Introduction
Alzheimer's disease (AD) is a neurodegenerative
disease which is incurable with higher occurrence in elder people
(1). With ever-increasing elder
populations in both developed and developing countries, AD has
become a huge public health problem and asocial and economic
burden. AD patients usually have neurological deficits including
the loss of memory and other cognitive abilities and a change in
their personality (2).
Histopathologically, AD is characterized by the presence of senile
plaques in the extracellular medium, intracellular inclusions of
neurofibrillary tangles (NFT) and loss of neurons. Amyloid β (Aβ)
is a major component of senile plaques. It is widely accepted that
the accumulation of Aβ is essential for AD pathogenesis (3). Aβ is associated with neuronal cell
death, NFT formation, inflammation and oxidative damage in AD
(3). Aβ-induced neurotoxicity
inhibits hippocampal neurogenesis and synaptic plasticity,
resulting in brain dysfunction in cognition, learning and memory
(1). Therefore, Aβ is proposed as
the most important causative factor of AD. Numerous studies have
been performed to inhibit the formation of Ab and to attenuate its
toxicity.
Neurotrophic factors, together with essential
proteins secreted by neurons and glial cells, support neuronal
survival, proliferation and maturation as well as maintain the
homeostasis of peripheral and central nervous system (4). Brain-derived neurotrophic factor
(BDNF) is a major central neurotrophic factor in the central
nervous system (4). Reduction of
BDNF was reported in the brains of cognitive impairment patients,
including AD patients (5,6). As BDNF involves in neuronal survival
and neuronal functions, it is proposed that the reduction of BDNF
is associated with the pathogenesis of AD (4). Studies performed in vitro and
in vivo suggested that BDNF inhibited Aβ-induced
neurotoxicity (7–9).
The reduction of BDNF in the brains of AD patients
is possibly due to the epigenetic modulations. Epigenetic
modulations refer to the regulation of gene expression by
manipulating DNA-related proteins rather than DNA, including
acetylation, methylation, phosphorylation, ubiquitinylation,
carbonylation and glycosylation. Epigenetic modulations commonly
occur at histones, remodeling chromatin between relatively ‘open’
and ‘closed’ forms (10).
Significant decreased acetylation of histone was reported in both
AD transgenic mice and postmortem human brains (11). The level of histone acetylation is
regulated by acetyltransferase and histone deacetylase (HDAC). An
increase of HDAC2 was observed in the hippocampus of AD patients
(12). The electrostatic
interaction between positively charged histones and negatively
charged DNA leads to a condensed and repressive chromatin structure
(10,11). The acetylation of histones
attenuates their positive charges, resulting in a reduced affinity
of histones to DNA and a free chromatin for recruiting
transcription factors to genes (10,11).
HDACs can remove acetyl groups from histones, which conversely
produces a condensed chromatin structure and repressed gene
expressions (13). Aβ induces
epigenetic changes by promoting HDAC2 expression, resulting in BDNF
reduction (14). In AD transgenic
mouse models, cortical expression of BDNF and BDNF-mediated TrkB
retrograde trafficking are inhibited when neuronal culture
submitted to Aβ peptides (2).
The traditional anesthesia (e.g.,
isoflurane-mediated anesthesia) increases the risk of AD (15–17).
Some traditional anesthetic agents are associated with AD
pathogenesis (18). Behavioral
tests in rodent models showed that a transient exposure to
anesthetic drug isoflurane caused a lasting impairment to the
cognition (15,16,19).
A cell co-cultured study showed that isoflurane interfered
astrocytes to support neuronal growth, which may explain the
association between the anesthesia application and AD pathogenesis
(20). Dexmedetomidine (Dex), a
specific agonist of α2-adrenoceptor, has been used as a new type of
clinical anesthetic agent. Dex recently attracts numerous
attentions because of its multiple neuroprotective actions in
experimental and pre-clinical studies (21,22).
Additionally, Dex has been found to inhibit the postoperative
cognitive dysfunction induced by isoflurane, which indicates the
potential protection of Dex against cognitive function impairment
and even AD (23). The mechanisms
underlying neuroprotective actions remain largely undefined,
however, a study of the protective effect of Dex against septic
acute kidney injury revealed an important molecular basis that Dex
blocked the transport of HDAC2 and HDAC5 to nuclear and inhibited
the acetylation of histone H3 (24).
Therefore, we hypothesized that Dex increases BDNF
production by manipulating HDAC2 and HDAC5 and consequently
attenuate Aβ cytotoxicity. BDNF is mainly produced by neurons and
astrocytes. This study prepared primary neurons and astrocytes and
further investigated our hypothesis on these cells.
Materials and methods
Ethics statement
The present study was approved by the Ethics
Committee of Xiangya School of Medicine (Hunan, China).
Preparation of primary neurons and
astrocytes
Primary neurons and astrocytes were acquired from
1–2 days' neonatal Sprague Dawley rats according to a previous
protocol (25). Eight Rat pups
(1–2 days; Central South University, Changsha, China) were housed
in vivariums that was maintained at fixed temperature (22–23°C) and
moisture (70%), with a 12-hour light on/off cycle. They sucked milk
from the maternal rat. All procedures were adhered to National
Institutes of Health Guidelines for the care and use of animals
(26). Rat pups were euthanatized,
immersed in 75% alcohol for 5 min, and then washed with D-Hank's.
The brain of neonatal rats was removed in a sterile field. The
cerebral cortex and hippocampus were isolated on ice for the
preparation of primary astrocytes and neurons, respectively. After
removing meninges and blood vessels, the cerebral cortex and
hippocampus were cut into a cubic millimeter pieces and digested by
2 mg/ml papain and 0.05 mg/ml DNAase in serum-free medium at 37°C
for 30 min. The tissue suspension was gently pipetted to disperse
the cells and filtered through 100 µM sterile filter. Neocortical
cells were plated in dishes at a density of 4×105
cells/ml in Dulbecco's modified Eagle medium with 10% fetal bovine
serum (FBS; both Hyclone Laboratories Inc., Logan, Utah, USA),
epidermal growth factor (10 ng/ml), penicillin (50 U/ml), and
streptomycin (50 U/ml; all Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). When astrocytes reached confluence, microglia were
removed by shaking at 200 rpm for at least 24 h. Relatively pure
neuronal cultures from hippocampus tissues were infused into a
poly-lysine-treated plate. After 4 h cultivation, the medium was
removed and new neurobasal-A medium with B27 (1%), glutamine (0.5
mmol/l), penicillin (50 U/ml), streptomycin (50 U/ml) and cytosine
arabinoside (3 µM; all Sigma-Aldrich; Merck KGaA) was added in the
plate. The cytosine arabinoside was added to inhibit glial
proliferation, but cytosine arabinoside was removed from the media
before cells were treated with Aβ and Dex to eliminate the
interference. The neurons and astrocytes were initially cultured
for 2 days (passage 0, day 2; P0D2) and subcultured for 4 days
(passage 1, day 4; P1D4). Neurons and astrocytes at the second
passage (P2D4) were used for further study.
