Introduction
UbiA prenyltransferase domain-containing protein 1
(UBIAD1), additionally referred to as transitional epithelial
response gene, was first identified as a tumor suppressor in
bladder cancer (1–3). The UBIAD1 gene is located on
chromosome 1p36 and encodes a protein of 338 amino acids, harboring
the UbiA isopentyl transferase domain (1). Previous studies demonstrated that
UBIAD1 exhibits specific subcellular localization, and is expressed
in the mitochondria (4), Golgi
(5) and endoplasmic reticulum
(6). UBIAD1 is conserved across
different species, including zebrafish and humans (7). UBIAD1 converts menadione to MK-4, a
principal form of vitamin K in humans (6). In addition, UBIAD1 catalyzes the
non-mitochondrial coenzyme Q10 (CoQ10) in zebrafish (5), which serves an important role in
producing endothelial nitric oxide (eNOS) and nitric oxide (NO)
(8). Therefore, UBIAD1 has
critical functions in maintaining cellular homeostasis.
UBIAD1 has been demonstrated to be involved in a
variety of human diseases. For instance, naturally occurring
mutations in the UBIAD1 gene have been causally linked to Schneider
lens corneal dystrophy, a genetic autosomal dominant disease that
is caused by abnormal cholesterol and phospholipid metabolism
(2). Furthermore, UBIAD1 functions
as a modifier of serine/threonine-protein kinase PINK1,
mitochondrial, and this mutation is associated with Parkinson's
disease (9). UBIAD1 may also act
as a tumor suppressor through negatively regulating the Ras-mitogen
activated protein kinase (MAPK) signal transduction pathway
(1,10). At present, a limited number of
studies have elucidated the role of UBIAD1 in cardiovascular
disease. In zebrafish, there is specific evidence that UBIAD1 is
cardioprotective against oxidative stress by mediating CoQ10
synthesis (5), and that a UBIAD1
mutant causes cardiac edema (7).
Whether UBIAD1 is involved in cardiac pathophysiology in mammals is
not yet known.
Cardiac hypertrophy is a principal factor leading to
cardiac muscle disorders. The exact mechanisms of cardiac
hypertrophy remain unclear; however, multifactorial mechanisms,
including genetics and environmental cues, likely serve a role in
pathogenesis. Multiple lines of evidence have suggested that NO is
involved in the pathophysiology of many diseases, including cardiac
hypertrophy (11). For instance,
increased levels of NO have been reported to reduce cardiomyocyte
hypertrophy progression (12).
Similarly, overexpression of eNOS was demonstrated to inhibit
cardiac hypertrophy in mice (13);
however, suppression of eNOS activity promotes cardiac hypertrophy,
as evidenced by eNOS deficient mice (14). In addition, decreased levels of
eNOS are associated with angiotensin II receptor knockout-induced
cardiac hypertrophy (15).
Collectively, these findings suggest the important roles of eNOS-NO
signaling in the pathogenesis and progression of cardiac
hypertrophy.
As mentioned above, UBIAD1 has been associated with
a number of human diseases. However, the role of UBIAD1 in cardiac
hypertrophy has not yet been investigated. Given that UBIAD1 is an
important regulator of eNOS-NO signaling and CoQ10 synthesis, which
significantly contribute to the pathogenesis of cardiovascular
diseases (16), it was
hypothesized that UBIAD1 functions in the development of cardiac
hypertrophy. In the present study, the expression of UBIAD1 and its
downstream signaling molecules was measured in the hearts of
spontaneously hypertensive rats (SHRs) and age-matched control
Wistar-Kyoto (WKY) rats using various molecular approaches. The
findings of the present study offer novel insight into the
mechanisms underlying the development of hypertensive cardiac
hypertrophy, and provide novel insight for the prevention and
treatment of hypertension.
Materials and methods
Reagents
UBIAD1 antibody (cat. no. sc-377013) and GAPDH
antibody (cat. no. sc-32233) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). CoQ10 antibody (cat. no.
17812-1-AP), eNOS antibody (cat. no. 20116-1-AP) were purchased
from Wuhan Sanying Biotechnology (Wuhan, China). Goat-antibody
rabbit secondary antibodies (cat. no. A0216) and goat-antibody
mouse secondary antibodies (cat. no. A0208) were purchased from
Beyotime Institute of Biotechnology (Shanghai, China). Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) and DAB reaction kits were purchased from Beyotime
Institute of Biotechnology (Shanghai, China), and the polymerase
chain reaction (PCR) kits were purchased from Takara Bio, Inc.
(Otsu, Japan).
Animals
A total of 48 eight-week male rats (24 SHR and 24
WKY) were obtained from the Beijing Vital River Laboratory Animal
Technology Co. Ltd. (Beijing, China). At the start of the study,
the rats were randomly divided into groups for three different
ages: 8, 16 and 32 weeks (8 rats/group). After the different
periods of time, subsequent experiments were performed. Rats were
housed in an environment with a temperature of 22–27°C, humidity
50±10% and 12/12 h light/dark cycle. All rats had free access to
water and regular chow. All animal protocols complied with and were
approved by the Ethics Committee of Jinzhou Medical University.
Blood pressure measurement
Blood pressure was measured using a non-invasive
tail cut-off multi-channel blood measurement device (Xinhua
Surgical Instrument Co., Ltd., Zibo, China). Blood pressure
measurements were repeated three times, and the mean value was
taken following the final blood pressure measurement.
Measurement of cardiac function
parameters
Animals were anesthetized with an intraperitoneal
injection of chloral hydrate (300 mg/kg), which was prepared in
sodium chloride to a final concentration of 15%. Following the
induction of anesthesia, rats were placed on a warming pad.
Catheters were inserted into the femoral and subclavian arteries
and connected to the multi-conductive physiological recorder
pressure transducer (ADInstruments Pty Ltd., Sydney, Australia).
Following stabilization, the following cardiac functional
parameters were measured: Maximal increase rate of left ventricular
pressure (+dp/dt max), maximal drop rate of left ventricular
pressure (-dp/dt max), left ventricular end diastolic pressure
(LVEDP), and left ventricular systolic pressure (LVSP). No signs of
peritonitis were observed following anesthesia administration.
Determination of cardiac hypertrophic
index
Rats were sacrificed following cardiac function
measurements, and body weight (BW) and left ventricular weight
(LVW) were subsequently measured. The cardiac hypertrophic index
was defined as the ratio of LVW to BW, which represented the degree
of ventricular hypertrophy.
