Introduction
Osteopetrosis is a rare, heritable bone disease that
is characterized by increased bone density due to failure of bone
resorption by osteoclasts (1).
Osteopetrosis is also known as marble bone or Albers-Schönberg
disease. This disease was first reported in 1904 by the German
radiologist Heinrich Albers-Schönberg (1,2).
Osteopetrosis is currently considered a monogenic disease and is
transmitted through various inheritance patterns, including
autosomal recessive, autosomal dominant or X-linked (2–4). On
the basis of the aforementioned inheritance patterns, researchers
have described several distinct types of osteopetrosis, including
infantile malignant autosomal recessive osteopetrosis (ARO),
intermediate osteopetrosis (IO), which can be either recessive or
dominant, and autosomal dominant osteopetrosis (ADO) (1,2,5).
ARO is the most severe form of the disease, whereas
ADO is considered the mildest form. Individuals with ADO usually
exhibit mild symptoms, including high bone density, increased bone
fracture, scoliosis, arthritis and osteomyelitis. These phenotypes
become evident during late childhood or adulthood (1,5,6). ADO
can be further subdivided into two forms, including ADO-I, which is
caused by a low-density lipoprotein receptor-related protein 5
mutation (7) and ADO-II, which is
caused by a chloride voltage-gated channel 7 (CLCN7)
mutation (3). Individuals with ARO
exhibit apparent phenotypes in early infancy, including frequent
bone fracture, compression of cranial nerves and bone marrow
failure, thereby leading to severe anemia and life-threatening
events, hepatosplenomegaly, brain abnormalities, epilepsy and
developmental delay (1,5,8). IAO
is an intermediate form of osteopetrosis. Several cases of IAO have
been reported (9) and IAO can be
transmitted through autosomal dominant and autosomal recessive
inheritance (2,9,10).
The symptoms for IAO that occur during childhood include risk of
fracture, mild anemia, renal tubular acidosis and calcification
(9,10).
At least 10 disease-causing genes have been
identified to date, which account for 70% of total osteopetrosis
cases (1,2). Among these genes, CLCN7 and T
cell immune regulator 1 (TCIRG1) are the most frequently
mutated genes in human osteopetrosis (11). Monogenic mutations of CLCN7
can cause recessive IAO (10),
ADO-II (12) and ARO (13), whereas monogenic mutations of
TCIRG1 can cause recessive IAO (4) and ARO (14).
In the present study, exome sequencing, Sanger
sequencing and microsatellite marker mapping were performed on a
family with ADO-II. The results revealed two novel heterozygous
mutations in CLCN7 and TCIRG1. These data indicated
the digenic inheritance of osteopetrosis and highlighted the
importance of next-generation sequencing to diagnose human
osteopetrosis.
Materials and methods
Ethics statement
The present study was approved by the Ethics
Committee of The Hunan Children's Hospital (Changsha, China). The
study was carried out according to the principles of the 2008
Edition of the Declaration of Helsinki. Prior to participating in
the study, written informed consent was obtained from all subjects
or guardians of children.
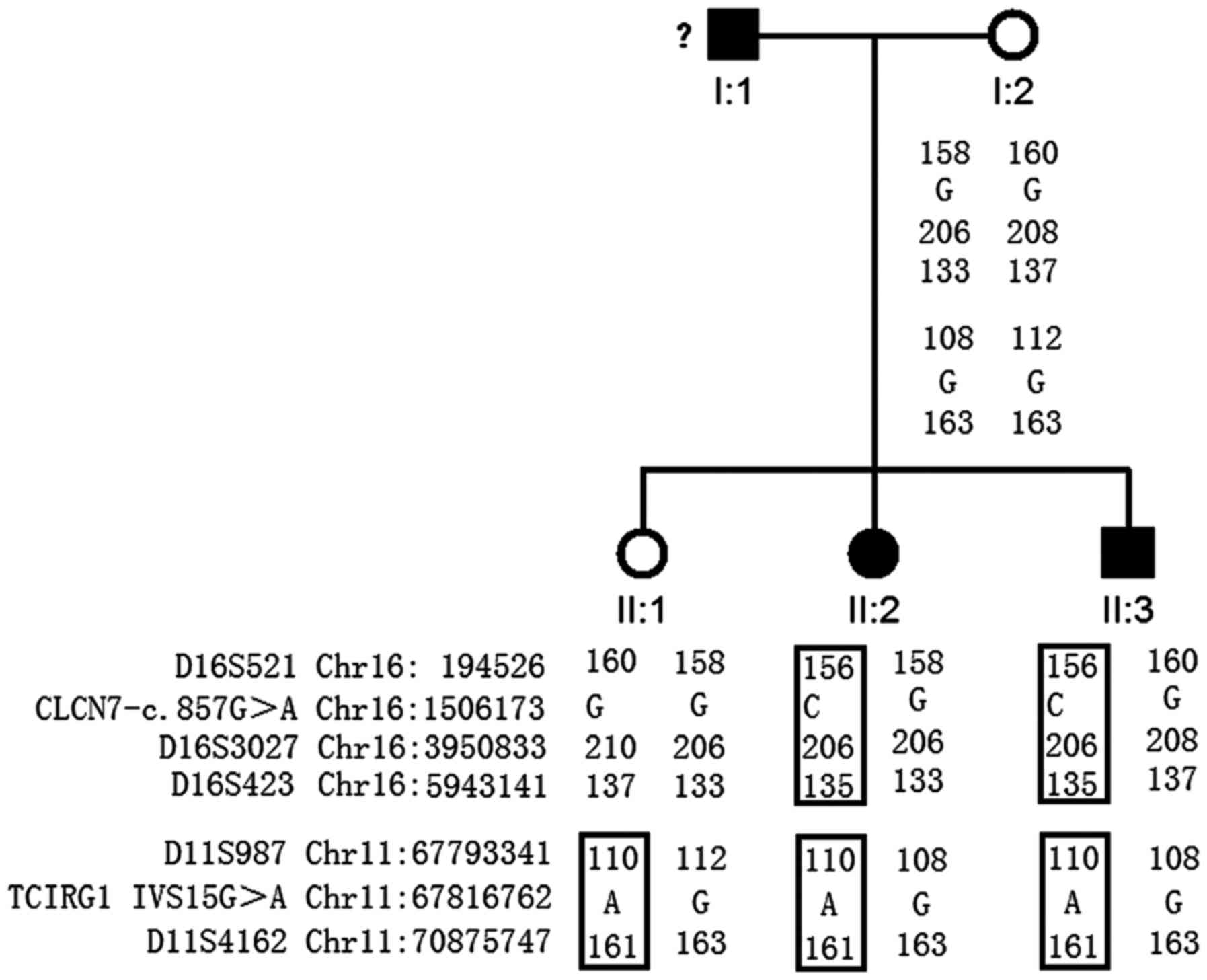
Study subjects
A Han Chinese family residing in the rural areas of
Hunan province with osteopetrosis was recruited for this study
(Fig. 1). In addition, 496 ethnic-