Immunocytochemistry (ICC)
Neurons and astrocytes in the cultures were
identified using ICC. Neurons and astrocytes were fixed in 4%
paraformaldehyde for 15 min, permeabilized and blocked in 0.1%
Triton X-100 with 5% normal goat serum (Sigma-Aldrich; Merck KGaA)
in phosphate-buffered saline (PBS) for 30 min at room temperature.
Neurons and astrocytes were then incubated with primary antibodies
against β-Tubulin III (1:200; T2200) and glial fibrillary acidic
protein (GFAP; 1:400 dilution; HPA056030; both Sigma-Aldrich) for
1–4 h at room temperature, respectively. Samples were washed three
times in PBS, followed by the incubation in horseradish peroxidase
(HRP)-labeled anti-IgG secondary antibody (1:2,000) for 1 h and DAB
(both Sigma-Aldrich; Merck KGaA) for 3 min at room temperature.
After additional washes, samples were stained using hematoxylin and
imaged using a microscope (Axio Imager 2; Zeiss AG, Oberkochen,
Germany).
Cell treatments
Monomeric Amyloid β-protein (Aβ, Human; 1–42,
Aβ1-42; Peptide Institute, Inc., Osaka, Japan) was
incubated for 24 h at 37°C to allow self-aggregation and
oligomerization. The primary neurons and astrocytes were incubated
with Aβ (at dosages of 0, 3, 10 and 30 nM) for 24 h for the
following cytotoxicity test. To investigate the protective effects
of Dex against Aβ and the underlying mechanisms, the cells were
simultaneously treated with Aß and Dex (Sigma-Aldrich; Merck KGaA),
Recombinant Human/Murine/Rat BDNF protein (ab9794) or Trichostatin
A (ab120850; both Abcam, Cambridge, UK), a selective inhibitor of
HDACs for 24 h.
MTT assay
Neurons and astrocytes were cultured in 96-well
plates. MTT solution (Nanjing KeyGEN Biotech, Nanjing, China) was
added after the cell treatments. The formazan crystals in cells was
dissolved sufficiently after the incubation with dimethylsulfoxide
for 4 h at 37°C. The absorbance was determined with a Microplate
Reader (ELX-800; BioTek Instruments, Inc., Winooski, VT, USA) at
570 nm.
Cell apoptosis assay
Neurons and astrocytes were plated at
2×105 cells/well in chamber culture slides and incubated
overnight. After the cell treatments, the cells were rinsed with
PBS and double-stained with Annexin V-FITC and propidium iodide
according to the manufacturer's instruction (Solarbio Science &
Technology Co., Ltd., Beijing, China). The apoptosis rate was
determined using a dual laser flow cytometer (Becton Dickinson, San
Jose, CA, USA) with the ModFitLT software (Verity Software House,
Topsham, ME, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total mRNA was extracted from treated neurons and
astrocytes using TRIzol reagent following the manufacturer's
protocol (Takara, Dalian, China). The mRNA was reverse-transcribed
into cDNA using the PrimeScript RT Reagent Kit (Takara). RT-qPCR
was performed in the iQ5 Multi-color Real-Time PCR Detection System
(Bio-Rad, Hercules, CA, USA) using SYBR Premix Ex Taq II (Takara).
The thermocycling conditions were as follows: Initial denaturation
at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C
for 15 sec, annealing at 60°C for 1 min and extension at 72°C for 1
min. The RT-qPCR primer sequences for BDNF and 18S
rRNA were as follows: BDNF forward,
5′-AGCTGAGCGTGTGTGACAGT-3′ and reverse, 5′-ACCCATGGGATTACACTTGG-3′;
18S rRNA forward, 5′-GCAATTATTCCCCATGAACG-3′ and reverse,
5′-GGCCTCACTAAACCATCCAA-3′. A melting curve analysis of the
amplified products was performed at the end of each PCR cycle. The
comparative Cq method (27) was
used to quantify the expression of BDNF using 18S
rRNA as the normalization control.
Western blot analysis
Neurons and astrocytes were seeded in culture plates
at a density of 1 × 106 cells/well. After the cell
treatments, the cells were harvested and washed with PBS. Cells
were lysed in RIPA buffer (Promega, Madison, WI, USA) containing
protease inhibitor cocktail (Roche, Basal, Switzerland). Nuclear
protein was extracted using a Nuclear Extract Kit according to the
manufacturer's instructions (Active Motif, Carlsbad, CA, USA). The
concentrations of total protein in the whole-cell lysate and
nuclear protein extracts were determined by the Bradford assay. An
equal amount (20 µg) of protein in each sample was separated by 10
or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and then transferred to a polyvinylidene difluoride
membrane (Millipore, Bedford, MA, USA). The membrane was incubated
with 5% (w/v) skim milk in PBS (pH 7.4) containing 0.05% Tween-20
(PBS-T) at room temperature for 1 h, washed with PBS-T and then
probed with anti-BDNF antibody (1:800 dilution; ab108319),
anti-GAPDH antibody (1:1,500 dilution; ab9485), anti-HDAC2 antibody
(1:1,000 dilution; ab16032), anti-HDAC5 antibody (1:1,000 dilution;
ab1439), anti-acetyl-histone H3 antibody (1:800 dilution; ab47915)
and anti-histone H2A antibody (1:1,000 dilution; ab88770; all
Abcam) for 2 h at room temperature. The membrane was washed in
PBS-T and then incubated with HRP-conjugated goat anti-rabbit IgG
secondary antibody (dilution 1:1,500; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) for 1 h at room temperature. Reactive
proteins were detected using Enhanced Chemiluminescent and
SuperSignal™ Chemiluminescent substrates (Pierce,
Rockford, IL, USA).