Hematoxylin and eosin (H&E)
staining
Rat left ventricular myocardium was collected, fixed
in 10% neutral formic acid solution at room temperature for 24–48
h, embedded in wax and cut into 5 µm thick sections. H&E
staining was performed according to a widely used standard
protocol; 0.8% hematoxylin staining for 5 min at room temperature,
and 0.35% eosin staining for 3 min at room temperature. Cardiac
myocardial structures, including myocardial cell morphology and
myocardial fiber arrangement, were viewed under an ordinary light
microscope (Olympus BX53; Camera system DP73; magnification,
×200).
Immunohistochemistry
Cardiac sections were dewaxed using the following
procedure: 60°C for 2 h, followed by two rounds of xylene for 15
min each. Subsequently, sections were placed in the gradient
alcohol and prepared with dewaxed water as follows: In anhydrous
ethanol I and II with for 5 min each, sequentially in 95, 85 and
75% ethanol with for 2 min each. Sections were subsequently placed
in distilled water for 2 min and in PBS for 5 min. Antigen
retrieval was performed by boiling the dewaxed slides in antigen
retrieval buffer (10 mM sodium citrate; PH 6.0) for 10 min,
followed by natural cooling and three washes with PBS (5 min/wash).
To quench the non-specific background signals, slides were
incubated with hydrogen peroxide (H2O2) for 5
min at room temperature, then washed three times with PBS. The
slides were blocked in 1% bovine serum albumin (cat. no. A600903;
Sangon Biotech Co. Ltd., Shanghai, China) at room temperature for 1
h, washed with PBS, and incubated with an anti-UBIAD1 antibody
(1:50 dilution) at 4°C overnight. Thereafter, the slides were
washed with PBS and incubated with horseradish
peroxidase-conjugated donkey anti-goat immunoglobulin G antibody
(cat. no. A0181; Beyotime Institute of Biotechnology; 1:200) for 30
min at 37°C. Staining was visualized with DAB and observed under an
ordinary light microscope (Olympus BX53; Camera system: DP73;
magnification, ×200). In total, five randomly selected fields per
section were scored and quantified using Image-Pro Plus 5.0 image
analysis software (National Institutes of Health, Bethesda, MD,
USA).
TUNEL staining
Myocardial cell apoptosis was assessed by TUNEL
staining according to the protocol provided by the manufacturer
(Beyotime Institute of Biotechnology). The number of apoptotic
cardiomyocytes was scored in five randomly selected fields under a
microscope (magnification, ×400). Apoptotic index was calculated as
the number of apoptotic cells/number of cardiomyocytes ×100.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the left ventricles
with TRIzol® reagent (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturer's protocol.
Purified RNA was dissolved in DEPC water and frozen at −80°C
following the determination of RNA concentration. RNA (1 µg) was
used for the RT reaction in a final volume of 20 µl using
PrimeScript RT Enzyme Mix (Takara Bio, Inc.) at 37°C for 15 min and
85°C for 5 sec. All primers were designed based on gene sequences
using primer 5.0 software (Primer Premier 5.0; Premier Biosoft
International, Palo Alto, CA USA), and the primer sequences used
are listed in Table I. qPCR was
performed using SYBR Premix Ex Taq (Takara Bio, Inc.) in a final
volume of 50 µl on an ABI7500 amplifier. The PCR conditions were as
follows: 95°C for 30 sec, 40 cycles of 95°C for 5 sec, 60°C for 30
sec and 72°C for 30 sec, and 4°C for 5 min. Relative quantification
was performed using the reference gene GAPDH as an internal
control. The relative expression was assessed and converted to fold
changes using the 2−ΔΔCq method (17).
 | Table I.Primer sequences used in the
quantitative polymerase chain reaction. |
Table I.
Primer sequences used in the
quantitative polymerase chain reaction.
| Gene name | Direction | Sequence
(5′-3′) |
|---|
| UBIAD1 | Forward |
AACGACTGTCCCGAGCAA |
|
| Reverse |
CGGCACAACCCACCAA |
| ANF | Forward |
AGCCGAGACAGCAAACA |
|
| Reverse |
GCCTGGGAGCCAAAA |
| CoQ10 | Forward |
GACCATAATGCCTCACC |
|
| Reverse |
ATGCGTTCATCACCAA |
| eNOS | Forward |
GCAGAGGAGTCCAGCGAACA |
|
| Reverse |
TGGGTGCTGAGCTGACAGAGTA |
| GAPDH | Forward |
GAGGCTCTCTTCCAGCCTTC |
|
| Reverse |
AGGGTGTAAAAGCAGCTCA |
Western blotting
Total protein was purified from rat hearts in
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology) and stored at −80°C. Protein concentrations were
determined using a bicinchoninic acid protein assay kit. Proteins
(20 µg/lane) were separated by 10% SDS-PAGE, transferred to
polyvinylidene difluoride membranes and blocked in 5% fat-free dry
milk for 1 h at room temperature. The membrane was subsequently
incubated with UBIAD1 (1:1,000), CoQ10 (1:1,000), Enos (1:1,000)
and GAPDH (1:500) primary antibodies at 4°C overnight, followed by
three washes with Tris-buffered saline with 0.1% Tween and another
incubation with goat-antibody rabbit or goat-anti-mouse horseradish
peroxidase conjugated-secondary antibodies (1:5,000) for 1 h at
room temperature. Protein bands were visualized using an enhanced
chemiluminescence detection system (GE Healthcare Life Sciences,
Shanghai, China).
Determination of serum and myocardial
NO content
Serum samples were collected from animals under
anesthesia. Following collection, the serum was stored at 4°C for 1
h, and subsequently centrifuged at 625 × g at 4°C for 10 min. Serum
and myocardial samples were diluted in PBS to a final concentration
of 2 µg/µl. Samples were boiled for 5 min and centrifuged at 10,005
× g at 4°C for 5 min. Supernatants were collected for NO
measurement (Griess method) using the Total Nitric Oxide
Measurement kit (cat. no. S0023; Beyotime Insitute of
Biotechnology). The standard NO curve was prepared by diluting 10
mM KNO2 to 1, 10, 20, and 50 µmol/l with PBS (pH 7.4),
respectively. Supernatants were incubated with lactate
dehydrogenase (LDH) Buffer and LDH at 37°C for 30 min.