and region-matched controls were included. These controls were
selected from patients (10–16 years old, 263 males and 233 females)
who came to Hunan Children's Hospital for GTG banding between June
2016 and July 2017; individuals with skeletal disorders were
excluded. The family comprised five individuals, including two
affected siblings (II:2 and II:3), an unaffected mother (I:2) and
an unaffected sibling (II:1; Fig.
1). The father (I:1) did not participate in the study. For each
participant, genomic DNA was extracted from peripheral blood (2–5
ml in sodium heparin tubes) using a DNA isolation kit (cat. no.
D3392-02; Omega Bio-Tek, Inc., Norcross, GA, USA) according to the
manufacturer's instructions.
Whole exome sequencing
Briefly, 3 µg genomic DNA from II:2 and II:3 was
fragmented with a bath sonicator (duty cycle, 10%; intensity, 5;
cycles per burst, 200; 3 min for 25°C; Covaris, Inc., Woburn, MA,
USA) and prepared following Illumina Genomic DNA Sample Prep kit
(cat. no. FC-102-1001; Illumina, Inc., San Diego, CA, USA)
according to the manufacturer's protocol, to generate
adapter-ligated DNA library. The prepared DNA was amplified using
ligation-mediated polymerase chain reaction. Phusion Master Mix.
The following thermocycling conditions were used: 98°C for 30 sec,
followed by 12 cycles of 98°C for 10 sec, 65°C for 30 sec and 72°C
for 30 sec; and 72°C for 5 min. Next, DNA was captured using
NimbleGen 2.1M HD array (Roche Applied Science, Madison, WI, USA)
according to the manufacturer's protocol. The captured library was
sequenced on a HiSeq™ 2000 Sequencing system (2×90 bp end reads;
Illumina, Inc.) and the Illumina base calling softwareCASAVA
(version 1.7) was used to convert raw bcf files to fastq file with
default parameters. After trimming the low quality bases (Phred
score <20), the obtained clean reads were mapped to the
reference human genome (hg19; genome.ucsc.edu) using the Burrows-Wheeler Aligner
(version 0.7.2; bio-bwa.sourceforge.net) with up to two mismatches.
The alignment files (.bam) were generated using SAMtools
(samtools.sourceforge.net) and reads with
low mapping quality (<Q30) were filtered out. Duplicate reads
that may be derived from PCR artifacts were removed with Picard
(broadinstitute.github.io/picard/) using default parameters. Short
read alignment and annotation visualization were performed using
the Integrative Genomics Viewer (software.broadinstitute.org/software/igv/). The
percentage alignment of the clean reads to the exome regions was
obtained using custom Perl scripts on the basis of the alignment
files. Single nucleotide variants (SNVs), as well as short
insertions and deletions (indels), were detected by Genome Analysis
Toolkit (software.broadinstitute.org/gatk/). Comprehensive
annotations of all of the detected SNVs and indels were performed
by ANNOVAR (annovar.openbioinformatics.org/en/latest/). The
annotations included function implication (gene region, functional
effect, mRNA GenBank accession number, amino acid change, cytoband
etc.), and allele frequencies in dbSNP (ncbi.nlm.nih.gov/projects/SNP/), 1000 Genomes
(1000genomes.org), ESP6500 (evs.gs.washington.edu/EVS/) and ExAc (exac.broadinstitute.org/). Damaging missense
mutations were predicted by SIFT (sift.bii.a-star.edu.sg/), PolyPhen-2 (genetics.bwh.harvard.edu/pph2/) and
MutationTaster (mutationtaster.org/).
