bp | Tm, °C | Products, bp |
|---|
| JNK1 F |
AGTTATTGAACAGCTCGGAA | 20 | 52.6 | 217 |
| JNK1 R |
TTTGGACGCATCTATCACC | 19 | 53.7 |
|
| p53 F |
CAGAGTTGTTAGAAGGCCCAGAG | 23 | 60.2 | 136 |
| p53 R |
TGAGAAGGGACGGAAGATGAC | 21 | 58.9 |
|
| Bax F |
TCCACCAAGAAGCTGAGCGAG | 21 | 65.2 | 98 |
| Bax R |
GTCCAGGCCCATGATGGTTCT | 21 | 65.6 |
|
| Bcl-2 F |
TGAACCGGCATCTGCACAC | 19 | 64.4 | 96 |
| Bcl-2 R |
CGTCTTCAGAGACAGCCAGGAG | 22 | 63.8 |
|
| p38 F |
CGGCTTGCTCATGTCCTCAGAAC | 22 | 67.6 | 214 |
| p38 R |
GGAGGGCGGCTGCACATACAC | 21 | 69.3 |
|
| NF-κB F |
ACGATCTGTTTCCCCTCATC | 20 | 58.9 | 241 |
| NF-κB R |
TGCTTCTCTCCCCAGGAATA | 20 | 59.8 |
|
| Caspase-3 F |
GACGACAGGGTGCTACGAT | 22 | 56.9 | 193 |
| Caspase-3 R |
TTTCCTTACGCTCTGACTGA | 19 | 55.6 |
|
| β-actin F |
GGAGATTACTGCCCTGGCTCCTAGC | 25 | 60.1 | 155 |
| β-actin R |
GGCCGGACTCATCGTACTCCTGCTT | 25 | 62.0 |
|
Western blot analysis
Protein concentrations were determined using a
bicinchoninic acid (BCA) protein assay kit (Thermo Fisher
Scientific, Inc.). Equal amounts of total cellular protein (40
µg/lane) were resolved by 15% SDS-PAGE, subsequently
electro-transferred onto a polyvinylidene fluoride membrane (cat.
no. IPVH00010; EMD Millipore, Billerica, MA, USA), and blocked with
5% non-fat dry milk in TBS/Tween (TBST) solution (20 mM Tris-HCl,
137 mM NaCl and 0.1% Tween-20, pH 7.4) for 2 h at room temperature.
The membranes were incubated with rabbit monoclonal or polyclonal
antibodies (all at 1:1,000 dilution) against phosphorylated (p)-p53
(cat. no. WL01813; Wanleibio, Co., Ltd.), phosphorylated (p)-JNK
(cat. no. WL02504; Wanleibio Co., Ltd.), Bcl-2-associated X protein
(Bax; cat. no. A00183; Wuhan Boster Biological Technology, Ltd.,
Wuhan, China), B-cell lymphoma 2 (Bcl-2; cat. no. WL01556;
Wanleibio Co., Ltd.), p-mitogen-activated protein kinase 11 (p-p38;
cat. no. WLP1576; Wanleibio Co., Ltd.), nuclear factor
κ-light-chain-enhancer of activated B-cells (NF-κB; cat. no.
WL01980; Wanleibio Co., Ltd.) or cleaved caspase-3 (cat. no.
WL01992; Wanleibio Co., Ltd.) diluted in TBST overnight at 4°C. The
membranes were also probed with β-actin antibody (cat. no. WL01845;
Wanleibio Co., Ltd.) as an internal control. The blots were
subsequently incubated with the corresponding horseradish
peroxidase-conjugated secondary antibody (goat-anti-rabbit
immunoglobulin G antibody; cat. no. WLA023; Wanleibio Co., Ltd.;
1:5,000 dilution) for 1 h at room temperature. Immunoreactive
proteins were detected using enhanced chemiluminescence western
blotting substrate (Thermo Fisher Scientific, Inc.). Densitometric
analysis was performed using Gel-Pro Analyzer 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA).
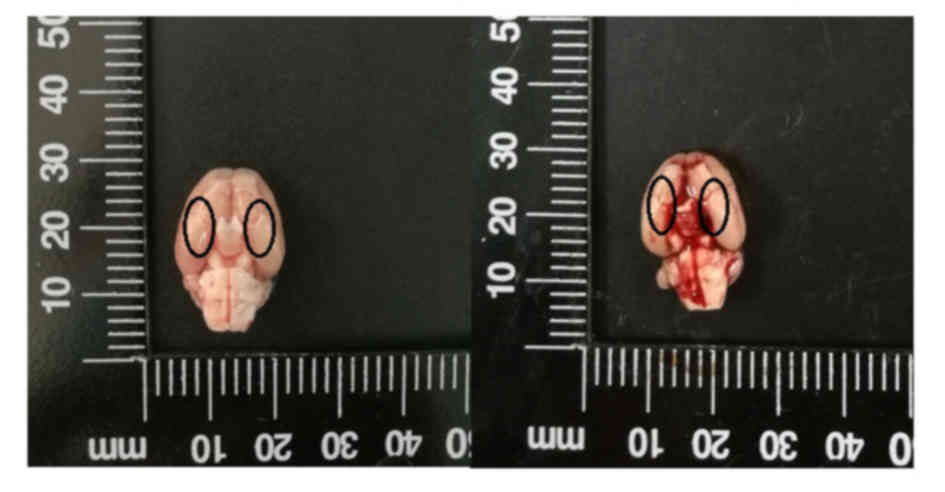
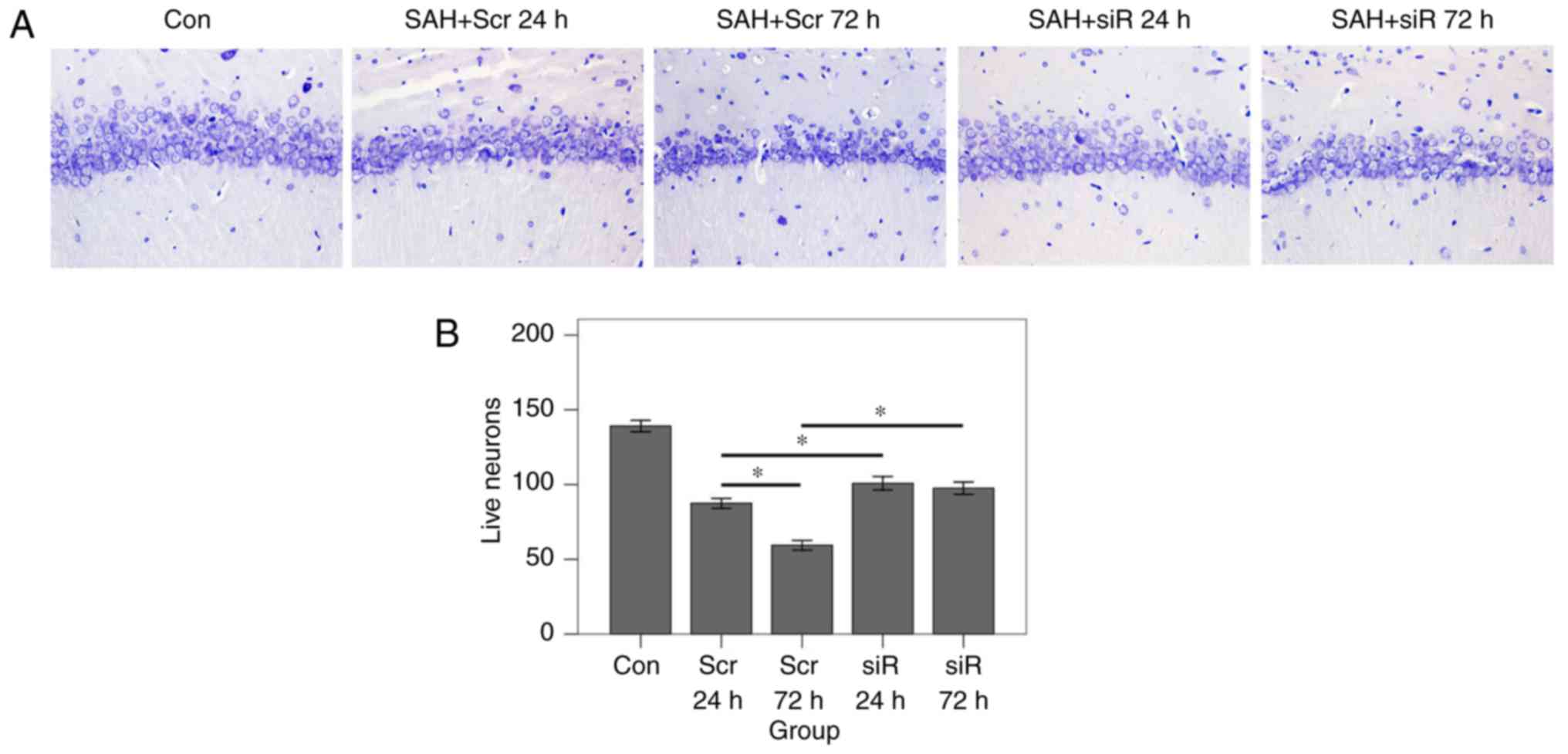
Histological assay
Previously fixed rat brains were dehydrated using