Subsequently, Griess Reagent I and Griess Reagent II were added
directly to the above solution and incubated for 10 min at room
temperature, followed by optical measurement. Optical density was
measured spectrophotometrically at an absorbance of 540 nm as
previously described (18).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Comparisons between two groups were performed with Student's
t-test, and comparisons among multiple groups were performed using
one-way analysis of variance followed by Fisher's Least Significant
Difference test. All statistical analyses were performed with SPSS
17.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
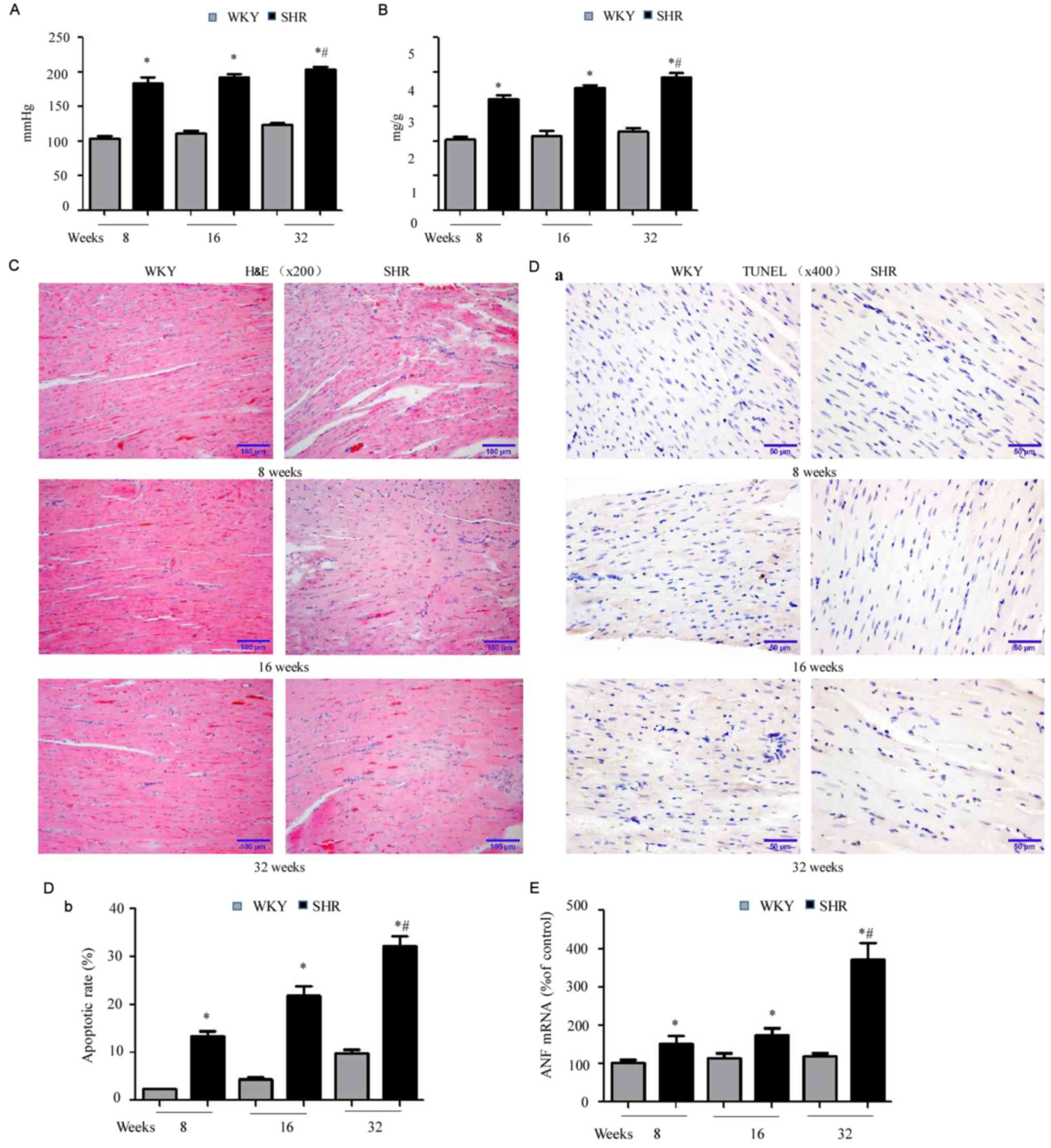
SHRs have higher blood pressure,
increased cardiac hypertrophic index and impaired cardiac
function
Initially, blood pressure and the cardiac
hypertrophic index was compared between SHR and WKY rats at 8, 16
and 28 weeks of age. In every age group, SHRs exhibited higher
blood pressure (Fig. 1A;
*P<0.05 vs. WKY) and increased cardiac hypertrophic index
(Fig. 1B; *P<0.05 vs. WKY).
Blood pressure in SHRs increased with aging (#P<0.05
vs. SHRs at 8 and 16 weeks). In addition, SHRs had a higher LVEDP,
as well as a lower LVSP, +dv/dt and -dv/dt, compared with
age-matched WKY rats (Table II),
indicating cardiac functional impairment in SHRs. Consistent with
the above findings, H&E staining demonstrated disorganized
sarcomeres in the hearts of SHRs, compared with well-organized
myocardial tissue in age-matched WKY rats (Fig. 1C). Subsequently, myocardial cell
apoptosis was assessed with TUNEL staining, which revealed
increased apoptosis in SHR hearts, compared with age-matched WKY
hearts (Fig. 1D-a and -b;
*P<0.05). In addition, increased myocardial cell apoptosis was
observed with increasing age in SHRs (#P<0.05 vs. 8
and 16 weeks). In agreement with the above observations, RT-qPCR
demonstrated significant upregulation of atrial natriuretic factor
(ANF), a cardiac disease marker, in the hearts of SHRs compared
with WKY rats. Furthermore, this upregulation increased with age in
SHRs (Fig. 1E; *P<0.05 vs. WKY;
#P<0.05 vs. 8 and 16 week SHRs). Collectively, these
findings supported previous studies (19,20)
demonstrating that SHRs exhibit age-associated hypertension and
cardiac hypertrophy, as well as impaired cardiac function,
accompanied by disorganized myocardium and increased cell
death.
| Table II.Cardiac function parameters in SHRs
and WKY rats. |
Table II.