Sanger sequencing
To validate the possible disease-causing variants,
family gDNA underwent PCR using the Goldstar® PCR kit
(cat. no. CW0655M; Jiangsu Kangwei Century Biotechnology Co., Ltd.,
Beijing, China) and Sanger sequencing. The following primers were
used: CLCN7, forward 5′-GACTCGGTTGTCCTGAAAGC-3′, reverse
5′-GGCTGGACCCAAGTACACTG-3′ (predicted size, 371 bp); and
TCIRG1, forward, 5′-CAGTGAGCACACCTCCCTCT-3′, and reverse,
5′-GTGAGGGTCTGCCTCCATAA-3′ (predicted size, 472 bp). The following
PCR conditions were used: 94°C for 5 min; followed by 35 cycles of
94°C for 30 sec, 60°C for 30 sec and 72°C for 40 sec; and a final
extension at 72°C for 5 min. The BigDye® Terminator v3.1
Cycle Sequencing kit (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) was used for the Sanger sequencing
according to manufacturers protocol. The amplified PCR products
were purified by 70% ethanol and then were run on an Applied
Biosystems™ 3500 Series Genetic Analyzer (Applied
Biosystems).
Microsatellite marker analysis
Five informative microsatellite markers, namely
D16S521, D16S3027, D16S423, D11S987 and D11S4162, were used for
segregation analysis. This genotyping method was described
previously (15). A total of 0.8
µl PCR products, 0.2 µl GeneScan™ 500 LIZ™ dye Size Standard