graded ethyl alcohol (70, 80, 90 and 100%), embedded in paraffin,
sliced into 5 µm sections, and stained with 0.5% Cresyl Violet
(cat. no. 71044080; Sinopharm Chemical Reagent Co., Ltd., Beijing,
China) at room temperature for 10 min. The assessed region was in
the inferior basal temporal lobe (portrayed by the area of black
ovals in Fig. 2). Cresyl
Violet-stained neuronal cell bodies (Nissl bodies) were counted. A
total of 5 random fields at magnification, ×400 in each coronary
section were selected, and the mean number of intact neurons in the
five fields of view was calculated. A total of 4 sections from each
animal were used for quantification. The final average number of
the 4 sections was regarded as the data value for each sample.
Neurological scoring
The scores of appetite, activity and neurological
deficits of the rats (Table II)
were recorded as examinations of their behavioral activity, as
described previously (19), which
were performed by an investigator blinded to the study groups at 24
and 72 h following SAH. The scores of 6 rats in each group were
calculated.
| Table II.Behavior and activity scores. |
Table II.
Behavior and activity scores.
| Category | Behavior | Score |
|---|
| Appetite | Finished meal | 0 |
|
| Left meal
unfinished | 1 |
|
| Scarcely ate | 2 |
| Activity | Walked and reached
at least three corners of the cage | 0 |
|
| Walked with some
stimulations | 1 |
|
| Almost always lying
down | 2 |
| Motor deficit | No deficit | 0 |
|
| Unstable walk | 1 |
|
| Impossible to
walk | 2 |
Survival analysis
Overall survival was recorded as the time from the
date of the rats being subjected to blood injection into the
pre-chiasmatic cistern to the date of mortality. The observational
period was 30 days.
Statistical analysis
Data were recorded as the mean ± standard deviation.
SPSS 17.0 was used to perform statistical analysis (SPSS, Inc.,
Chicago, IL, USA). One-way analysis of variance (ANOVA), followed
by either the least significant difference (LSD) test (homogeneity
of variance) or the Games-Howell test (non-homogeneity of
variance), was used to analyze differences between groups.
Neurobehavioral scores were analyzed with non-parametric tests
(Kruskal-Wallis tests, followed by Mann-Whitney U tests for
comparison between groups). Kaplan-Meier curves and log-rank tests
were employed for survival analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
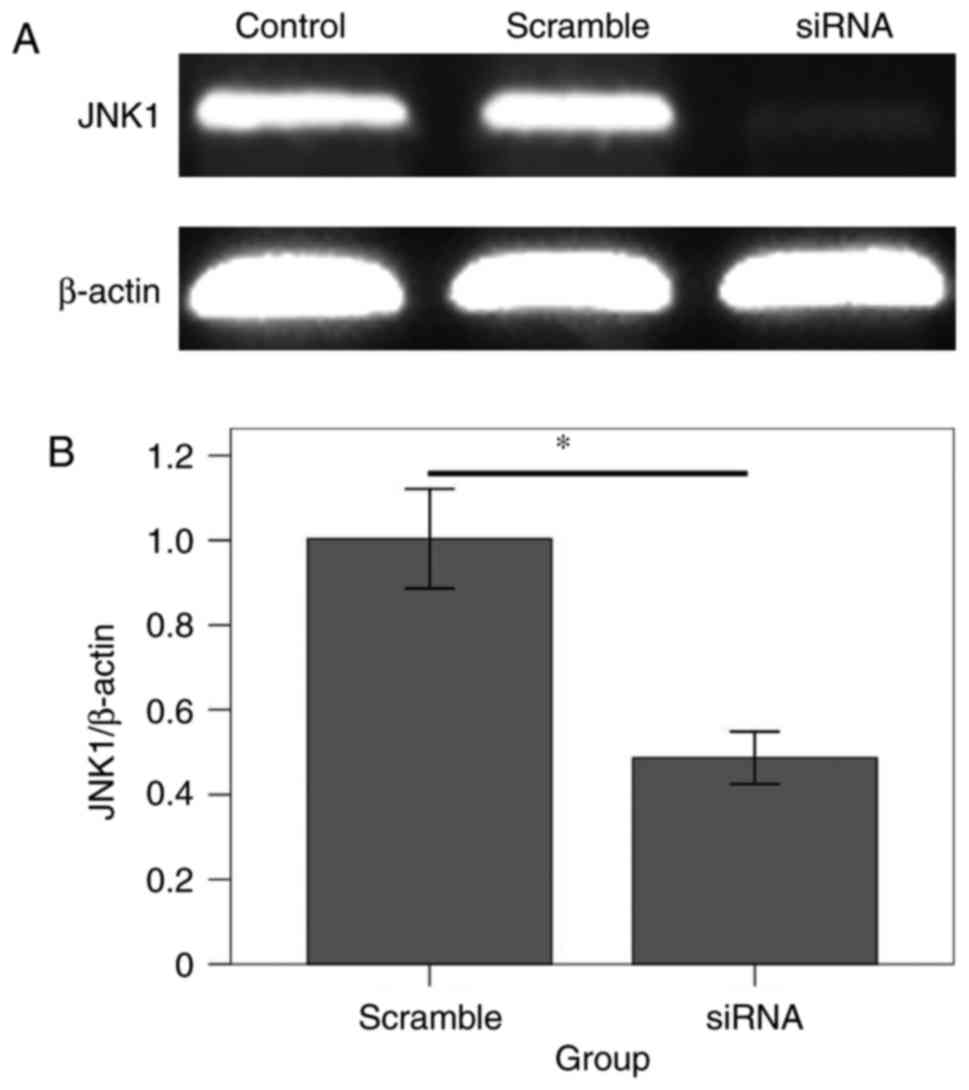
JNK1 siRNA downregulates JNK1
transcription in rats
At 24 h following the in vivo injection of
the transfected reagents, the RT-qPCR results (Fig. 1) revealed that the JNK1 transcript
level in the siRNA group was significantly decreased compared with
that in the scramble group (F=279.36; P<0.001), which indicated
that the in vivo RNAi rat model had been successfully
established.