Cardiac function parameters in SHRs
and WKY rats.
| A, 8 weeks |
|---|
|
|---|
| Experimental
group | LVSP, mmHg | LVEDP, mmHg | +dv/dt max,
mmHg/msec | -dv/dt max,
mmHg/msec |
|---|
| WKY | 127±5.3 | 2.53±0.2 | 5.32±1.12 | 5.15±1.13 |
| SHR | 97±3.8a |
6.65±0.5a |
4.35±0.72a |
4.12±0.75a |
|
| B, 16
weeks |
|
| Experimental
group | LVSP,
mmHg | LVEDP,
mmHg | +dv/dt max,
mmHg/msec | -dv/dt max,
mmHg/msec |
|
| WKY | 122±4.6 | 2.97±0.5 | 4.95±0.95 | 4.44±0.94 |
| SHR | 91±3.6a |
7.43±0.6a |
3.79±0.68a |
3.38±0.71a |
|
| C, 28
weeks |
|
| Experimental
group | LVSP,
mmHg | LVEDP,
mmHg | +dv/dt max,
mmHg/msec | -dv/dt max,
mmHg/msec |
|
| WKY | 118±3.7 | 3.74±0.8 | 4.56±0.86 | 4.37±0.79 |
| SHR | 86±2.9a,b |
8.37±0.5a,b |
2.56±0.62a,b |
2.32±0.58a,b |
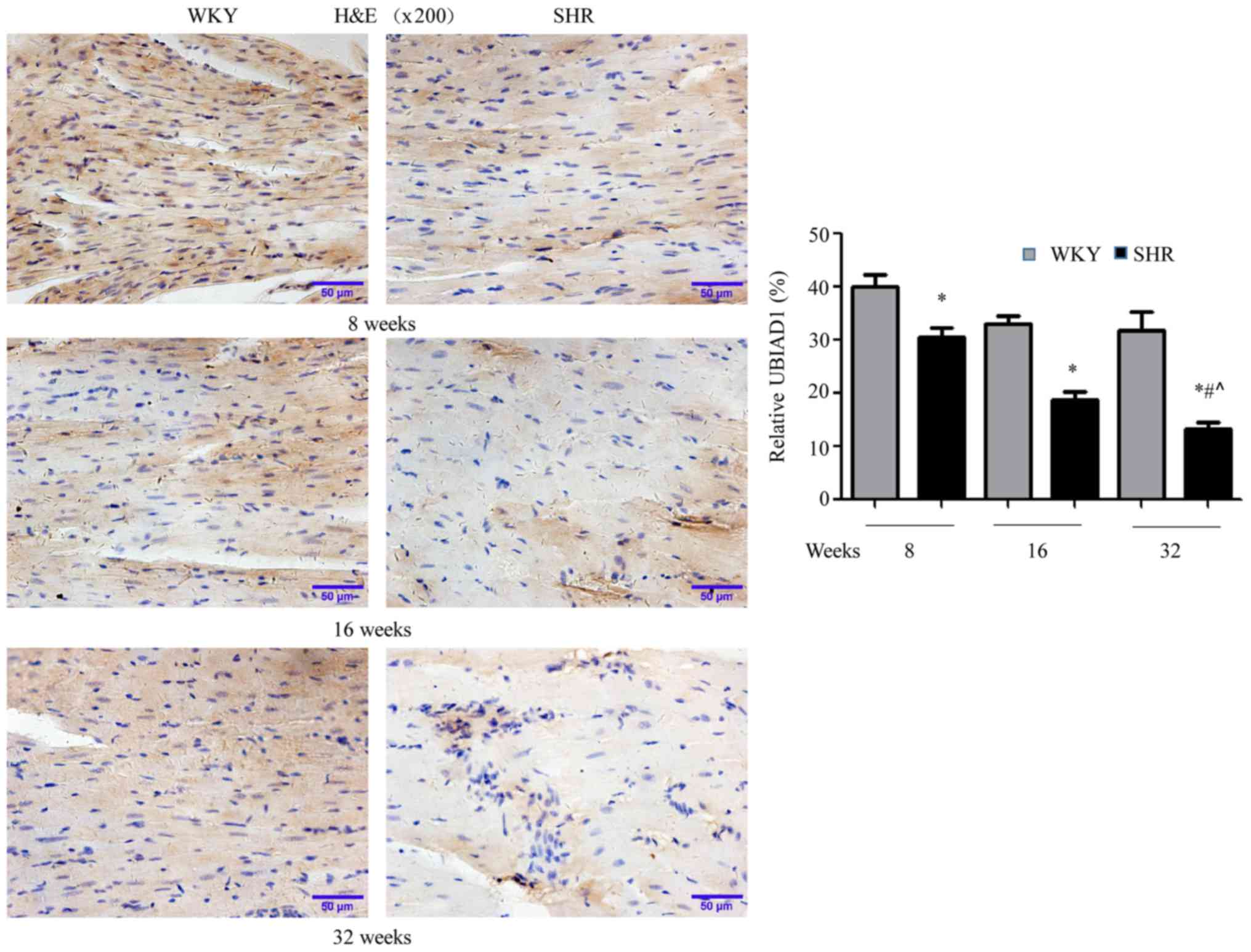
UBIAD1 expression decreases in SHR
hearts
To determine the potential involvement of UBIAD1 in
hypertensive cardiac hypertrophy, the expression of UBIAD1 in the
hearts of SHR and age-matched WKY rats was initially evaluated by
immunohistochemistry. As presented in Fig. 2, UBIAD1 staining was weaker in SHR
hearts compared with WKY hearts, at each age examined (*P<0.05
vs. WKY). Additionally, UBIAD1 expression in SHR hearts decreased
with aging (#P<0.05 vs. SHR at 8 and 16 weeks). These
data demonstrated that UBIAD1 expression is downregulated in SHR
hearts, compared with age-matched WKY hearts, and that this
downregulation increased over time.
UBIAD1, CoQ10 and eNOS expression
decrease in SHR hearts
UBIAD1 has been observed to be involved in mediating
CoQ10 activity, which mediates eNOS expression (5). Therefore, the gene and protein
expression of CoQ10, eNOS and UBIAD1 was measured using RT-qPCR and
western blotting, respectively. In line with the
immunohistochemical findings, UBIAD1 mRNA expression was
downregulated in SHR hearts compared with WKY rat hearts, and this
downregulation was age-dependent in SHRs (Fig. 3A; *P<0.05 vs. WKY;
#P<0.05 vs. SHRs at 8 and 16 weeks). CoQ10 (Fig. 3B) and eNOS (Fig. 3C) expression was significantly
lower in the SHR hearts compared to WKY hearts at 8, 16 and 28
weeks (P<0.05 vs. WKY; #P<0.05 vs. SHRs at 8 and
16 weeks). Furthermore, a similar trend in UBIAD1, CoQ10 and eNOS
protein expression was observed (Fig.