ELISA assay
Cell culture supernatants were collected. BDNF
Emax® ImmunoAssay System kit was used to determine the
BDNF concentrations (Promega), according to the manufacturer's
instructions. The plates were washed after the reaction with 100 µl
of o-phenylenediamine substrate (Promega) added to each well.
Plates were incubated for 30 min at room temperature, after which
50 µl/well of 4 N sulfuric acid was added. The microplate reader
was used to quantify the absorbance at 490 nm.
Statistical analysis
All data from at least three independent experiments
were presented as the mean ± standard deviation. The statistical
analysis was performed using one-way analysis of the variance
followed by Dunnett's post hoc test using SPSS version 10.0
software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
Dex attenuates Aβ-induced toxicity in
neurons and astrocytes
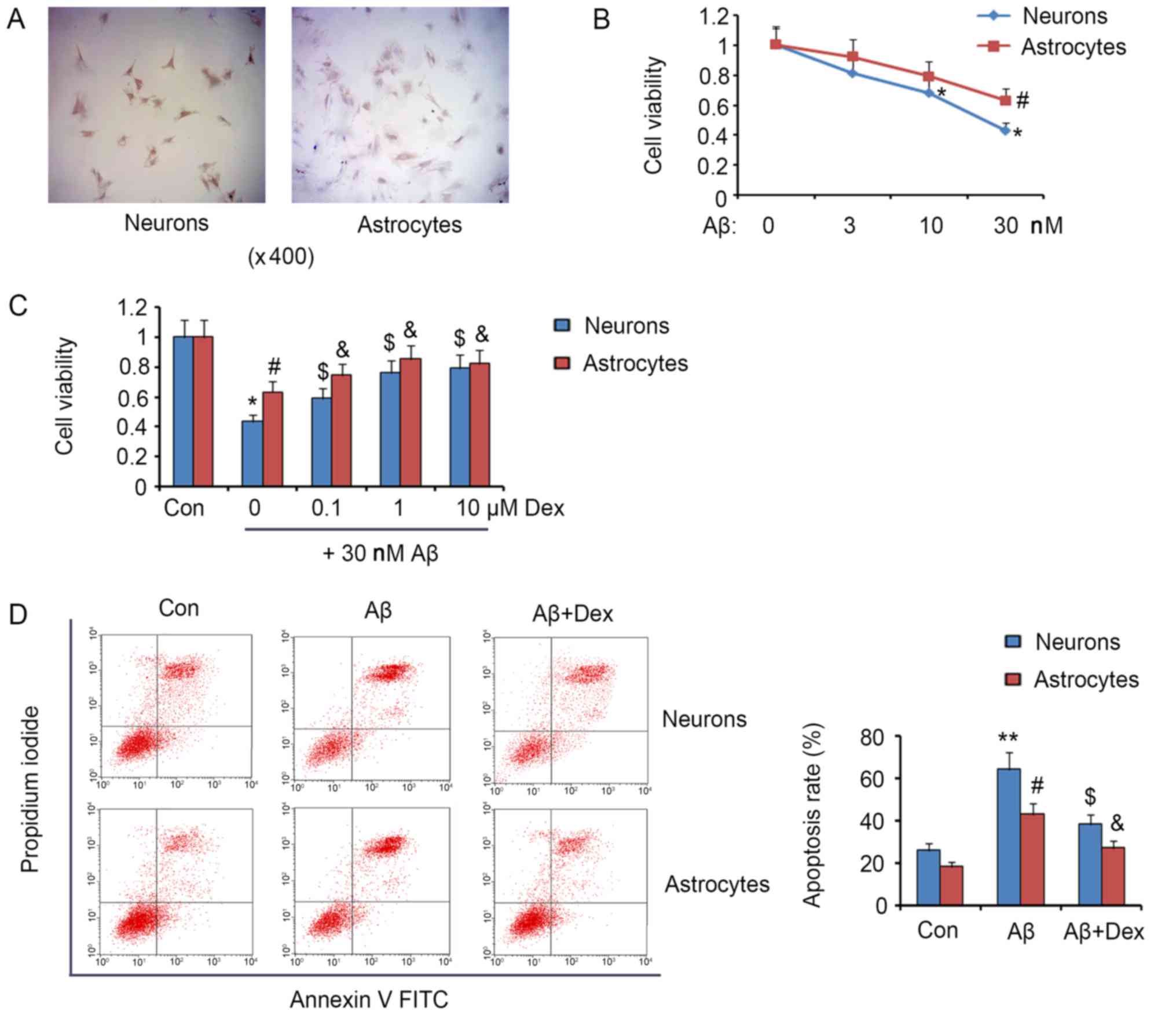
Primary neurons and astrocytes were respectively
isolated from the hippocampus and cerebral cortex tissues of 1–2
days' neonatal Sprague Dawley rats. The staining of β-Tubulin III
and GFAP in ICC assay confirmed the successful isolation of neurons
and astrocytes, respectively (Fig.
1A). The exposure of neurons to 10 nM Aβ resulted in a
reduction in the cell viability (P<0.05, Fig. 1B), agreeing to the reported
toxicity of Aβ on neurons and astrocytes. The viability of both
neurons and astrocytes was attenuated by 30 nM Aβ (P<0.05). As
shown in Fig. 1C, 0.1 µM Dex
reversed the reduction of both neuron and astrocyte viability
caused by 30 nM Aβ (P<0.05). The reversion effect detected under
1 µM Dex treatment was stronger compared to 0.1 µM Dex treatment
(P<0.05). But no further improvement was observed under 10 µM
Dex treatment. The pro-apoptotic effect of Aβ takes the major
responsibility for the toxicity. Analysis of flow cytometer showed
that 30 nM Aβ significantly increased the apoptosis rate of neurons
and astrocytes at approximate 2.5-fold (P<0.01) and 2.2-fold
(P<0.05) compared to the control, respectively (Fig. 1D). Co-treatment with 1 µM Dex and
30 nM Aβ decreased apoptosis rate of neurons from 64 to 38%
(P<0.05), and astrocytes from 43 to 27% (P<0.05). Although
Dex attenuated Aβ-induced toxicity in neurons and astrocytes, the
treatment with Dex, alone, at a suitable range of concentrations
had no significant effect on viability and apoptosis of neurons and
astrocytes (data not shown).