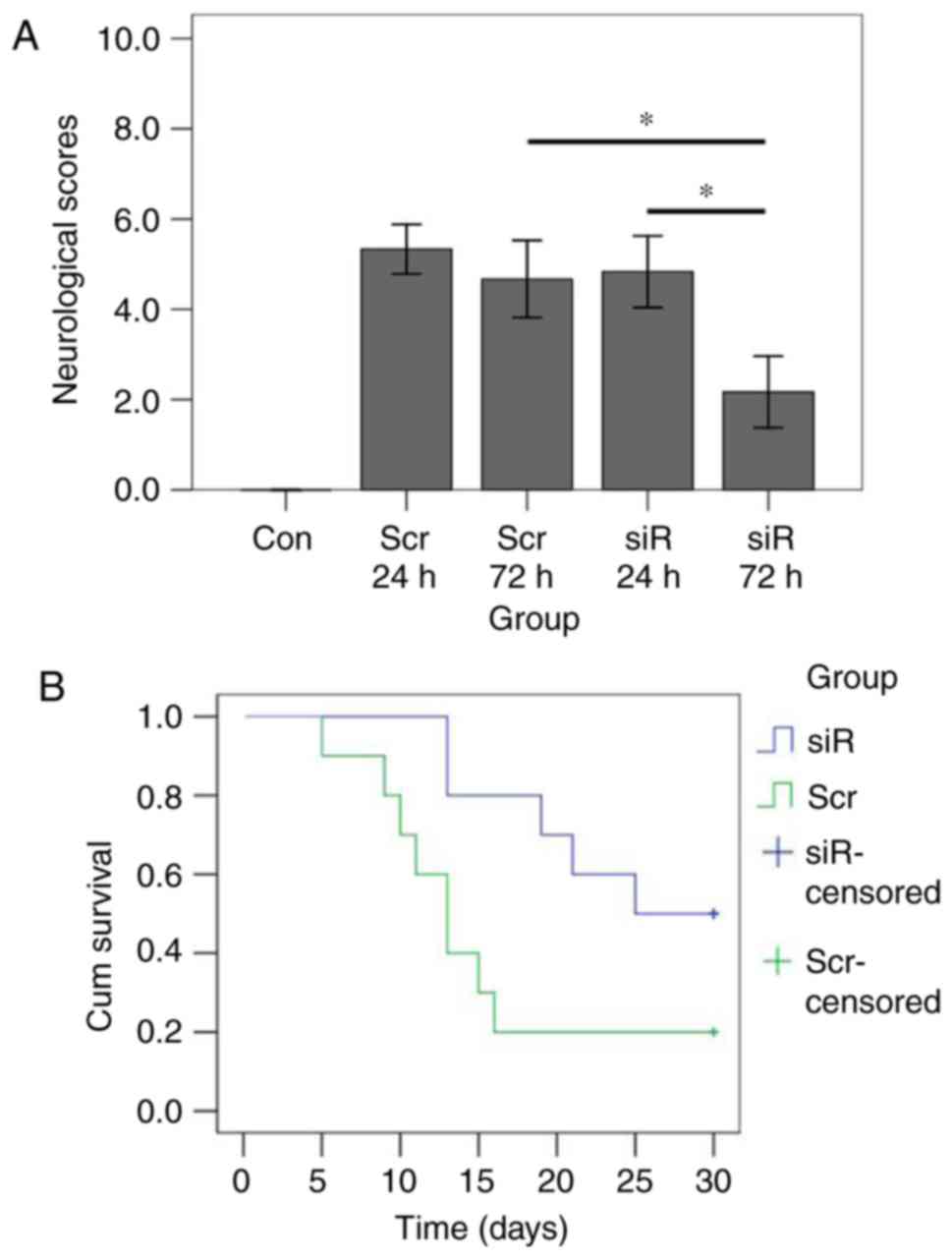
JNK1 inhibition improves the
neuro-rehabilitation and survival times of rats
The SAH rat models were successfully constructed as
previously described by Zhang et al (20). The inferior basal temporal lobe was
stained with blood, indicating that the adjacent brain tissue, in
particular the hippocampus, may be injured by SAH-induced
inflammatory responses (Fig. 2).
To evaluate the effect of JNK1 inhibition on neuroprotection, the
mean neurodeficit scores of 6 randomly chosen rats in each group
were recorded and analyzed. In addition, survival analyses were
also performed. The results demonstrated that there were
significant differences in neurological scores among the groups
(Kruskal-Wallis χ2=24.198, P<0.001; Fig. 3). The mean score at 72 h was
significantly decreased compared with that at 24 h following SAH in
the siRNA group (P=0.003). By contrast, in the scramble group, the
mean score at 24 h did not reveal any significant difference
compared with that at 72 h (P=0.118). At 24 h following SAH, the
scramble and the siRNA groups exhibited similar mean scores
(P=0.206). At 72 h, the mean score in the siRNA group was
significantly decreased compared with that in the scramble group
(P=0.003). Kaplan-Meier curve analysis revealed that the median
survival time in the siRNA group was significantly increased
compared with that in the scramble group (log-rank P=0.038).