3D). Taken together, these results demonstrated that
downregulated UBIAD1 expression in SHR hearts was associated with
decreased CoQ10 and eNOS expression.
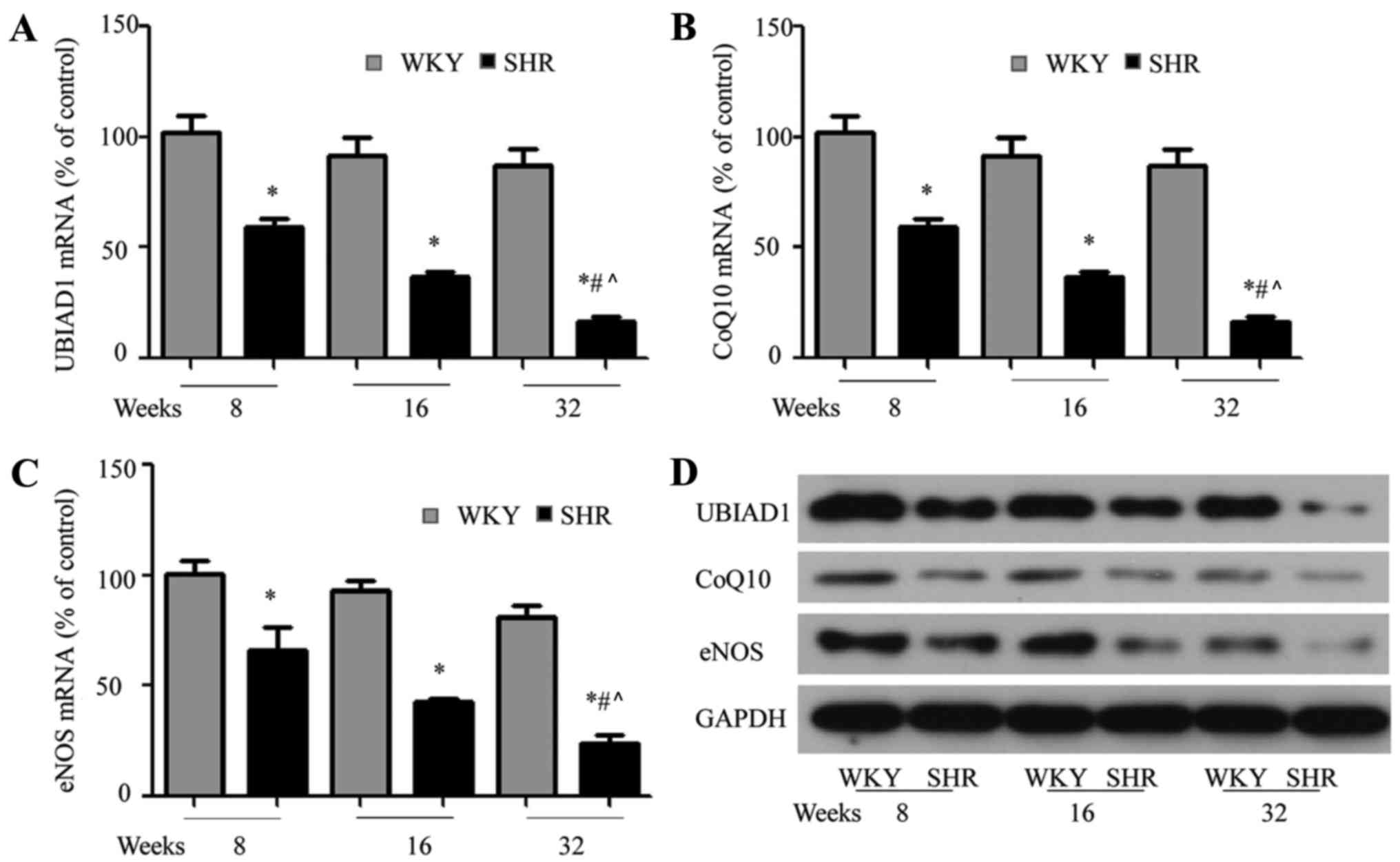 | Figure 3.UBIAD1, CoQ10 and eNOS expression is
decreased in SHR hearts. (A) Quantitative polymerase chain reaction
demonstrated that UBIAD1, (B) CoQ10 and (C) eNOS mRNA expression
was decreased in SHR hearts, compared with age-matched WKY rat
hearts. (D) Western blotting demonstrated that UBIAD1, CoQ10 and
eNOS protein expression was decreased in SHR hearts, compared with
WKY rat hearts. Each experiment was performed in triplicate.