(Thermo Fisher Scientific, Inc.) and 9 µl Hi-Di Formanmide (Thermo
Fisher Scientific, Inc.) were detected on an Applied
Biosystems™ 3500 Series Genetic Analyzer (Applied
Biosystems). Cyrillic program (version 2; www.apbenson.com/cyrillic-downloads) was used for
constructing haplotypes.
Validation of TCIRG1 splicing mutation
on the mRNA level
Total RNA was extracted from peripheral blood
mononuclear cells obtained from patient II:3 and control I:2 using
the RNAiso Plus kit (Takara Biotechnology Co., Ltd., Dalian,
China). cDNA was synthesized from 1 µg RNA using PrimeScript™ II
1st Strand cDNA Synthesis kit (Takara Biotechnology Co., Ltd.)
according to the manufacturer's protocol. PCR was performed using
cDNA as a template with primers specific for exons 13–14, forward,
5′-CTATTTGGAGCCTGGCTGCC-3′ and exon 15, reverse,
5′-CGTCAGGCAGGTCCAGCAACC-3′ (predicted size, 497 bp). The amplified
PCR products were run on a 1% agarose gel (100 V; 40 min). The
forward primer was used as the sequencing primer. The relevant PCR
and Sanger sequencing conditions are the same as those mentioned
above.
Results
Clinical data
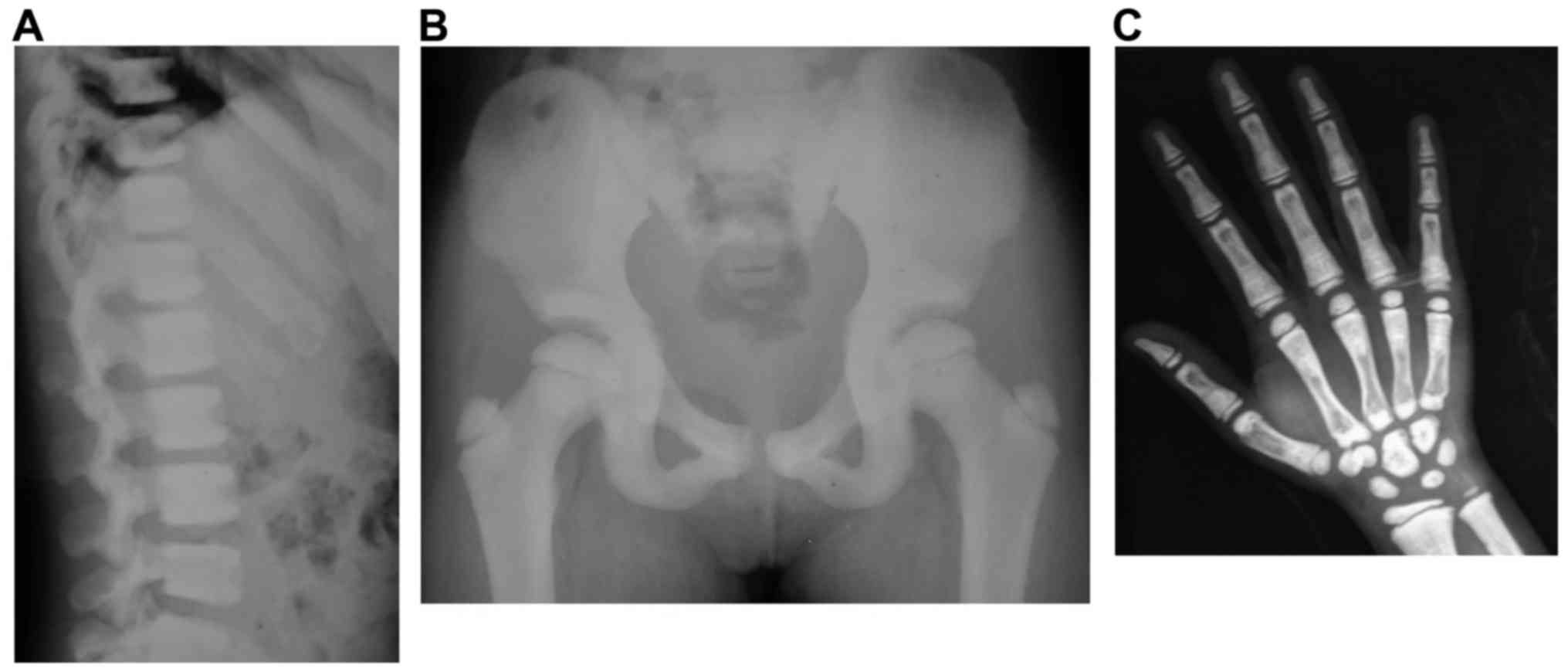
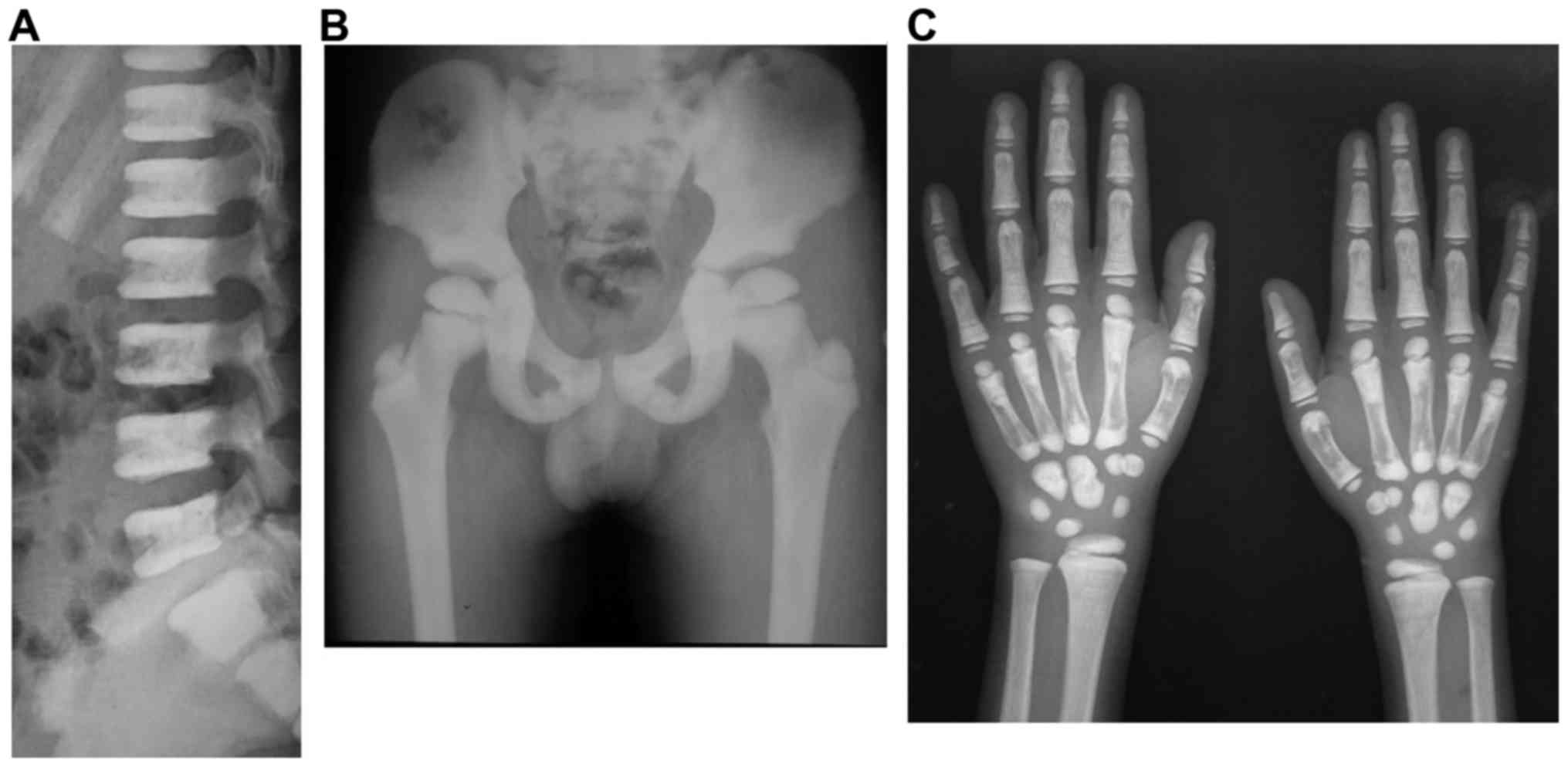
The X-ray results revealed that the two patients,
II:2 and II:3, exhibited ‘sandwich vertebrae’ and
‘bone-within-bone’ appearance (Figs.