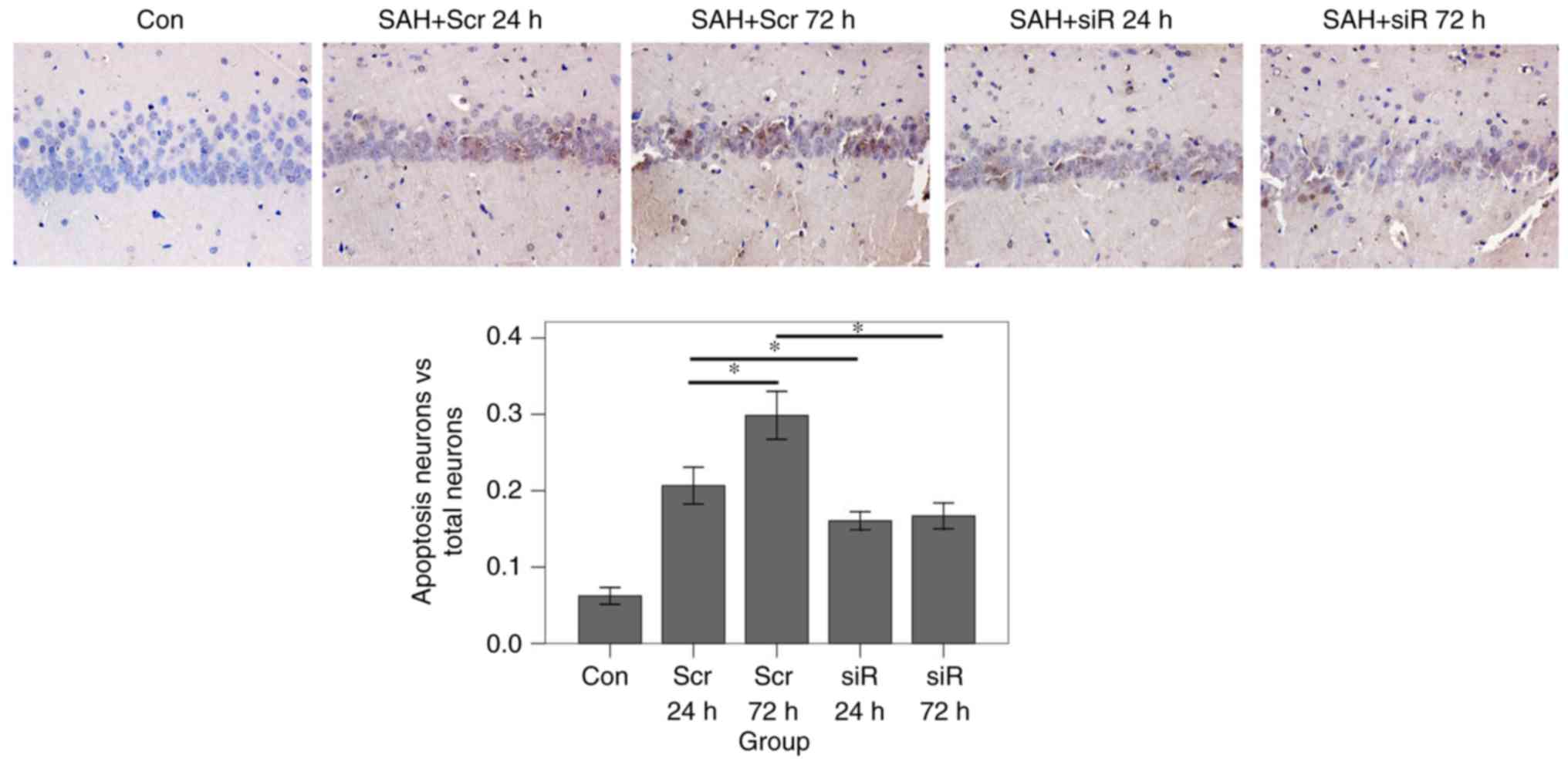
JNK1 downregulation decreases the
level of cell apoptosis in the hippocampus
Hippocampus tissues were harvested for pathological
analysis. TUNEL assays were employed to detect the rates of
apoptosis. The results of the TUNEL assay (Fig. 4) demonstrated that the apoptosis
ratio in the scramble 72 h group was significantly increased
compared with that in the scramble 24 h group (P<0.001).
However, in the siRNA group, the apoptosis ratio at 72 h after SAH
was almost at an identical level with the ratio at 24 h after SAH
(P=0.963). At 24 and 72 h, the apoptosis ratio in the RNAi group
was significantly decreased compared with that in the scramble
group (after 24 h, P=0.010; after 72 h, P<0.001). Nissl staining
was subsequently performed to reveal the numbers of intact neurons
in the hippocampus area. As demonstrated in Fig. 5, at 24 and 72 h following SAH, the
number of intact neurons in the RNAi group was significantly
increased compared with that in the scramble group (P<0.001). In
the scramble group, the number of neurons at 72 h following SAH was
markedly decreased compared with the number present at 24 h after
SAH (P<0.001). By contrast, in the RNAi group, the numbers of
living neurons at the 2 time points were at an almost identical
level (P=0.220).
JNK1 RNAi downregulates the expression
levels of apoptosis-associated genes
To investigate the biological mechanism of decreased
apoptosis levels via JNK1 gene silencing, RT-qPCR and western blot
analysis assays were performed. The results of the RT-qPCR revealed
that, at 24 and 72 h, the JNK1 transcript levels in the RNAi group
were significantly decreased compared with those in the control
group (at 24 h, P<0.001; at 72 h, P<0.001; Fig. 6). By contrast, the JNK1 transcript
levels in the scramble group were at an almost identical level
compared with those in the control group (at 24 h, P=0.500; at 72
h, P=0.757). At the 2 time points, the JNK1 transcript level in the
RNAi group was decreased compared with that in the scramble group
(at 24 h, P<0.001; at 72 h, P<0.001). The p53 transcript
levels in all groups were similar (P=0.398). At 24 h, the Bax
transcript levels in the scramble and RNAi groups were
significantly increased compared with that in the control group
(scramble group, P<0.001; RNAi group, P<0.001); the Bax
transcript level in the scramble group was also significantly
increased compared with that in the RNAi group (P<0.001). After
72 h, the Bax transcript level in the scramble group was
significantly increased compared with that in the control group
(P<0.001), although the transcript level in the RNAi group was
almost identical with that in the control group (P=0.771); in
addition, the Bax transcript level in the scramble group was
significantly increased compared with that in the RNAi group
(P<0.001). In the RNAi group, the Bcl-2 transcript levels at 24
and 72 h were significantly increased compared with that in the
control group (P<0.001 for the 2 time points). The Bcl-2
transcript level in the scramble group at 24 h was significantly
decreased compared with that in the control group (P<0.001), but
the transcript level at 72 h was almost identical with that of the
control group (P=0.074). At 24 h, the Bcl-2 transcript level in the
RNAi group was significantly increased compared with that in the
scramble group (P<0.001). At 24 h, the p38 transcript levels in
the scramble and the RNAi groups were significantly increased
compared with that in the control group (P<0.001). At 72 h, the
p38 transcript level in the scramble and the RNAi groups were
significantly increased compared with that in the control group
(P<0.001). At the 2 time points (24 or 72 h), the p38 transcript
levels in the RNAi group was almost identical with that of the
scramble group (at 24 h, P=0.825; at 72 h, P=0.546). At 24 h, the
NF-κB transcript levels in the scramble and the RNAi groups were
significantly increased compared with that in the control group
(P<0.001). At 72 h, the NF-κB transcript levels in the scramble
and the RNAi groups were significantly increased compared with that
in the control group (scramble group: P=0.001; RNAi group,
P<0.001). Irrespective of whether the data at 24 or 72 h were
being compared, the NF-κB transcript level in the RNAi group was
almost at the same level as that of the scramble group (at 24 h,
P=0.739; at 72 h, P=0.469). Finally, at 24 h the caspase-3
transcript level in the scramble and the RNAi groups were
significantly increased compared with that in the control group
(P<0.001). At 72 h, the caspase-3 transcript levels in the
scramble and the RNAi groups were significantly increased compared
with that in the control group (P<0.001). At the 2 time points
(24 or 72 h), the caspase-3 transcript level in the RNAi group was
at a very similar level to that of the scramble group (at 24 h,
P=0.534; at 72 h, P=0.345).