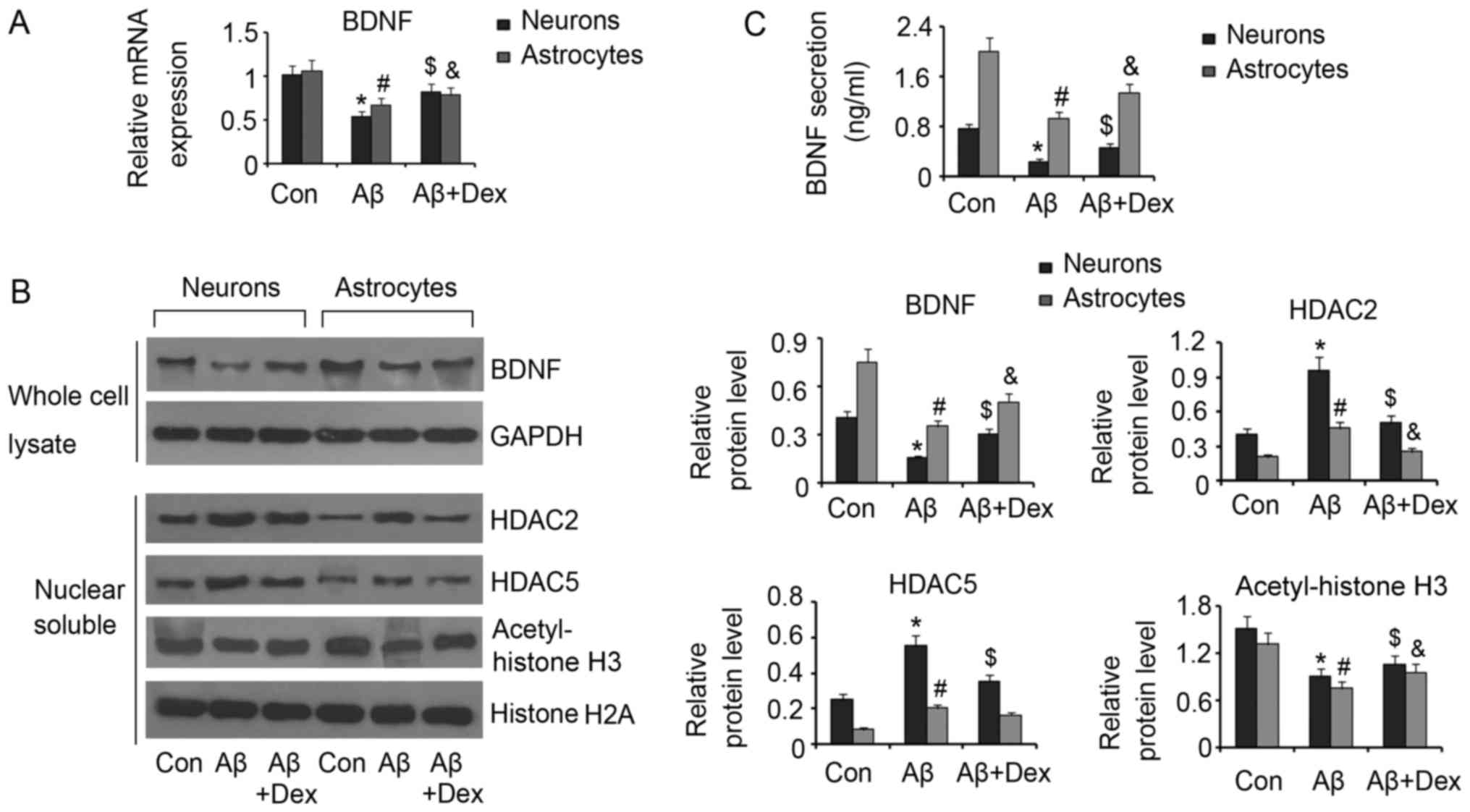
Dex inhibits Aβ-induced reduction of
BDNF in neurons and astrocytes
The importance of BDNF in supporting the
proliferation and development of neurons and astrocytes has been
established by previous studies. However, Aβ has inhibitory effect
on BDNF production. In this study, 30 nM Aβ decreased BDNF
mRNA levels in both neurons and astrocytes (P<0.05, Fig. 2A). However, 1 µM Dex inhibited the
reduction of BDNF mRNA levels in neurons and astrocytes
(P<0.05) compared to the control. Western blot assay also showed
decreased BDNF protein levels in neurons and astrocytes by Aβ
(P<0.05, Fig. 2B), but Dex
rescued the reduction of BDNF protein (P<0.05, Fig. 2B). It has known that BDNF
expression is notably impacted by the acetylation degree of histone
H3. Aβ has been reported to promote to HDACs accumulation in cell
nucleus. In addition, Aβ increased HDAC2 and HDAC5 protein levels
in the cell nucleus in both neurons and astrocytes (P<0.05).
Consequently, acetylated histone H3 decreased under Aβ treatment
(P<0.05). Dex inhibited the increase of HDAC2 and HDAC5 protein
levels in the cell nucleus in neurons under Aβ treatment (P<0.05
vs. Aβ group). In astrocytes, Dex only inhibited Aβ-induced
increase of HDAC2 (P<0.05 vs. Aβ group). As a result,
Aβ-promoted acetylation of histone H3 in neurons and astrocytes was
inhibited by Dex (P<0.05 vs. Aβ group). Besides, Aβ decreased
BDNF concentrations in neurons and astrocytes (P<0.05; Fig. 2C), which was reversed by Dex
treatment (P<0.05; Fig.
2C).
BDNF mediates the protective effect of
Dex against Aβ
Trichostatin A, a selective inhibitor of HDACs, was
added to neurons and astrocytes with Dex and Aβ to further
investigate the effect of Dex against Aβ-promoted BDNF production
by inhibiting HDAC2 and HDAC5. Treatment with 0.2 µm Trichostatin A
increased BDNF mRNA expression that was reduced by Aβ
(P<0.05 vs. Aβ group, Fig. 3A).
Moreover, extracellular concentration of BDNF increased by
Trichostatin A in Aβ-treated neurons (P<0.01 vs. Aβ group,
Fig. 3B) and astrocytes (P<0.05
vs. Aβ group). Although our data herein demonstrated that Dex
reversed the reduced production of BDNF by Aβ, it remained unclear
whether the protective effect of Dex against Aβ is associated to
BDNF functions. As Trichostatin A could also increase BDNF
production and secretion by Aβ-treated neurons and astrocytes,
Trichostatin A theoretically has similar protective effect with
Dex. Co-treatment with Trichostatin A (0.2 µm) and Aβ (30 nm)
increased viabilities of neurons and astrocytes compared with Aβ
treatment alone (both P<0.05; Fig.