2 and 3). II:2 had two
fractures, whereas II:3 had no fractures. The laboratory test
results revealed that II:3 exhibited mild anemia (Table I). The symptoms of II:2 and II:3
were consistent with those of ADO-II. The father, I:1, did not
participate in the study; however, as described by I:2, I:1 had two
fractures during childhood. Therefore, I:1 was likely also a
patient with osteopetrosis.
 | Table I.Laboratory test results for
II:2.a |
Table I.
Laboratory test results for
II:2.a
| Variable | II:2 | Normal range |
|---|
| Red-cell count
(1012)/l | 4.05 (↓) | 4.09–5.74 |
| Hemoglobin
(g/l) | 115 (↓) | 120-140 |
| Hematocrit (%) | 0.334 (↓) | 0.38–0.51 |
| Mean corpuscular
volume (fl/cell) | 82.5 (↓) | 86-100 |
Genetic findings
Whole exome sequencing was successfully performed on
II:2 and II:3. Firstly, common variants present in public and
in-house databases were filtered out. Given that II:2 and II:3
exhibited ‘bone-within-bone’ phenotypes, genes involved in
osteopetrosis were investigated (3). Two heterozygous variants,
chr11-67816762 G>A and chr16-1506173 G>C, were detected in
these two patients among 10 osteopetrosis genes (data not shown).
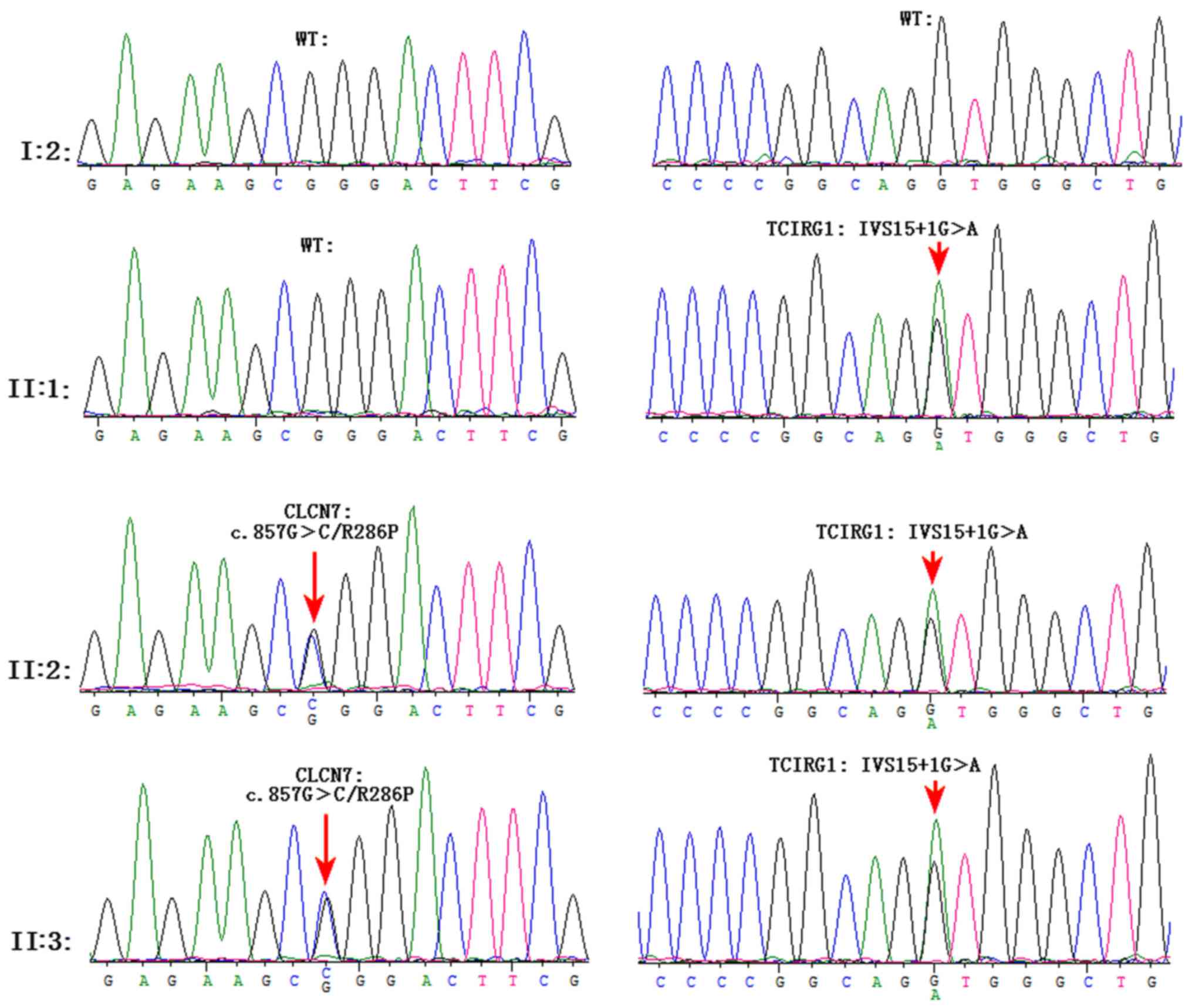
The Sanger sequencing results confirmed the two mutations (Fig. 4).