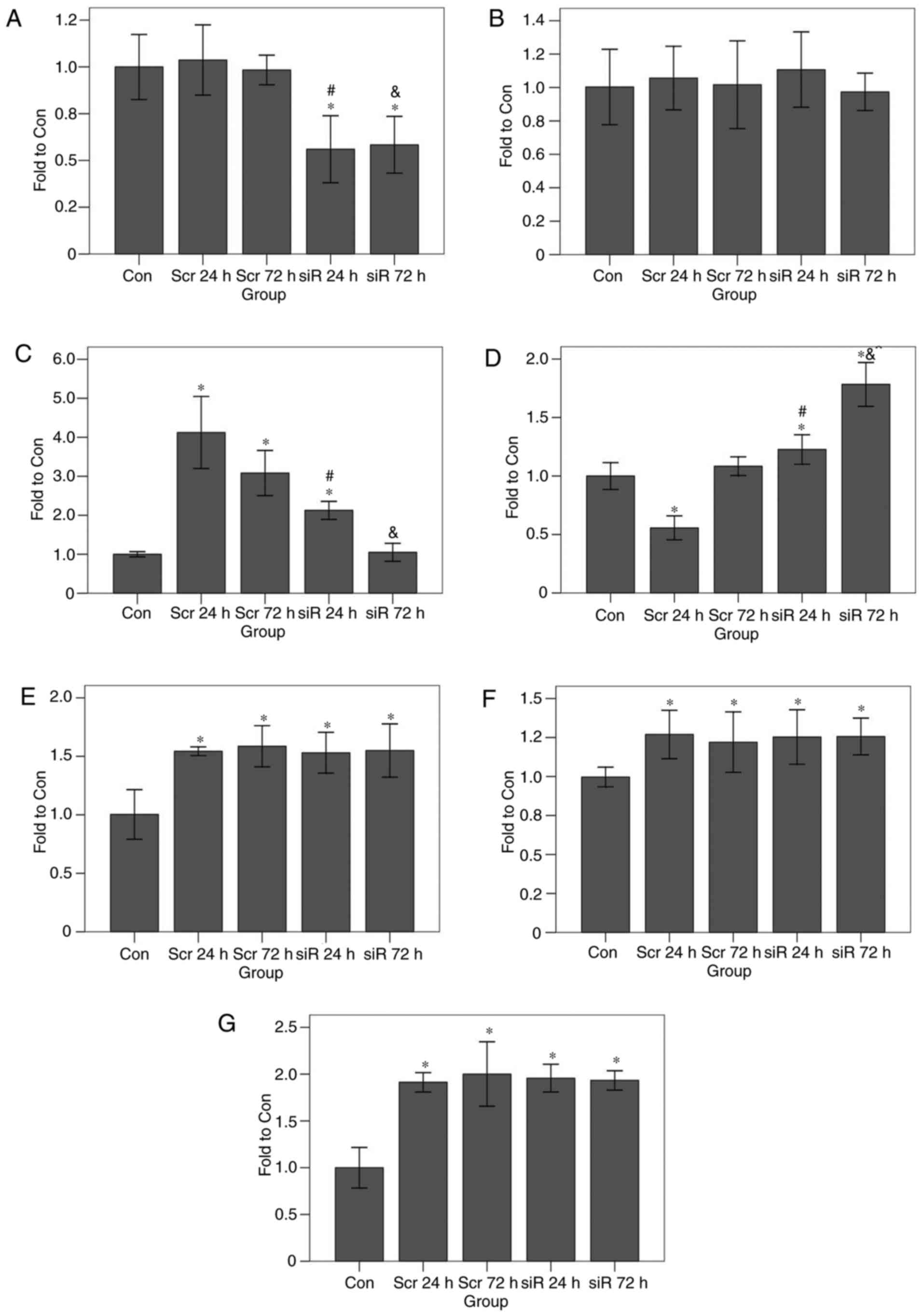 | Figure 6.Effects of JNK1 inhibition on
transcription levels of apoptosis-associated genes as determined by
reverse transcription quantitative polymerase chain reaction. JNK1
inhibition led to an increase in the Bcl-2 transcript level and a
decrease in the Bax transcript level, but no significant effect on
any other genes was observed. The transcript levels for (A) c-Jun
terminal kinase 1, (B) tumor protein 53, (C) Bax, (D) Bcl-2, (E)
mitogen-activated protein kinase 11, (F) nuclear factor
κ-light-chain-enhancer of activated B-cells, and (G) caspase-3 are
presented. *P<0.05 vs. Con group; #P<0.05 vs. the
Scr 24 h group; &P<0.05 vs. the Scr 72 h group.
Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; Con,
control; siR, siRNA; Scr, scramble. |
Representative bands and correlated histograms of
the western blot analysis experiments are presented in Fig. 7. The p-JNK expression levels in the
scramble group after 24 (P<0.001) and 72 h (P=0.009) were
significantly increased compared with that in the control group.
The levels of the same protein in the RNAi group at 24 (P=0.008)
and 72 h (P=0.001) were significantly decreased compared with that
in the control group. At 24 and 72 h, the p-JNK expression levels
in the RNAi group were significantly decreased compared with that
in the scramble group (P<0.001 for the 2 time points). The p-p53
expression levels in the scramble 24 h (P<0.001) and scramble 72
h (P=0.009) groups were significantly increased compared with that
in the control group. The expression levels of the same protein in
the RNAi 24 h (P=0.001) and RNAi 72 h (P=0.001) groups were
significantly decreased compared with that in the control group. At
24 and 72 h, the p-p53 expression levels in the RNAi group were
significantly decreased compared with that in the scramble group
(P<0.001 for the 2 time points). The Bax protein expression
levels in the scramble 24 h, scramble 72 h, and RNAi 24 h groups
were all significantly increased compared with that in the control
group (all P<0.001). However, the expression level of the same
protein in the RNAi 72 h group was not significantly different from
that in the control group (P=0.268). At 24 and 72 h, the Bax
expression levels in the RNAi group were significantly decreased
compared with that in the scramble group (P<0.001 for the 2 time
points). The Bcl-2 protein expression levels in the scramble 72 h,
the RNAi 24 h, and the RNAi 72 h groups were all significantly
increased compared with that in the control group (all P<0.001).