3C), suggesting similar protective effect with Dex. In
addition, we added recombinant BDNF protein to Aβ-treated neurons
and astrocytes, to further verify the role of BDNF in the
protection of Dex against Aβ. As the biological activity of the
recombinant BDNF protein produced by Escherichia coli is
lower than that of BDNF secreted from neurons and astrocytes, we
added relatively high levels of recombinant BDNF protein in the
cell medium. Recombinant BDNF protein at 0.1 µm had significantly
lower effect on improving viability of neurons and astrocytes
(P>0.05 vs. Aβ group; Fig. 3D).
However, 1 and 10 µm recombinant BDNF significantly increased
viabilities of neurons and astrocytes (P<0.05 vs. Aβ group).
Flow cytometer results showed that 10 µm recombinant BDNF
significantly lowered apoptosis rates of neurons and astrocytes
that was increased by Aβ (P<0.05 vs. Aβ group). Treatment with
Trichostatin A inhibited Aβ-induced apoptosis of neurons (P<0.05
vs. Aβ group), but had no significant effect on apoptosis rate of
astrocytes.
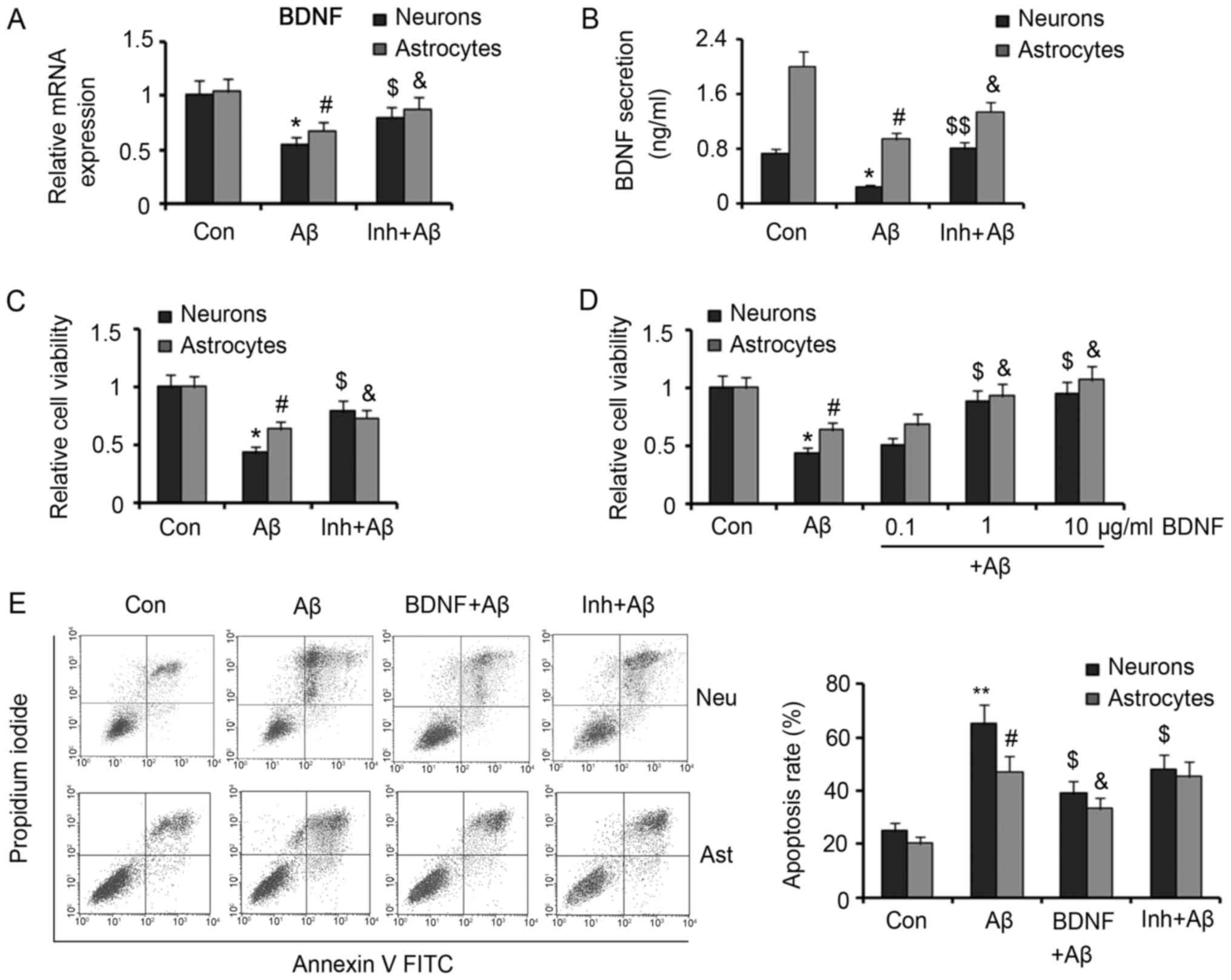 | Figure 3.Protective effect of Trichostatin A
and recombinant BDNF protein against Aβ in neurons and astrocytes.
Trichostatin A was added to neurons and astrocytes treated with 30
µM Aβ. (A) Reverse transcription-quantitative polymerase chain
reaction and (B) ELISA assays detected BDNF mRNA expression and
secretion, respectively. (C) Viabilities of the treated neurons and
astrocytes were measured. (D) Different doses of recombinant BDNF
protein were added to neurons and astrocytes that were treated with
30 nM Aβ. Then the cell viability was assessed. (E) The isolated
neurons and astrocytes were incubated with Aβ alone or in
combination with Trichostatin A or recombinant BDNF protein. Then,
the apoptotic rate was assessed by flow cytometry. *P<0.05 and
**P<0.01 vs. neuron control; #P<0.05 vs. astrocyte
control; $P<0.05 and $$P<0.01 vs.
neuron Aβ group; &P<0.05 vs. astrocyte Aβ group.