*P<0.05 vs. age matched WKY rats; #P<0.05 vs. SHRs
at 8 weeks; ^P<0.05 vs. SHR at 16 weeks. UBIAD1, UbiA
prenyltransferase containing 1; CoQ10, coenzyme Q10; eNOS,
endothelial nitric oxide synthase; SHRs, spontaneously hypertensive
rats; WKY, Wistar-Kyoto. |
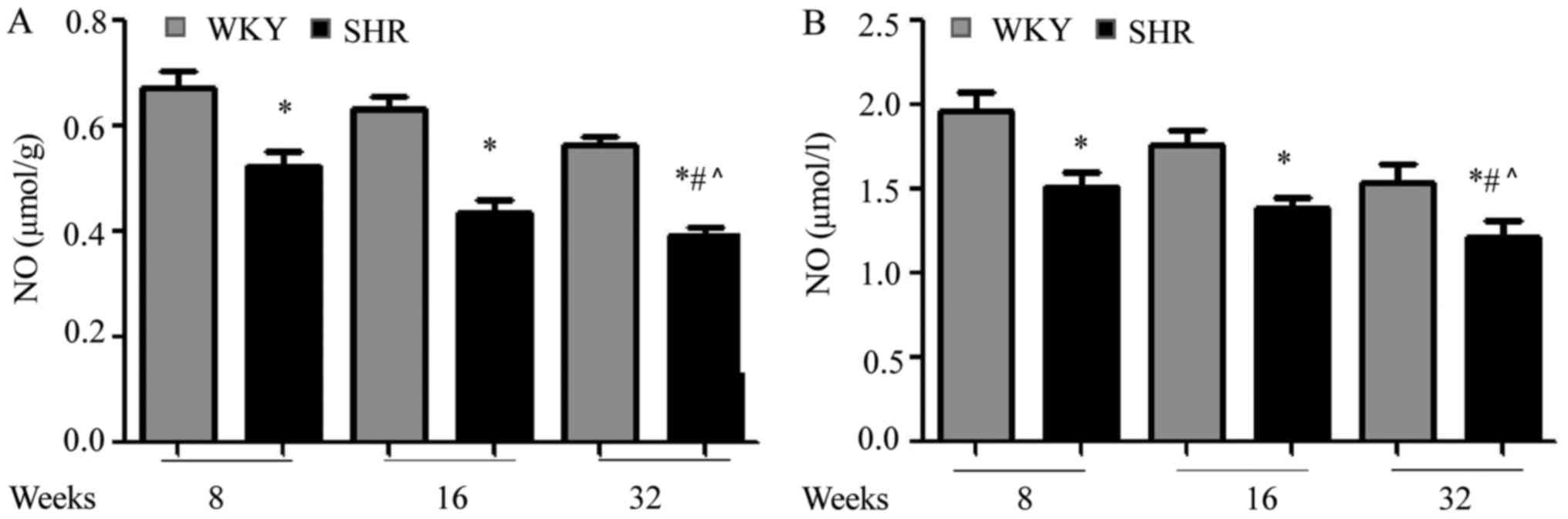
Circulating and myocardial NO are
decreased in SHR
As eNOS is an important regulator of NO production
(21), the NO content in the serum
and myocardial tissue of SHR and WKY rats was determined. As
present in Fig. 4A, it was
demonstrated that NO content was decreased in SHR hearts, compared
with age-matched WKY hearts (*P<0.05 vs. WKY), in agreement with
the eNOS results. The NO content in the SHR hearts also decreased
with aging (#P<0.05 vs. SHRs at 8 and 16 weeks).
Further, it was demonstrated that serum NO levels were
significantly lower in SHRs, compared with WKY rats (Fig. 4B; *P<0.05 vs. WKY), and that NO
was further reduced with aging (#P<0.05 vs. SHRs at 8
and 16 weeks). Therefore, it was demonstrated that NO expression in
the hearts and serum of SHRs was substantially downregulated, and
that this effect further increases with aging.
Discussion
Although UBIAD1 has been reported to be involved in
a number of human diseases (1,2,4–6), its
potential contribution to cardiovascular disorders remains unclear.
In the present study, it was confirmed that SHR exhibited
age-associated increases in blood pressure and myocardial
apoptosis, accompanied by cardiac dysfunction deterioration.
Furthermore, increased expression of ANF, a cardiac disease marker,
was detected. Immunohistochemical analysis, RT-qPCR and western
blotting also revealed decreased UBIAD1 expression in SHR hearts,
compared with the control group. This decrease was associated with
decreased myocardial CoQ10 and eNOS expression. In addition, serum
and myocardial tissue NO expression levels were significantly lower
in SHRs. Furthermore, expression of the aforementioned factors
decreased in SHRs in an age-dependent manner. Given the role of
UBIAD1 in eNOS and CoQ10 signaling regulation (5), the data obtained in the present study
indicated that UBIAD1 had critical functions in decreasing eNOS,
CoQ10 and NO expression in hypertensive cardiac hypertrophy.
The principal finding of the present study was that
myocardial UBIAD1 expression was significantly downregulated in
SHRs, and that this downregulation was associated with age. A
previous study suggested that high UBIAD1 expression in human heart
tissue was indicative of its potential role in heart disease
(7). Indeed, as a biosynthetic
enzyme for both vitamin K2 and MK-4 (6), a UBIAD1 mutant caused a number of
phenotypes, including cardiac edema in zebrafish, which were
rescued by re-repressing wild-type human UBIAD1 (7). UBIAD1−/− mouse embryos die
before embryonic day 10.5; however, it remains unclear if these
mutants exhibited cardiac structural defects prior to death
(22). To the best of our
knowledge, the present study was the first to demonstrate that
UBIAD1 expression is decreased in hypertension-associated
hypertrophic hearts. This merits further examination, in order to
determine whether UIBAD1 expression is altered in other myocardial
disease states, including ischemia/reperfusion and
pressure-overload induced cardiac hypertrophy. In addition, it
would be of great interest to investigate if UBIAD1 expression is
altered in human cardiac muscle disorders and heart failure.
Another question that requires addressing is how
UBIAB1 expression is regulated in hearts. A recent study suggested
that transcriptional repressor protein YY1, which is additionally a
ubiquitously expressed factor like UBIAB1, is a positive regulator
of UBIAD1 expression (23). YY1
serves an important role in early cardiac development, cardiac
muscle disorders and heart failure (24–26).
YY1 additionally offers protection against pathological hypertrophy
(27). Therefore, the role of YY1
in regulating UBIAD1 expression in hypertrophic myocardium warrants
further research.
In the present study, while a decrease in UBIAD1
expression in the SHRs was observed, it remains unclear whether
this decrease was the consequence of hypertrophy or if it was an
underlying cause. It could also not be concluded that decreased
UBIAD1 expression directly exacerbated cardiac dysfunction in SHRs
with age. In the future, the effects of UBIAD1 on the earlier
stages of SHR prior to the development of overt cardiac hypertrophy
will be used, as well as a cardiac-specific UBIAD1 knockout to
further address these questions.
In addition to its role in vitamin K synthesis,
UBIAD1 catalyzes the biosynthesis of CoQ10 and mediates eNOS
activity in zebrafish and humans. The role for CoQ10 in cardiac
hypertrophy and heart failure has been well investigated clinically
and in animal models (28). CoQ10
expression is deficient in a number of cardiac diseases (29). Consistently, CoQ10 pretreatment is
thought to be protective against isoproterenol-induced rat model of
cardiac hypertrophy (30), and
previous clinical studies have demonstrated the beneficial effects
of CoQ10 administration in patients with heart failure (31,32).
In the present study, it was demonstrated that CoQ10 expression was
substantially decreased in hypertension-induced hypertrophic
myocardial tissue, which was accompanied by decreased expression of
its downstream effector, eNOS (33,34).