The expression level of the same protein in the scramble 24 h group
was significantly decreased compared with that in the control group
(P<0.001). At 24 and 72 h, the Bax expression levels in the RNAi
group were significantly increased compared with that in the
scramble group (both P<0.001). The p-p38 protein expression
levels in the scramble 24 h, the scramble 72 h, the RNAi 24 h and
the RNAi 72 h groups were all significantly increased compared with
that in the control group (all P<0.001). No significant
differences were identified among the four experimental groups
(F=1.771; P=0.230). The NF-κB-p65 expression levels in the scramble
24 h (P=0.019), the scramble 72 h (P=0.023), the RNAi 24 h
(P=0.004), and the RNAi 72 h (P=0.001) groups were all
significantly increased compared with that in the control group.
Again, no significant differences were identified among the four
experimental groups (F=1.042, P=0.412). Finally, at 24 and 72 h the
cleaved caspase-3 protein expression levels in the scramble group
were significantly increased compared with that in the control
group (P=0.001 for the 2 time points). The expression levels of the
same protein in the RNAi 24 h (P=0.040) and the RNAi 72 h (P=0.029)
groups were significantly decreased compared with that in the
control group. At 24 and 72 h, the cleaved caspase-3 expression
level in the RNAi group was significantly decreased compared with
that in the scramble group (P<0.001 for the two time points). In
the scramble group, the cleaved caspase-3 level at 72 h was
significantly increased compared with that at 24 h (P=0.021).
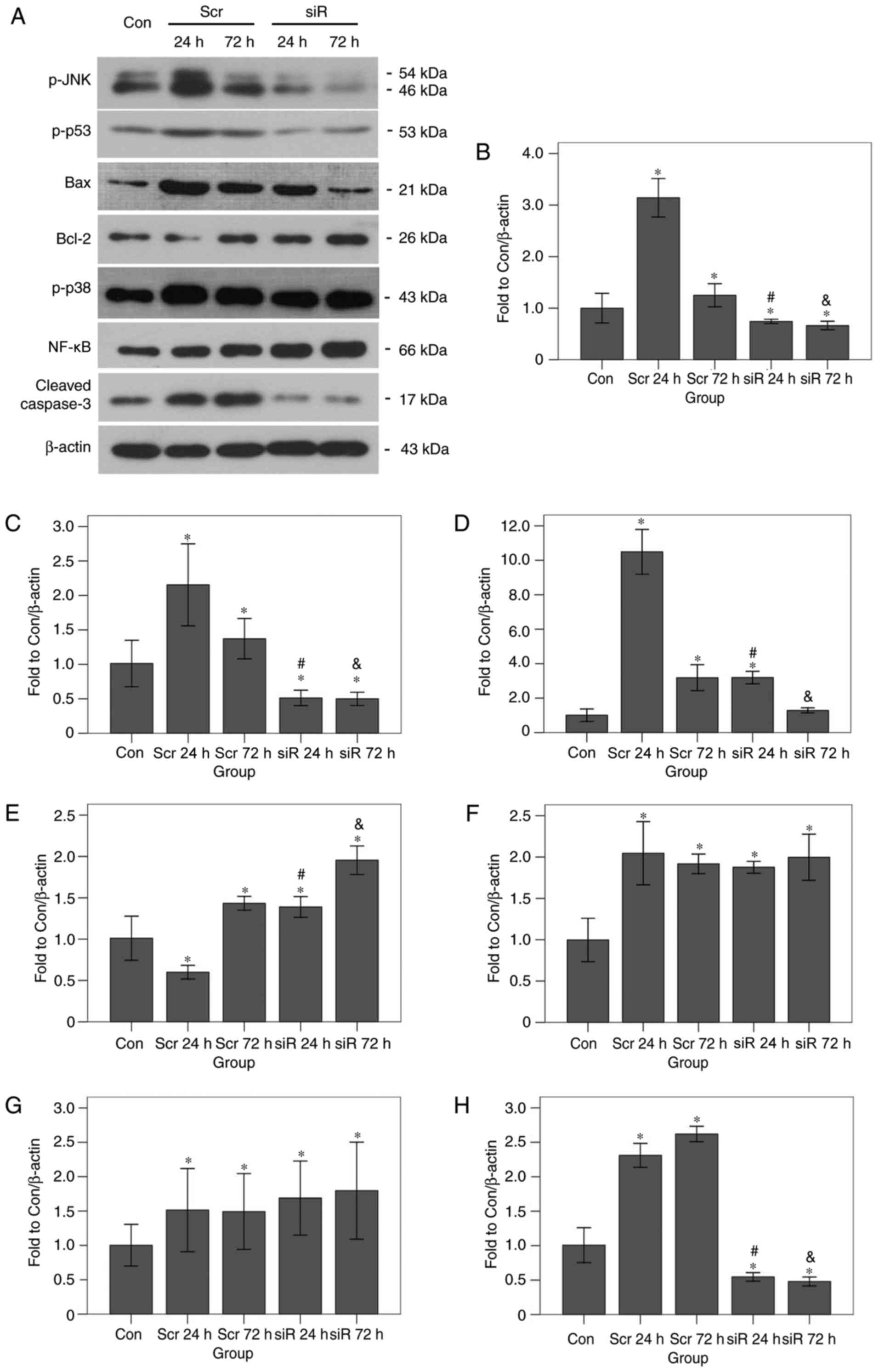 | Figure 7.Effects of JNK1 inhibition on the
protein expression levels of apoptosis-associated genes as
determined by western blot analysis. (A) Representative bands are
presented. JNK1 inhibition led to an increase in Bcl-2 expression
and a decrease in the protein expression levels of p-p53, Bax and
cleaved caspase-3, but had no effect on the expression of p-p38 or
NF-κB. Data are presented for (B) p-JNK1, (C) p-p53, (D) Bax, (E)
Bcl-2, (F) p-p38, (G) NF-κB, and (H) Cleaved caspase-3. *P<0.05
vs. the Con group, #P<0.05 vs. the Scr 24 h group;
&P<0.05 vs. the Scr 72 h group. p,
phosphorylated; p38, mitogen-activated protein kinase 11; p53,
tumor protein 53; JNK1, c-Jun terminal kinase 1; Bcl-2, B-cell
lymphoma 2; Bax, Bcl-2-associated X protein; NF-κB, nuclear factor
κ-light-chain-enhancer of activated B-cells; Con, control; siR,
siRNA; Scr, scramble. |
Discussion
In the present study, the mRNA transcript of JNK1
was first downregulated in an aSAH rat model, and any ethological
and histological variations were subsequently investigated. The
period from 24 to 72 h following SAH is considered to be an
important phase for EBI, during which SAH induces a prominent
impairment of the clinical behavioral function (20). The present study demonstrated that
JNK1 inhibition attenuated the neurodeficit scores of SAH rats at
72 h. Additionally, TUNEL assays revealed that JNK1 inhibition
reversed the increase in levels of apoptotic cells in the
hippocampus. According to the Nissl staining results, it was also
identified that the RNAi group exhibited a markedly increased
number of living neurons compared with the scramble group at 24 and
72 h following SAH. In the RNAi group, the number of living neurons
at 72 h were not markedly decreased compared with that at 24 h,
indicating that neuron apoptosis had been interrupted.