Dex, dexmedetomidine; Aβ, β-Amyloid; BDNF, brain-derived
neurotrophic factor; HDAC, histone deacetylase; Inh, the inhibitor
of HDACs, Trichostatin A; FITC, fluorescein isothiocyanate; Neu,
neurons; Ast, astrocytes. |
Discussion
Aβ-induced neurotoxicity and neuroinflammation play
an important role in AD pathogenesis. Aβ causes neuronal cell death
in the development of AD, resulting in cognitive and behavioral
deficits (28,29). The neuronal cell death is closely
related to Aβ-induced oxidative stress and consequent activation of
pro-apoptosis signaling, because antioxidative substances and
inhibition of apoptosis signaling, alone or in combination, prevent
the cell death (30–32). Astrocytes, a major glial cell type
in the brain, participate in the neuropathogenic mechanisms after
chronic exposure to Aβ (30). Aβ
can activate astrocytes and then induce the release of inflammatory
and neurotoxic molecules, contributing to chronic neuroinflammation
and neuronal death (30). It is
noteworthy that activated astrocytes have increased levels of the
three essential components for Aβ production: amyloid precursor
protein, β-secretase and γ-secretase, indicating a vicious circle
between Aβ production and astrocyte activation (30). The effect of Aβ on growth
performance of astrocytes remains controversial. Some studies
report that Aβ1-42 is toxic to primary cultures of
astrocytes and suppresses the viability (20,33),
while no significant effect of Aβ1-42 on proliferation
of astrocytes was observed by Baglietto-Vargas et al
(34). Non-aggregated
Aβ25-35 even promotes primary astrocyte proliferation
in vitro (35). In the
present study, Aβ1-42 inhibited the viability of both
neurons and astrocytes, and contributed to their apoptosis,
suggesting the toxicity of Aβ1-42 on both types of
cells.
Dex is one of the few anesthetics with
neuroprotective property. It has reported that Dex mitigates the
LPS-induced neuroinflammation in the hippocampus and cortex by
inhibiting the expression of IL1-β and TNF-α genes
(36). In addition, Dex protects
neuronal cells from cerebral ischemia injury (21). The protective mechanisms associate
the inhibition of matrix metalloproteinases, caspase-3 activation,
DNA fragmentation and cell apoptosis which are induced during
cerebral ischemia (21,36). A recent study reported decreased
incidence of postoperative cognitive dysfunction in Dex-treated AD
patients and aged people (23). As
Aβ is the most important causative factor of AD, the present study
investigated whether Dex could neutralize the toxicity of Aβ. Dex
attenuated the inhibitory effect of Aβ on viabilities of neurons
and astrocytes and reduced their apoptosis in response to Aβ,
suggesting the potential protective effect of Dex against AD.
BDNF participates in the regulation of neuronal
survival, differentiation and synaptic plasticity as well as
influences formation and consolidation of memory (4,37).
Treatment with exogenous BDNF or overexpression of BDNF by
adenovirus vector shows protective effect against the apoptosis of
neurons and astrocytes (38–40).
Human and preclinical studies suggest that the loss of BDNF is
involved in AD. Inhibited BDNF expression in brain leads to
cognitive decline in people with AD and mild cognitive impairment
and older adults (41).
Application of exogenous BDNF decreased Aβ production in primary
neurons by regulating intracellular trafficking and cleavage of
amyloid precursor protein (42).
Moreover, BDNF rescued Aβ-mediated neuronal toxicity in animal
models, remitting Aβ-induced deficits in spatial learning and
memory (43). Intriguingly, Aβ can
in turn downregulate the expression of BDNF in the brain (44) and block BDNF signaling by impairing
axonal transport (45). Aβ at
sublethal concentrations interferes with the BDNF-induced
activation of the Ras/ERK andPI3K/Akt pathways, resulting increased
vulnerability of neurons and inhibited BDNF protection against DNA
damage- and trophic deprivation-induced apoptosis (2).
The reduction of BDNF by Aβ may be relevant to the
epigenetic modification. Aβ induces the expression of HDAC2and
increases the nuclear translocation of HDACs in human neurons.
HDAC2 can bind the promoter region of BDNF exon IV and contribute
to the histone H3 deacetylation, resulting in the suppressed
expression of BDNF (14). The
regulatory mechanism was also studied by using the selective
inhibitor of HDACs to rescue the Aβ-induced suppression of BDNF
expression and increase the BDNF production. Dex inhibits the
expression of HDAC2 and HDAC5 in LPS-stimulated rat renal tubular
epithelial NRK52E cells which leads to the septic acute kidney
injury (24). However, the effect
of Dex on HDAC2 and HDAC5 in neurons and astrocytes has never been
reported. In the present study, Dex decreased HDAC2 and HDAC5
accumulation in the cell nucleus in neurons and astrocytes that
were subjected to Aβ, resulting in an increased acetylation of
histone H3. Additionally, Dex inhibited Aβ-induced reduction in
BDNF, suggesting that Dex protect cells against Aβ toxicity by
promoting BDNF production. This hypothesis was supported by
protective effects from recombinant BDNF protein. The protective
effect of many other agents, such as Smilagenin and Aripiprazole,
against Aβ is also dependent on BDNF (7,46).
Class I HDAC inhibitors are believed to have positive effects on
neurite outgrowth, synaptic plasticity, and neurogenesis in adult
brain (47). In previous report,
Trichostatin A, a pan-HDAC inhibitor, promoted functional recovery
from stroke in mice when used in the delayed phase (48). Trichostatin A increased the
production of BDNF and serotonin, and further protected the
neuronal cell damage caused by some toxic agents (49–51).
Although Trichostatin A also protects neurons and astrocytes by
increasing BDNF in the present study, it has some side effects as a
pan-inhibitor of HDACs and is limitedly used (11). In the present study, Trichostatin A
failed to suppress Aβ-induced astrocytes' apoptosis, which may
explain its side effects on the beneficial functions of BDNF.
In summary, this study provided a novel evidence
that Dex can antagonize the toxicity of Aβ on neurons and
astrocytes. It also revealed that the protective mechanism is
particularly relevant to the promoted BDNF expression by inhibiting
the HDAC2- and HDAC5-induced deacetylation of histone H3. This
study laid a foundation for further study of Dex protection against
Aβ on neuron and astrocytein animal models and pre-clinical
researches.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article, and are also available from the
corresponding author on reasonable request.