It has been well documented that NO is catalyzed by eNOS, and is
released by vascular endothelial cells and cardiomyocytes where it
functions as a vasodilator (11).
eNOS is widely expressed in vascular endothelial cells,
cardiomyocytes and platelets and serves an important role in the
maintenance of cardiovascular homeostasis, predominantly through
regulating NO expression (35). NO
has a variety of biological activities, including platelet
aggregation inhibition, cardiac function regulation, vascular
smooth muscle relaxation (36). NO
has critical functions in vascular tone maintenance and blood
pressure regulation, as evidenced by the findings that inhibition
of NO synthesis increases blood pressure in healthy humans
(37). It has been reported that
hypertensive patients have decreased serum NO levels and eNOS gene
expression (38). In line with
previous findings, the present study demonstrated that circulating
and myocardial NO levels were decreased in SHRs compared with
age-matched control WKY rats, and that this decrease was
exacerbated with aging. Given the role of UBIAD1 in mediating CoQ10
activity, the findings of the present study collectively pointed to
the potential contribution of the UBIAD1-CoQ10-eNOS-NO axis in the
pathogenesis of hypertensive cardiac hypertrophy, by which reduced
levels of UBIAD1 resulted in insufficient expression of CoQ10,
subsequently diminishing eNOS expression and NO levels. This
hypothesis should be further investigated in future studies.
Previous studies have suggested that UBIAD1
regulates apoptosis through multiple mechanisms. For instance,
UBIAD1 was reported to promote apoptosis through mediating Golgi
function in a number of human cancer cell lines (39), and another study indicated that
UBIAD1 induces apoptosis in bladder tumor cells through regulating
cellular cholesterol (40).
Consistent with the above findings, the present study revealed that
decreased UBIAD1 expression was accompanied by increased apoptosis
in SHR hearts. However, a direct link between these two factors
remains to be established.
Certain limitations of the present study require
acknowledgement. As mentioned above, future studies must be
performed to reveal the causative relationship between decreased
UBIAD1 expression and the pathogenesis of hypertension-related
cardiac hypertrophy. One way to address this would be to rescue
cardiac function and hypertrophy by administering UBIAD1 to SHRs.
Another limitation was that a direct link between decreased UBIAD1
expression and decreased CoQ10 and eNOS levels was not established.
Given that a previous study using UBIAD1 knockout mice suggested
that UBIAD1 may not be a major regulator of CoQ10 expression in
mice (22), establishment of such
direct evidence is important to understand the exact function of
UBIAD1 pin the development and progression of hypertensive cardiac
hypertrophy.
In conclusion, it was demonstrated that UBIAD1
expression was decreased in SHR hearts in an age-dependent manner,
compared with those of age-matched controls. Altered UBIAD1
expression was accompanied by reduced CoQ10, eNOS, and NO
expression in SHRs. Given the well-recognized benefits offered by
CoQ10 for cardiovascular diseases, UBIAD1 may represent a potential
therapeutic target for the clinical treatment of
hypertension-induced cardiac hypertrophy.
Acknowledgements
The authors would like to thank Mrs Song Ying of the
Central Laboratory of the First Affiliated Hospital of Jinzhou
Medical University (Jinzhou, China) for the experimental guidance
and technical support.
Funding
The present study was supported by the Youth Project
of Liaoning Provincial Department of Education (grant no.
JYTQN201714).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
BY performed the experiments. JW designed the
experiments and supervised the study. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All animal protocols complied with and were approved
by the Ethics Committee of Jinzhou Medical University (Jinzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McGarvey TW, Nguyen T, Puthiyaveettil R,
Tomaszewski JE and Malkowicz SB: TERE1, a novel gene
affecting growth regulation in prostate carcinoma. Prostate.
54:144–155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Orr A, Dubé MP, Marcadier J, Jiang H,
Federico A, George S, Seamone C, Andrews D, Dubord P, Holland S, et
al: Mutations in the UBIAD1 gene, encoding a potential
prenyltransferase, are causal for Schnyder crystalline corneal
dystrophy. PLoS One. 2:e6852007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McGarvey TW, Nguyen T, Tomaszewski JE,
Monson FC and Malkowicz SB: Isolation and characterization of the
TERE1 gene, a gene down-regulated in transitional cell carcinoma of
the bladder. Oncogene. 20:1042–1051. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nickerson ML, Kostiha BN, Brandt W,
Fredericks W, Xu KP, Yu FS, Gold B, Chodosh J, Goldberg M, Lu DW,
et al: UBIAD1 mutation alters a mitochondrial
prenyltransferase to cause Schnyder corneal dystrophy. PLoS One.
5:e107602010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mugoni V, Postel R, Catanzaro V, De Luca
E, Turco E, Digilio G, Silengo L, Murphy MP, Medana C, Stainier DY,
et al: Ubiad1 is an antioxidant enzyme that regulates eNOS activity
by CoQ10 synthesis. Cell. 152:504–518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakagawa K, Hirota Y, Sawada N, Yuge N,
Watanabe M, Uchino Y, Okuda N, Shimomura Y, Suhara Y and Okano T:
Identification of UBIAD1 as a novel human menaquinone-4
biosynthetic enzyme. Nature. 468:117–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hegarty JM, Yang H and Chi NC:
UBIAD1-mediated vitamin K2 synthesis is required for vascular
endothelial cell survival and development. Development.
140:1713–1719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tousoulis D, Kampoli AM, Tentolouris C,
Papageorgiou N and Stefanadis C: The role of nitric oxide on
endothelial function. Curr Vasc Pharmacol. 10:4–18. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vos M, Esposito G, Edirisinghe JN, Vilain
S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B,
Meganathan R, et al: Vitamin K2 is a mitochondrial
electron carrier that rescues pink1 deficiency. Science.
336:1306–1310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xia Y, Wei X, Wu S, Wang B, Wang X and
Hong L: Down-regulation of TERE1/UBIAD1 activated Ras-MAPK
signalling and induced cell proliferation. Cell Biol Int Rep.
17:e000052010. View Article : Google Scholar
|
|
11
|
Zhan CD: The role of nitric oxide in the
prevention of myocardial hypertrophic response and its mechanisms.