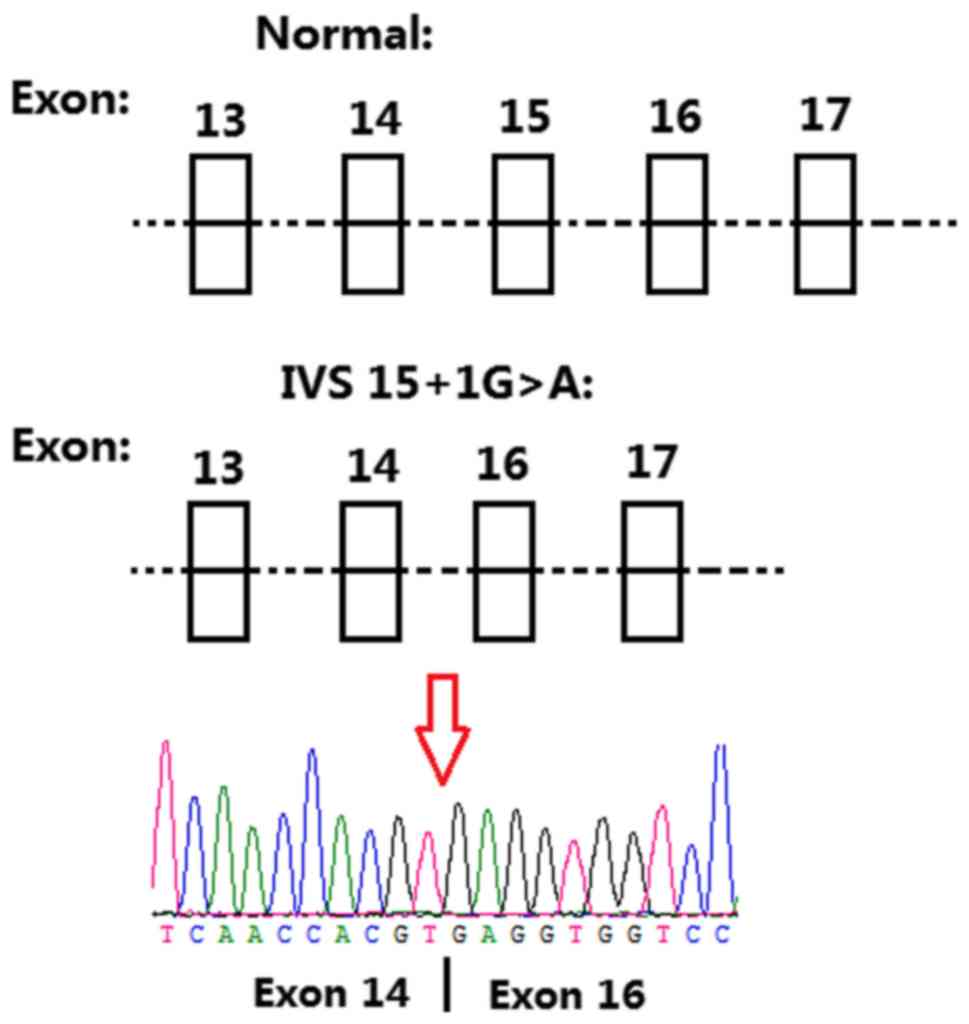
Variant chr11-67816762 G>A (IVS15+1G>A)
occurred on intron 15 (the splicing site of exon 15) in
TCIRG1 (NCBI reference sequence: NM_006019). In
silico analysis (Mutation taster, http://www.mutationtaster.org/) predicted that the
splice acceptor site was lost. Total RNA was extracted from whole
blood, cDNA was synthesized and amplification was performed with
the primers flanking exon 15 of TCIRG1. For patient II:3,
two bands at 497 and 283 bp, were visualized on agarose gel. For
control I:2, only one band was detected at 497 bp. Skipping of the
whole exon 15 was confirmed by PCR and validated by Sanger
sequencing (Fig. 5). Accordingly,
the mutated transcript led to a frameshift mutation from the amino
acid V558, which truncated the mutated protein at amino acid L614.
Variant chr16-1506173 G>C was a c.857G>C missense mutation in
exon 10 of CLCN7 (NCBI reference sequence: NM_001287;
Fig. 4). The c.857G>C missense
mutation changed codon 286 from CGG to CCG. This resulted in the
substitution of arginine by proline (p.R286P). The R286 position
was highly conserved (Fig. 6). Two
known CLCN7-disease mutations on the R286 position, p.R286W
and p.R286Q, have previously been reported (Table II).
| Table II.Genotype and phenotype of several
reported cases of osteopetrosis. |
Table II.