Authors' contributions
WM conceived and designed the study. YW and AJ
performed the experiments and wrote the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Xiangya School of Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kazim SF and Iqbal K: Neurotrophic factor
small-molecule mimetics mediated neuroregeneration and synaptic
repair: Emerging therapeutic modality for Alzheimer's disease. Mol
Neurodegener. 11:502016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tong L, Balazs R, Thornton PL and Cotman
CW: Beta-amyloid peptide at sublethal concentrations downregulates
brain-derived neurotrophic factor functions in cultured cortical
neurons. J Neurosci. 24:6799–6809. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Frost GR and Li YM: The role of astrocytes
in amyloid production and Alzheimer's disease. Open Biol. 7(pii):
1702282017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benussi L, Binetti G and Ghidoni R: Loss
of neuroprotective factors in neurodegenerative dementias: The end
or the starting point? Front Neurosci. 11:6722017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peng S, Wuu J, Mufson EJ and Fahnestock M:
Precursor form of brain-derived neurotrophic factor and mature
brain-derived neurotrophic factor are decreased in the pre-clinical
stages of Alzheimer's disease. J Neurochem. 93:1412–1421. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Buchman AS, Yu L, Boyle PA, Schneider JA,
De Jager PL and Bennett DA: Higher brain BDNF gene expression is
associated with slower cognitive decline in older adults.
Neurology. 86:735–741. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park SY, Shin HK, Lee WS, Bae SS, Kim K,
Hong KW and Kim CD: Neuroprotection by aripiprazole against
β-amyloid-induced toxicity by P-CK2α activation via inhibition of
GSK-3β. Oncotarget. 8:110380–110391. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arancibia S, Silhol M, Moulière F, Meffre
J, Höllinger I, Maurice T and Tapia-Arancibia L: Protective effect
of BDNF against beta-amyloid induced neurotoxicity in vitro and in
vivo in rats. Neurobiol Dis. 31:316–326. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang L, Fang Y, Lian Y, Chen Y, Wu T,
Zheng Y, Zong H, Sun L, Zhang R, Wang Z and Xu Y: Brain-derived
neurotrophic factor ameliorates learning deficits in a rat model of
Alzheimer's disease induced by aβ1-42. PLoS One. 10:e01224152015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang XJ and Seto E: Lysine acetylation:
Codified crosstalk with other posttranslational modifications. Mol
Cell. 31:449–461. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu X, Wang S, Yu L, Jin J, Ye X, Liu Y
and Xu Y: HDAC3 negatively regulates spatial memory in a mouse
model of Alzheimer's disease. Aging Cell. 16:1073–1082. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sen A, Nelson TJ and Alkon DL: ApoE4 and
Aβ oligomers reduce BDNF expression via HDAC nuclear translocation.
J Neurosci. 35:7538–7551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang J and Zhuang S: Epigenetics in acute
kidney injury. Curr Opin Nephrol Hypertens. 24:351–358.
2015.PubMed/NCBI
|
|
14
|
Wang BY, Zhong Y, Zhao Z and Miao Y:
Epigenetic suppression of hippocampal BDNF mediates the memory
deficiency induced by amyloid fibrils. Pharmacol Biochem Behav.
126:83–89. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xie Z, Dong Y, Maeda U, Alfille P, Culley
DJ, Crosby G and Tanzi RE: The common inhalation anesthetic
isoflurane induces apoptosis and increases amyloid beta protein
levels. Anesthesiology. 104:988–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang S, Hu X, Guan W, Luan L, Li B, Tang
Q and Fan H: Isoflurane anesthesia promotes cognitive impairment by
inducing expression of β-amyloid protein-related factors in the
hippocampus of aged rats. PLoS One. 12:e01756542017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Evered L, Scott DA and Silbert B:
Cognitive decline associated with anesthesia and surgery in the
elderly: Does this contribute to dementia prevalence? Curr Opin
Psychiatry. 30:220–226. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bilotta F, Qeva E and Matot I: Anesthesia
and cognitive disorders: A systematic review of the clinical
evidence. Expert Rev Neurother. 16:1311–1320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu J, Bie B and Naguib M: Epigenetic
manipulation of brain-derived neurotrophic factor improves memory
deficiency induced by neonatal anesthesia in rats. Anesthesiology.
124:624–640. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ryu YK, Khan S, Smith SC and Mintz CD:
Isoflurane impairs the capacity of astrocytes to support neuronal
development in a mouse dissociated coculture model. J Neurosurg
Anesthesiol. 26:363–368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu YJ, Wang DY, Yang YJ and Lei WF:
Effects and mechanism of dexmedetomidine on neuronal cell injury
induced by hypoxia-ischemia. BMC Anesthesiol. 17:1172017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen M, Wang S, Wen X, Han XR, Wang YJ,
Zhou XM, Zhang MH, Wu DM, Lu J and Zheng YL: Dexmedetomidine exerts
neuroprotective effect via the activation of the PI3K/Akt/mTOR
signaling pathway in rats with traumatic brain injury. Biomed
Pharmacother. 95:885–893. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou C, Zhu Y, Liu Z and Ruan L: Effect of
dexmedetomidine on postoperative cognitive dysfunction in elderly
patients after general anaesthesia: A meta-analysis. J Int Med Res.
44:1182–1190. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hsing CH, Lin CF, So E, Sun DP, Chen TC,
Li CF and Yeh CH: α2-Adrenoceptor agonist dexmedetomidine protects
septic acute kidney injury through increasing BMP-7 and inhibiting
HDAC2 and HDAC5. Am J Physiol Renal Physiol. 303:F1443–F1453. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dong D, Mao Y, Huang C, Jiao Q, Pan H, Ma
L and Wang R: Astrocytes mediated the nootropic and neurotrophic
effects of Sarsasapogenin-AA13 via upregulating brain-derived
neurotrophic factor. Am J Transl Res. 9:4015–4025. 2017.PubMed/NCBI
|
|
26
|
Institute for laboratory animal research:
Guide for the care and use of laboratory animals. 8th. Washington
(DC): National Academies Press: 2011
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koseoglu MM, Ozdilek BA, Djakbarova U and
Gulusur A: Targeting ras activity prevented amyloid beta-induced
aberrant neuronal cell cycle re-entry and death. Curr Alzheimer
Res. 13:1267–1276. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suganthy N, Malar DS and Devi KP:
Rhizophora mucronata attenuates beta-amyloid induced
cognitive dysfunction, oxidative stress and cholinergic deficit in
Alzheimer's disease animal model. Metab Brain Dis. 31:937–949.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hettiarachchi NT, Boyle JP, Dallas ML,
Al-Owais MM, Scragg JL and Peers C: Heme oxygenase-1 derived carbon
monoxide suppresses Aβ1-42 toxicity in astrocytes. Cell Death Dis.