Consequently, we hypothesized that the quick recovery of
neurological function at 72 h in the RNAi group had resulted from
the interruption of cascaded cell apoptosis. In the majority of
cases, neuronal cell death due to aSAH would be experienced as a
hypoxic process caused by numerous pathological events, including
blood-brain barrier dysfunction, edema, inflammation and oxidative
cascades (21). Thereafter, it was
inferred that the downregulation of JNK1 may strengthen the hypoxic
endurance of the neurons. These results are comparable with those
from a study comprising a JNK1 inhibitor that led to an improvement
in the survival time of neurons in an EBI model via the suppression
of Nur77-dependent apoptosis pathways (15). However, that study lacked data
associated with any effects observable 24 h following SAH. The
present study has provided additional information regarding the
protective effects of JNK1 inhibition on EBI at 72 h following SAH.
Under the identical conditions, the survival times of the SAH rats
were also recorded. Upon JNK1 inhibition, a clear improvement in
the survival times of the SAH rats was observed, indicating that
the interruption of the EBI process may improve the resistance of
neurons to subsequent effects of SAH.
To elucidate the molecular mechanism, RT-qPCR and
western blot analyses were performed. The results revealed that SAH
led to an increase in the transcription and protein expression
levels of the phosphorylated proteins of JNK1, p38 and NF-κB, which
are all associated with promoting pro-apoptotic and
pro-inflammatory cellular signaling pathways, leading to enhanced
SAH-induced inflammatory responses and poorer SAH outcomes
(22,23). It has been demonstrated that
decreasing the activation of these pathways simultaneously with a
specific transforming growth factor β-activated kinase 1 inhibitor,
5Z−7-oxozeaenol, effectively prevented neuronal apoptosis
and attenuated neurological deficits in an SAH model (20). However, the effects of inhibiting
the pathways separately has not been described extensively. In the
present study, it was demonstrated that JNK1 inhibition did not
affect the phosphorylation of p38 and NF-κB. It was suggested that
the JNK1 signaling pathway may be independent of the other 2
pathways in the SAH model, rather than JNK1 being positioned
downstream of them, as has been suggested by certain studies
(24,25). Additional studies are required to
gain novel information concerning the detailed interactions among
the 3 pathways. p53 has emerged as one of the most important
anti-apoptosis targets in SAH (26). The present study demonstrated that
the expression of p-p53 was synchronized with that of p-JNK1,
although transcription of the p53 gene was not affected by JNK1
inhibition, indicating that JNK1 and p53 interact at the level of
phosphorylation, a result that is consistent with the mechanisms
operating in oxidative stress-induced apoptosis (13). Once p53 signaling is inhibited, at
least one apoptotic pathway will be blocked. The downregulation of
Bax protein and upregulation of Bcl-2 observed in the present study
suggested deactivation of the mitochondrial apoptotic pathway,
accompanied by the downregulation of cleaved caspase-3. As
mentioned above, the levels of cleaved caspase-3 in the RNAi group
were significantly decreased compared with that in the scramble
group at the same time point. Caspase-3 may be cleaved either via
the intrinsic apoptotic pathway (mitochondrial pathway) or via the
extrinsic apoptotic pathway (death receptor pathway) (27). Based on the results in the present
study, it may be possible that JNK1 inhibition prevented the
mitochondrial apoptotic pathway from exerting a role in the overall
EBI process.
The association between JNK and p53 has previously
been investigated in a model of DNA damage caused by cisplatin, a
chemotherapeutic drug used for cancer treatment (28). In the process of cisplatin-induced
DNA damage, p53 protein levels accumulated gradually, and may have
been regulated by JNK. It was suggested that JNK1 functions as a
negative regulator in the process (13). Furthermore, an additional study
investigating signaling in a model of fibronectin fragment-mediated
apoptosis verified this conclusion (29). However, in the present study, the
opposite phenomenon was observed, suggesting that JNK1 may interact
with p53 differently according to the prevailing circumstances. In
fact, a study examining deoxycholic acid-induced liver cell
apoptosis also revealed that JNK may directly or indirectly
modulate p53 expression and positively affect apoptosis. The
possible mechanism suggested was of p53 stabilization and its
increased ability to elicit apoptosis, which resulted from
JNK-induced p53 phosphorylation and attenuation of the interaction
of p53 with mouse double minute 2 homolog (28,30).
Even though a putative preliminary mechanism was
suggested by the results from the present study, there were certain
limitations. Although the method of pre-chiasmatic cistern
injection provided a reasonably homogenous animal model, it was not
fully able to simulate the in vivo situation of aSAH, as the
intracranial closeness was broken. The method of internal carotid
artery puncture may provide a closer approximation to the in
vivo conditions of aSAH, according to previous studies
(31,32); however, in the present study the
quantity of hemorrhage and severity of subsequent complications
would not be easy to maintain at the same level. Consequently,
these 2 approaches will continue to be employed according to the
preferences of the specific study, until improved methods are
devised.
In conclusion, in the present study the expression
of JNK1 was downregulated with in vivo RNAi technology,
which revealed that JNK1 inhibition improves the neurological
scores and survival times of SAH rats. The mechanism that has been
proposed involves an interruption of neuronal apoptosis in the EBI
process. JNK1 inhibition may contribute to this process by
decreasing p53 phosphorylation and deactivating the downstream
mitochondrial apoptotic pathway.
Acknowledgements
Not applicable.
Funding
The present study was supported by Special funds for
Scientific and Technological Innovation Talents Research from
Harbin Science and Technology Bureau (grant no. 2014RFXGJ015) and
the National Natural Science Foundation of China (grant no.
81302177).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors participated in the design,
interpretation of the studies and analysis of the data and review
of the manuscript. GQL, XFL and WY designed the protocol of
experiments. XHL constructed the aSAH animal model and conducted
the in vivo transfection protocol. ZYW collected the brain
tissue and conducted the TUNEL assays and Nissl staining. DYM
conducted the RT-qPCR and western blot analysis assays. YW recorded
the behavioral scores and survival data. GQL and WY wrote the
manuscript. XFL performed the statistical analysis.
Ethics approval and consent to
participate
All procedures were approved by Harbin Medical
University Animal Care and Use Committee and in accordance with the
Guide for the Care and Use of Laboratory Animals by the National
Institute of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Danière F, Gascou G, Menjot de Champfleur
N, Machi P, Leboucq N, Riquelme C, Ruiz C, Bonafé A and Costalat V:
Complications and follow up of subarachnoid hemorrhages. Diagn
Interv Imaging. 96:677–686. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Macdonald RL, Pluta RM and Zhang JH:
Cerebral vasospasm after subarachnoid hemorrhage: The emerging
revolution. Nat Clin Pract Neurol. 3:256–263. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Macdonald RL, Higashida RT, Keller E,
Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A,
et al: Clazosentan, an endothelin receptor antagonist, in patients
with aneurysmal subarachnoid haemorrhage undergoing surgical
clipping: A randomised, double-blind, placebo-controlled phase 3
trial (CONSCIOUS-2). Lancet Neurol. 10:618–625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sehba FA, Hou J, Pluta RM and Zhang JH:
The importance of early brain injury after subarachnoid hemorrhage.