Genotype and phenotype of several
reported cases of osteopetrosis.
| Studies | Mutation | Phenotype | (Refs.) |
|---|
| Cleiren et
al (2001) |
CLCN7-p.R286W | ADO-II | (12) |
| Frattini et
al (2003) |
CLCN7-p.R286Q | ADO-II | (21) |
| Present study |
CLCN7-p.R286P; TCIRG1-IVS
15+1G>A | ADO-II | Present study |
| Yu et al
(2014) |
CLCN7-IVS20-1G>T;
TCIRG1-Y303X; TCIRG1-R670X | ARO | (22) |
Sanger sequencing was conducted to analyze the
distribution of the two mutations in the family. I:2 carried none
of these mutations, II:1 only carried the TCIRG1 (IVS15+1
G>A) mutation, and II:2 and II:3 carried both mutations
(Fig. 1). Furthermore, the two
mutations, TCIRG1-IVS15+1G>A and
CLCN7-c.857G>A, were undetected in the 496 region and
ethnic-matched controls (data not shown).
Microsatellite marker analysis was performed in the
family, of which five markers were informative. The haplotype
analysis results confirmed that the two mutations were not
transmitted from the unaffected mother (Fig. 1).
Discussion
Monogenic mutations of CLCN7 and
TCIRG1 have previously been fully documented as the cause of
human osteopetrosis (2); however,
osteopetrosis is a heterogeneous disease (16). Human osteopetrosis exhibits high
phenotypic and genotypic diversity (3,17,18);
for example, individuals with a CLCN7 mutation can be
asymptomatic or symptomatic, with symptoms ranging from very mild
to severe (3,17,18).
This is consistent with the fact that the penetrance of human
osteopetrosis is not complete (19). Although several studies have
suggested the presence of modifier genes for human osteopetrosis,
including a modifier gene on chromosome 9q21-9q22 (16,20),
the precise mechanism of the phenotypic diversities of human
osteopetrosis requires further investigation.
To the best of our knowledge, the present study was
the first to describe two heterogeneous mutations occurring in
CLCN7 and TCIRG1 in a single family with
osteopetrosis. It has been proposed that both mutations are
disease-causing mutations due to the following characteristics: i)
The TCIRG1 mutation (IVS15+1G>A) was a novel splicing
mutation, which has not been previously reported, and the splice
acceptor site was lost as predicted by MutationTaster. In addition,
detection of mRNA expression indicated that the whole exon 15 of
TCIRG1 was skipped, and the predicted protein was truncated.
ii) The CLCN7 mutation (p.R286P) was also a novel missense
mutation, and MutationTaster also predicted that CLCN7 was a
disease-causing mutation. In addition, the R286 position of
CLCN7 was highly conserved among various species and two
known disease mutations on the R286 position, p.R286W and p.R286Q,
have previously been reported (14,21).
The present study proposed the possibility of
digenic inheritance of osteopetrosis, where CLCN7-p.R286P
combined with TCIRG1-IVS 15+1G>A cause ADO-II. This
proposition is supported by a previous study; Yu et al
(22) described an ARO case with
three mutations, including two compound heterozygous mutations in
TCIRG1, p.Y303X and p.R670X, and one splicing mutation in
CLCN7 (IVS 20-1G>T). For the ARO case (22), TCIRG1-p.Y303X and
CLCN7 IVS 20-1G>T mutations were passed on from the
asymptomatic mother of the patient, and the TCIRG1-p.R670X
mutation was transmitted from the asymptomatic father of the
patient. In addition, the compound heterozygous nonsense mutations
of TCIRG1 were sufficient to cause ARO; however, it may not
be a coincidence to have mutations on two rare pathogenic genes.
This condition may be due to the fact that the mutations of
TCIRG1 and CLCN7 interacting with each other in the
two mutations exert a digenic inheritance (22).
In conclusion, the present study identified two
novel gene mutations in TCIRG1 and CLCN7 in a single
family, thereby suggesting the possibility of digenic inheritance
of osteopetrosis. This study also highlighted the importance of
next-generation sequencing for comprehensive molecular diagnosis of
osteopetrosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 31501017).
Availability of data and materials
The dataset used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YY and YZ planned the project, analyzed the whole
exome sequencing data and wrote the manuscript. YY and LL conceived
and designed the study. WY, JG, LZ, MT and YY carried out sample
collection, clinical diagnosis, Sanger sequencing and
microsatellite marker mapping. All authors critically revised,
reviewed and gave final approval of this manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of The Hunan Children's Hospital (Changsha, China). Prior
to participating in the study, all subjects and guardians of
children provided written informed consent.
Patient consent for publication
Informed consent was obtained from all subjects or
their guardians for the publication of patient data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sobacchi C, Schulz A, Coxon FP, Villa A
and Helfrich MH: Osteopetrosis: Genetics, treatment and new
insights into osteoclast function. Nat Rev Endocrinol. 9:522–536.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stark Z and Savarirayan R: Osteopetrosis.
Orphanet J Rare Dis. 4:52009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deng H, He D, Rong P, Xu H, Yuan L, Li L,
Lu Q and Guo Y: Novel CLCN7 mutation identified in a Han Chinese
family with autosomal dominant osteopetrosis-2. Mol Pain.