Sheng Li Ke Xue Jin Zhan. 31:322–324. 2000.(In Chinese). PubMed/NCBI
|
|
12
|
Wollert KC and Drexler H: Regulation of
cardiac remodeling by nitric oxide: Focus on cardiac myocyte
hypertrophy and apoptosis. Heart Fail Rev. 7:317–325. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ozaki M, Kawashima S, Yamashita T, Hirase
T, Ohashi Y, Inoue N, Hirata K and Yokoyama M: Overexpression of
endothelial nitric oxide synthase attenuates cardiac hypertrophy
induced by chronic isoproterenol infusion. Circ J. 66:851–856.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Flaherty MP, Brown M, Grupp IL, Schultz
JE, Murphree SS and Jones WK: eNOS deficient mice develop
progressive cardiac hypertrophy with altered cytokine and calcium
handling protein expression. Cardiovasc Toxicol. 7:165–177. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brede M, Roell W, Ritter O, Wiesmann F,
Jahns R, Haase A, Fleischmann BK and Hein L: Cardiac hypertrophy is
associated with decreased eNOS expression in angiotensin AT2
receptor-deficient mice. Hypertension. 42:1177–1182. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan B, Sun Y and Wang J: Depletion of ubiA
prenyltransferase domain containing 1 expression promotes
angiotensin II-induced hypertrophic response in AC16 human
myocardial cells via modulating the expression levels of coenzyme
Q10 and endothelial nitric oxide synthase. Mol Med Rep.
16:6910–6915. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ridnour LA, Sim JE, Hayward MA, Wink DA,
Martin SM, Buettner GR and Spitz DR: A spectrophotometric method
for the direct detection and quantitation of nitric oxide, nitrite,
and nitrate in cell culture media. Anal Biochem. 281:223–229. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nordborg C and Johansson BB: Morphometric
study on cerebral vessels in spontaneously hypertensive rats.
Stroke. 11:266–270. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Diez J, Panizo A, Hernández M, Vega F,
Sola I, Fortuño MA and Pardo J: Cardiomyocyte apoptosis and cardiac
angiotensin-converting enzyme in spontaneously hypertensive rats.
Hypertension. 30:1029–1034. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Förstermann U and Sessa WC: Nitric oxide
synthases: Regulation and function. Eur Heart J. 33:829–837,
837a-837d. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakagawa K, Sawada N, Hirota Y, Uchino Y,
Suhara Y, Hasegawa T, Amizuka N, Okamoto T, Tsugawa N, Kamao M, et
al: Vitamin K2 biosynthetic enzyme, UBIAD1 is essential
for embryonic development of mice. PLoS One. 9:e1040782014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Funahashi N, Hirota Y, Nakagawa K, Sawada
N, Watanabe M, Suhara Y and Okano T: YY1 positively regulates human
UBIAD1 expression. Biochem Biophys Res Commun. 460:238–244. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Beketaev I, Zhang Y, Kim EY, Yu W, Qian L
and Wang J: Critical role of YY1 in cardiac morphogenesis. Dev Dyn.
244:669–680. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stauffer BL, Dockstader K, Russell G,
Hijmans J, Walker L, Cecil M, Demos-Davies K, Medway A, McKinsey TA
and Sucharov CC: Transgenic over-expression of YY1 induces
pathologic cardiac hypertrophy in a sex-specific manner. Biochem
Biophys Res Commun. 462:131–137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sucharov CC, Dockstader K and McKinsey TA:
YY1 protects cardiac myocytes from pathologic hypertrophy by
interacting with HDAC5. Mol Biol Cell. 19:4141–4153. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sucharov CC, Mariner P, Long C, Bristow M
and Leinwand L: Yin Yang 1 is increased in human heart failure and
represses the activity of the human alpha-myosin heavy chain
promoter. J Biol Chem. 278:31233–31239. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar A, Kaur H, Devi P and Mohan V: Role
of coenzyme Q10 (CoQ10) in cardiac disease, hypertension and
Meniere-like syndrome. Pharmacol Ther. 124:259–268. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Muraro PA, Vergelli M, Kalbus M, Banks DE,
Nagle JW, Tranquill LR, Nepom GT, Biddison WE, McFarland HF and
Martin R: Immunodominance of a low-affinity major
histocompatibility complex-binding myelin basic protein epitope
(residues 111-129) in HLA-DR4 (B1*0401) subjects is associated with
a restricted T cell receptor repertoire. J Clin Invest.
100:339–349. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghule AE, Kulkarni CP, Bodhankar SL and
Pandit VA: Effect of pretreatment with coenzyme Q10 on
isoproterenol-induced cardiotoxicity and cardiac hypertrophy in
rats. Curr Ther Res Clin Exp. 70:460–471. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fotino AD, Thompson-Paul AM and Bazzano
LA: Effect of coenzyme Q10 supplementation on heart
failure: A meta-analysis. Am J Clin Nutr. 97:268–275. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sharma A, Fonarow GC, Butler J, Ezekowitz
JA and Felker GM: Coenzyme Q10 and heart failure: A
state-of-the-art review. Circ Heart Fail. 9:e0026392016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chew GT and Watts GF: Coenzyme Q10 and
diabetic endotheliopathy: Oxidative stress and the ‘recoupling
hypothesis’. QJM. 97:537–548. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsai KL, Huang YH, Kao CL, Yang DM, Lee
HC, Chou HY, Chen YC, Chiou GY, Chen LH, Yang YP, et al: A novel
mechanism of coenzyme Q10 protects against human endothelial cells
from oxidative stress-induced injury by modulating NO-related
pathways. J Nutr Biochem. 23:458–468. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Forstermann U and Münzel T: Endothelial
nitric oxide synthase in vascular disease: From marvel to menace.
Circulation. 113:1708–1714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bian K, Doursout MF and Murad F: Vascular
system: Role of nitric oxide in cardiovascular diseases. J Clin
Hypertens. 10:304–310. 2008. View Article : Google Scholar
|
|
37
|
Kang JH, Wiggs JL, Rosner BA, Haines J,
Abdrabou W and Pasquale LR: Endothelial nitric oxide synthase gene
variants and primary open-angle glaucoma: Interactions with
hypertension, alcohol intake and cigarette smoking. Arch
Ophthalmol. 129:773–780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Q, Youn JY and Cai H: Mechanisms and
consequences of endothelial nitric oxide synthase dysfunction in
hypertension. J Hypertens. 33:1128–1136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Wang D, Jing P, Wu Y, Xia Y, Chen
M and Hong L: A novel Golgi retention signal RPWS for tumor
suppressor UBIAD1. PLoS One. 8:e720152013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fredericks WJ, McGarvey T, Wang H, Lal P,
Puthiyaveettil R, Tomaszewski J, Sepulveda J, Labelle E, Weiss JS,
Nickerson ML, et al: The bladder tumor suppressor protein TERE1
(UBIAD1) modulates cell cholesterol: Implications for tumor
progression. DNA Cell Biol. 30:851–864. 2011. View Article : Google Scholar : PubMed/NCBI
|