Prog Neurobiol. 97:14–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuksel S, Tosun YB, Cahill J and Solaroglu
I: Early brain injury following aneurysmal subarachnoid hemorrhage:
Emphasis on cellular apoptosis. Turk Neurosurg. 22:529–533.
2012.PubMed/NCBI
|
|
6
|
Hasegawa Y, Suzuki H, Sozen T, Altay O and
Zhang JH: Apoptotic mechanisms for neuronal cells in early brain
injury after subarachnoid hemorrhage. Acta Neurochir Suppl.
110:43–48. 2011.PubMed/NCBI
|
|
7
|
Cahill J, Calvert JW and Zhang JH:
Mechanisms of early brain injury after subarachnoid hemorrhage. J
Cereb Blood Flow Metab. 26:1341–1353. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou C, Yamaguchi M, Colohan AR and Zhang
JH: Role of p53 and apoptosis in cerebral vasospasm after
experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab.
25:72–82. 2005. View Article : Google Scholar
|
|
9
|
Dawson VL and Dawson TM: Deadly
conversations: Nuclear-mitochondrial cross-talk. J Bioenerg
Biomembr. 36:287–294. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Loo G, Saelens X, van Gurp M,
MacFarlane M, Martin SJ and Vandenabeele P: The role of
mitochondrial factors in apoptosis: A Russian roulette with more
than one bullet. Cell Death Differ. 9:1031–1042. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dérijard B, Hibi M, Wu IH, Barrett T, Su
B, Deng T, Karin M and Davis RJ: JNK1: A protein kinase stimulated
by UV light and Ha-Ras that binds and phosphorylates the c-Jun
activation domain. Cell. 76:1025–1037. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gkouveris I and Nikitakis NG: Role of JNK1
signaling in oral cancer: A mini review. Tumour Biol.
39:10104283177116592017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fuchs SY, Adler V, Pincus MR and Ranai Z:
MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad
Sci USA. 95:10541–10546. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ferreira DM, Afonso MB, Rodrigues PM,
Simão AL, Pereira DM, Borralho PM, Rodrigues CM and Castro RE:
c-Jun N-terminal kinase 1/c-Jun activation of the p53/MicroRNA
34a/Sirtuin 1 pathway contributes to apoptosis induced by
deoxycholic acid in rat liver. Mol Cell Biol. 34:1100–1120. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dai YX, Zhang W, Zhou XM and Shi J:
Inhibition of c-Jun N-terminal kinase ameliorates early brain
injury after subaracnoid hemorrhage through inhibition of a Nur77
dependent pathway. Neurochem Res. 39:1603–1611. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals. Guide for the Care and Use of Laboratory Animals. National
Academies Press; Washington, DC: 2010, PubMed/NCBI
|
|
17
|
Shen H, Chen Z, Wang Y, Gao A, Li H, Cui
Y, Zhang L, Xu X, Wang Z and Chen G: Role of neurexin-1β and
neuroligin-1 in cognitive dysfunction after subarachnoid hemorrhage
in rats. Stroke. 46:2607–2615. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Z, Ma C, Meng CJ, Zhu GQ, Sun XB, Huo
L, Zhang J, Liu HX, He WC, Shen XM, et al: Melatonin activates the
Nrf2-ARE pathway when it protects against early brain injury in a
subarachnoid hemorrhage model. J Pineal Res. 53:129–137. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang D, Yan H, Li H, Hao S, Zhuang Z, Liu
M, Sun Q, Yang Y, Zhou M, Li K and Hang C: TGFβ-activated kinase 1
(TAK1) inhibition by 5Z-7-oxozeaenol attenuates early brain injury
after experimental subarachnoid hemorrhage. J Biol Chem.
290:19900–19909. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ayer R and Zhang J: Connecting the early
brain injury of aneurysmal subarachnoid hemorrhage to clinical
practice. Turk Neurosurg. 20:159–166. 2010.PubMed/NCBI
|
|
22
|
Kusaka G, Ishikawa M, Nanda A, Granger DN
and Zhang JH: Signaling pathways for early brain injury after
subarachnoid hemorrhage. J Cereb Blood Flow Metab. 24:916–925.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
You WC, Wang CX, Pan YX, Zhang X, Zhou XM,
Zhang XS, Shi JX and Zhou ML: Activation of nuclear factor-κB in
the brain after experimental subarachnoid hemorrhage and its
potential role in delayed brain injury. PLoS One. 8:e602902013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu GS: The fuctional interactions between
the p53 and MAPK signaling pathways. Cancer Biol Ther. 3:2156–2161.
2004. View Article : Google Scholar
|
|
25
|
Liu R, Wu CX, Zhou D, Yang F, Tian S,
Zhang L, Zhang TT and Du GH: Pinocembrin protects against
β-amyloid-induced toxicity in neurons through inhibiting receptor
for advanced glycation end products (RAGE)-independent signaling
pathways and regulating mitochondrion-mediated apoptosis. BMC Med.
10:1052012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cahill J, Calvert JW, Marcantonio S and
Zhang JH: p53 may play an orchestrating role in apoptotic cell
death after experimental subarachnoid hemorrhage. Neurosurgery.
60:531–545. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang G, Zhang W, Qin Q, Wang J, Zheng H,
Xiong W and Yuan J: Mono (2-ethylhexyl) phthalate induces apoptosis
in p53-silenced L02 cells via activation of both mitochondrial and
death receptor pathways. Environ Toxicol. 30:1178–1191. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Akaboshi M, Kawai K, Ujeno Y, Takada S and
Miyahara T: Binding characteristics of
(−)-(R)-2-aminomethylpyrrolidine
(1,1-cyclobutanedicarboxylato)-2-platinum (II) to DNA, RNA and
protein molecules in HeLa cells and its lethal effect: Comparison
with cis- and trans-diamminedichloroplatinums(II). Jpn J Cancer
Res. 85:106–111. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tafolla E, Wang S, Wong B, Leong J and
Kapila YL: JNK1 and JNK2 oppositely regulate p53 in signaling
linked to apoptosis triggered by an altered fibronectin matrix: JNK
links FAK and p53. J Biol Chem. 280:19992–19999. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ljungman M: Dial 9-1-1 for p53: Mechanisms
of p53 activation by cellular stress. Neoplasia. 2:208–225. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ying GY, Jing CH, Li JR, Wu C, Yan F, Chen
JY, Wang L, Dixon BJ and Chen G: Neuroprotective effects of
valproic acid on blood-brain barrier disruption and
apoptosis-related early brain injury in rats subjected to
subarachnoid hemorrhage are modulated by heat shock protein
70/matrix metalloproteinases and heat shock protein 70/AKT
pathways. Neurosurgery. 79:286–295. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yin J, Li R, Liu W, Chen Y, Zhang X, Li X,
He X and Duan C: Neuroprotective effect of protein phosphatase
2A/tristetraprolin following subarachnoid hemorrhage in rats. Front
Neurosci. 12:962018. View Article : Google Scholar : PubMed/NCBI
|