12:17448069166526282016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Palagano E, Blair HC, Pangrazio A,
Tourkova I, Strina D, Angius A, Cuccuru G, Oppo M, Uva P, Van Hul
W, et al: Buried in the middle but guilty: Intronic mutations in
the TCIRG1 gene cause human autosomal recessive osteopetrosis. J
Bone Miner Res. 30:1814–1821. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Del Fattore A, Cappariello A and Teti A:
Genetics, pathogenesis and complications of osteopetrosis. Bone.
42:19–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Waguespack SG, Koller DL, White KE,
Fishburn T, Carn G, Buckwalter KA, Johnson M, Kocisko M, Evans WE,
Foroud T and Econs MJ: Chloride channel 7 (ClCN7) gene mutations
and autosomal dominant osteopetrosis, type II. J Bone Miner Res.
18:1513–1518. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Van Wesenbeeck L, Cleiren E, Gram J, Beals
RK, Bénichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y,
et al: Six novel missense mutations in the LDL receptor-related
protein 5 (LRP5) gene in different conditions with an increased
bone density. Am J Hum Genet. 272:763–771. 2003. View Article : Google Scholar
|
|
8
|
Kornak U, Kasper D, Bösl MR, Kaiser E,
Schweizer M, Schulz A, Friedrich W, Delling G and Jentsch TJ: Loss
of the ClC-7 chloride channel leads to osteopetrosis in mice and
man. Cell. 104:205–215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pangrazio A, Pusch M, Caldana E, Frattini
A, Lanino E, Tamhankar PM, Phadke S, Lopez AG, Orchard P, Mihci E,
et al: Molecular and clinical heterogeneity in CLCN7-dependent
osteopetrosis: Report of 20 novel mutations. Hum Mutat.
31:E1071–E1080. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Balemans W, Van Wesenbeeck L and Van Hul
W: A clinical and molecular overview of the human osteopetroses.
Calcif Tissue Int. 77:263–274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Del Fattore A, Peruzzi B, Rucci N, Recchia
I, Cappariello A, Longo M, Fortunati D, Ballanti P, Iacobini M,
Luciani M, et al: Clinical, genetic, and cellular analysis of 49
osteopetrotic patients: Implications for diagnosis and treatment. J
Med Genet. 43:315–325. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cleiren E, Bénichou O, Van Hul E, Gram J,
Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama
T, et al: Albers-Schönberg disease (autosomal dominant
osteopetrosis, type II) results from mutations in the ClCN7
chloride channel gene. Hum Mol Genet. 10:2861–2867. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lam CW, Tong SF, Wong K, Luo YF, Tang HY,
Ha SY and Chan MH: DNA-based diagnosis of malignant osteopetrosis
by whole-genome scan using a single-nucleotide polymorphism
microarray: Standardization of molecular investigations of genetic
diseases due to consanguinity. J Hum Genet. 52:98–101. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mazzolari E, Forino C, Razza A, Porta F,
Villa A and Notarangelo LD: A single-center experience in 20
patients with infantile malignant osteopetrosis. Am J Hematol.
84:473–479. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li N, Yang Y, Bu J, Zhao C, Lu S, Zhao J,
Yan L, Cui L, Zheng R, Li J, et al: An autosomal dominant
progressive congenital zonular nuclear cataract linked to
chromosome 20p12.2-p11.23. Mol Vis. 12:1506–1510. 2006.PubMed/NCBI
|
|
16
|
Alam I, Gray AK, Chu K, Ichikawa S,
Mohammad KS, Capannolo M, Capulli M, Maurizi A, Muraca M, Teti A,
et al: Generation of the first autosomal dominant osteopetrosis
type II (ADO2) disease models. Bone. 59:66–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coudert AE, de Vernejoul MC, Muraca M and
Del Fattore A: Osteopetrosis and its relevance for the discovery of
new functions associated with the skeleton. Int J Endocrinol.
2015:3721562015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aggarwal S: Skeletal dysplasias with
increased bone density: Evolution of molecular pathogenesis in the
last century. Gene. 528:41–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang C, Zhang H, He JW, Gu JM, Hu WW, Hu
YQ, Li M, Liu YJ, Fu WZ, Yue H, et al: The virulence gene and
clinical phenotypes of osteopetrosis in the Chinese population: Six
novel mutations of the CLCN7 gene in twelve osteopetrosis families.
J Bone Miner Metab. 30:338–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chu K, Koller DL, Snyder R, Fishburn T,
Lai D, Waguespack SG, Foroud T and Econs MJ: Analysis of variation
in expression of autosomal dominant osteopetrosis type 2: Searching
for modifier genes. Bone. 37:655–661. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Frattini A, Pangrazio A, Susani L,
Sobacchi C, Mirolo M, Abinun M, Andolina M, Flanagan A, Horwitz EM,
Mihci E, et al: Chloride channel ClCN7 mutations are responsible
for severe recessive, dominant, and intermediate osteopetrosis. J
Bone Miner Res. 18:1740–1747. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu T, Yu Y, Wang J, Yin L, Zhou Y, Ying D,
Huang R, Chen H, Wu S, Shen Y, et al: Identification of TCIRG1 and
CLCN7 gene mutations in a patient with autosomal recessive
osteopetrosis. Mol Med Rep. 9:1191–1196. 2014. View Article : Google Scholar : PubMed/NCBI
|