8:e28842017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chay KO, Nam Koong KY, Hwang S, Kim JK and
Bae CS: NADPH oxidase mediates β-amyloid peptide-induced neuronal
death in mouse cortical cultures. Chonnam Med J. 53:196–202. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oguchi T, Ono R, Tsuji M, Shozawa H, Somei
M, Inagaki M, Mori Y, Yasumoto T, Ono K and Kiuchi Y: Cilostazol
suppresses Aβ-induced neurotoxicity in SH-SY5Y cells through
inhibition of oxidative stress and MAPK signaling pathway. Front
Aging Neurosci. 9:3372017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro
M, Iradi A, Obrador E, Mauricio MD, Vila JM, Marchio P and Valles
SL: WIN 55,212-2, agonist of cannabinoid receptors, prevents
amyloid β1-42 effects on astrocytes in primary culture. PLoS One.
10:e01228432015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baglietto-Vargas D, Sánchez-Mejias E,
Navarro V, Jimenez S, Trujillo-Estrada L, Gómez-Arboledas A,
Sánchez-Mico M, Sánchez-Varo R, Vizuete M, Dávila JC, et al: Dual
roles of Aβ in proliferative processes in an amyloidogenic model of
Alzheimer's disease. Sci Rep. 7:100852017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ohki EC, Langan TJ, Rodgers KR and Chou
RC: Non-aggregated Aβ25-35 upregulates primary astrocyte
proliferation in vitro. Front Cell Neurosci. 11:3012017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamanaka D, Kawano T, Nishigaki A, Aoyama
B, Tateiwa H, Shigematsu-Locatelli M, Locatelli FM and Yokoyama M:
Preventive effects of dexmedetomidine on the development of
cognitive dysfunction following systemic inflammation in aged rats.
J Anesth. 31:25–35. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen SD, Wu CL, Hwang WC and Yang DI: More
Insight into BDNF against Neurodegeneration: Anti-apoptosis,
anti-oxidation, and suppression of autophagy. Int J Mol Sci.
18(pii): E5452017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Uchida K, Nakajima H, Hirai T, Yayama T,
Chen K, Guerrero AR, Johnson WE and Baba H: The retrograde delivery
of adenovirus vector carrying the gene for brain-derived
neurotrophic factor protects neurons and oligodendrocytes from
apoptosis in the chronically compressed spinal cord of twy/twy
mice. Spine (Phila Pa 1976). 37:2125–2135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schizas N, König N, Andersson B,
Vasylovska S, Hoeber J, Kozlova EN and Hailer NP: Neural crest stem
cells protect spinal cord neurons from excitotoxic damage and
inhibit glial activation by secretion of brain-derived neurotrophic
factor. Cell Tissue Res. 372:493–505. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Spagnuolo MS, Donizetti A, Iannotta L,
Aliperti V, Cupidi C, Bruni AC and Cigliano L: Brain-derived
neurotrophic factor modulates cholesterol homeostasis and
Apolipoprotein E synthesis in human cell models of astrocytes and
neurons. J Cell Physiol. 11–Jan;2018.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tapia-Arancibia L, Aliaga E, Silhol M and
Arancibia S: New insights into brain BDNF function in normal aging
and Alzheimer disease. Brain Res Rev. 59:201–220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rohe M, Synowitz M, Glass R, Paul SM,
Nykjaer A and Willnow TE: Brain-derived neurotrophic factor reduces
amyloidogenic processing through control of SORLA gene expression.
J Neurosci. 29:15472–15478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Arancibia S, Silhol M, Mouliere F, Meffre
J, Höllinger I, Maurice T and Tapia-Arancibia L: Protective effect
of BDNF against beta-amyloid induced neurotoxicity in vitro and in
vivo in rats. Neurobiol Dis. 31:316–326. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Peng S, Garzon DJ, Marchese M, Klein W,
Ginsberg SD, Francis BM, Mount HT, Mufson EJ, Salehi A and
Fahnestock M: Decreased brain-derived neurotrophic factor depends
on amyloid aggregation state in transgenic mouse models of
Alzheimer's disease. J Neurosci. 29:9321–9329. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Poon WW, Blurton-Jones M, Tu CH, Feinberg
LM, Chabrier MA, Harris JW, Jeon NL and Cotman CW: β-Amyloid
impairs axonal BDNF retrograde trafficking. Neurobiol Aging.
32:821–833. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang R, Wang Z, Howson PA, Xia Z, Zhou S,
Wu E, Xia Z and Hu Y: Smilagenin attenuates beta amyloid
(25–35)-induced degeneration of neuronal cells via stimulating the
gene expression of brain-derived neurotrophic factor. Neuroscience.
210:275–285. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Takamatsu G, Katagiri C, Tomoyuki T,
Shimizu-Okabe C, Nakamura W, Nakamura-Higa M, Hayakawa T,
Wakabayashi S, Kondo T, Takayama C and Matsushita M: Tescalcin is a
potential target of class I histone deacetylase inhibitors in
neurons. Biochem Biophys Res Commun. 482:1327–1333. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tang Y, Lin YH, Ni HY, Dong J, Yuan HJ,
Zhang Y, Liang HY, Yao MC, Zhou QG, Wu HY, et al: Inhibiting
histone deacetylase 2 (HDAC2) promotes functional recovery from
stroke. J Am Heart Assoc. 6(pii): e0072362017.PubMed/NCBI
|
|
49
|
Fukui T, Asakura K, Hikichi C, Ishikawa T,
Murai R, Hirota S, Murate K, Kizawa M, Ueda A, Ito S and Mutoh T:
Histone deacetylase inhibitor attenuates neurotoxicity of
clioquinol in PC12 cells. Toxicology. 331:112–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Asaoka N, Nagayasu K, Nishitani N,
Yamashiro M, Shirakawa H, Nakagawa T and Kaneko S: Inhibition of
histone deacetylases enhances the function of serotoninergic
neurons in organotypic raphe slice cultures. Neurosci Lett.
593:72–77. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Suo H, Wang P, Tong J, Cai L, Liu J, Huang
D, Huang L, Wang Z, Huang Y, Xu J, et al: NRSF is an essential
mediator for the neuroprotection of trichostatin A in the MPTP
mouse model of Parkinson's disease. Neuropharmacology. 99:67–78.
2015. View Article : Google Scholar : PubMed/NCBI
|