Introduction
Breast cancer is a highly prevalent malignancy in
women that is associated with high rates of morbidity and mortality
worldwide, and these rates have continued to increase over the past
few years (1). Although breast
cancer is traditionally treated using various methods including
surgery, chemotherapy and endocrine therapy, recurrence and
metastasis still remain a serious issue in advanced stage patients
(2).
Calreticulin (CRT) is a multifunctional
calcium-binding protein that is predominantly expressed in the
endoplasmic reticulum (3). This
protein contains three functional domains: N-, P- and C-domains
(4). It is involved in a variety
of cellular processes, including protein folding, calcium
homeostasis and cell adhesion (5).
Extensive research has revealed that the expression of CRT is
markedly increased in various types of cancers (6). A previous study demonstrated that the
expression of CRT was positively associated with lymph node
metastasis and clinical stages in breast cancer (7). An earlier study reported
epithelial-mesenchymal transition (EMT)-like changes in the
cellular phenotype in CRT-overexpressing Madin-Darby canine kidney
cells (8). Furthermore, the mRNA
expression of the EMT marker E-cadherin was reduced when CRT was
overexpressed. Overexpressing CRT regulated EMT marker
characteristics in gastric cancer cells (9). These results suggested that there may
be a direct association between CRT and EMT.
Ecotropic viral integration site-1 (EVI-1) has a
critical role in oncogenesis as a transcription factor (10). Overexpression of EVI-1 has
frequently been observed in hematological malignancies (11–13)
and in several types of solid tumor (14–16).
However, very little is known about how EVI-1 regulates the
oncogenesis of breast cancer.
Previous studies have demonstrated that EVI-1 is a
master regulatory element in EMT (17,18).
Nayak et al (19) reported
that the expression of EVI-1 was strongly correlated with
E-cadherin and N-cadherin in stage IV of colon cancer. Whether
EVI-1 regulates the process of EMT in breast cancer via CRT remains
unknown. In the present study, the overexpression of EVI-1 promoted
cell proliferation, migration and invasion, and inhibited apoptosis
in breast cancer cells. In addition, EVI-1 positively regulated the
expression of CRT in breast cancer. Furthermore, a novel
mechanistic pathway was investigated for how EVI-1 induced CRT
activation in breast cancer. The results revealed that EVI-1 may be
a potential effective therapeutic target in breast cancer.
Materials and methods
Cell lines and culture
The human breast cancer cell line MDA-MB-231 was
purchased from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). Cells were cultured in L-15 (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; HyClone; GE Healthcare Life Sciences,
Logan, UT, USA) in a humidified atmosphere at 37°C without
CO2.
Cell transfection
MDA-MB-231 breast cancer cells were cultured in a
6-well plate for 24 h at 37°C and then transfected with plasmids
pcDNA3.1-EVI-1 and pSilencer-2.1-EVI-1 [EVI-1-short hairpin RNA
(shRNA)]; both synthesized by Jrdun Biotechnology Co., Ltd.,
Shanghai, China). The following primers were used to amplify the
EVI-1 sequence for cloning into the pcDNA vector: pcDNA3.1-EVI-1
forward, 5′-CCGGAATTCATGATCTTAGACGAATTTTACA-3′; pcDNA3.1-EVI-1
reverse, 5′-CGCGGATCCTCATACGTGGGGATAGCACTGGA-3′. The short hairpin
RNA sequence (shRNA) in pSilencer2.1-EVI-1 (EVI-1-shRNA) was
forward, 5′-CCTACGATCAGTCCTACCA-3′ and reverse,
5′-TGGTAGGACTGATCGTAGG-3′. For EVI-1 expression, 2.5 µg pcDNA3.1
control vector/pcDNA3.1-EVI-1 or pSilencer-2.1 control
vector/pSilencer-2.1-EVI-1 were transfected into cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) into 6-well plates (3×105 cells/well).
For CRT expression, a small interfering RNA (siRNA) targeting CRT
was used (synthesized by Jrdun Biotechnology Co., Ltd.), which had
the following sequence: siRNA-CRT, 5′-GGAGCAGUUUCUGGACGGATT-3′.
siRNA-NC was 5′-UUCUCCGAACGUGUCACGUTT-3′. siRNAs (90 pmol) were
transfected into MDA-MB-231 cells (6-well plates; 3×105
cells/well) using the Lipofectamine® RNAiMAX
Transfection Reagent (Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions (incubated for 1 day at 37°C).
Dual luciferase reporter system
The MDA-MB-231 cells were transfected
psiCHECK2-CRT-WT and pcDAN3.1 empty vector, psiCHECK2-CRT-WT (the
fragments of the promoter region of CRT) pcDAN3.1-EVI-1,
psiCHECK2-CRT-MUT and pcDAN3.1 empty vector, or pcDAN3.1-EVI-1 and
psiCHECK2-CRT-MUT using Lipofectamine® 2000 (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. For all plasmids, 2 µg was used fro transfection and
were synthesized by Jrdun Biotechnology Co., Ltd. After 48 h, the
luciferase activity was measured using a Dual-Luciferase reporter
assay system according to the manufacturer's instructions (Promega
Corporation, Madison, WI, USA). Firefly luciferase activities were
normalized by Renilla luciferase activities to control for
transfection efficiency.
Electrophoresis mobility shift assay
(EMSA)
EMSA was performed using the LightShift EMSA kit
(Pierce; Thermo Fisher Scientific, Inc.). Nuclear protein extracts
from breast cancer cells were prepared using NE-PER™ Nuclear and
Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Inc.).
Protein concentration was determined using the bicinchoninic acid
assay. Oligonucleotides were synthesized for the CRT EVI-1 binding
site and the mutation, and labeled with biotin by Beijing Genomics
Institute (Shenzhen, China). The oligonucleotide sequences were as
follows: EVI-1 binding site from CRT promoter region,
5′-GCTGGTTCTCAAATGCAAGATAAGAGCTGG-3′; and EVI-1 binding site-mutant
from CRT promoter region 5′-GCTGGTTCTCATCGATCTGATAAGAGCTGG-3′. The
binding reaction mixtures (2 µl 5X Gel shift buffer, 2 µl nuclear
extracts, 1 µl labeled DNA probe, nuclease-free water to 10 µl)
were incubated according to the manufacturer's instruction. For
competition experiments, the corresponding unlabeled probe (cold
probe) was used at 100-fold excess concentrations when compared
with the labeled probe in the binding reaction. Reaction products
were separated by 5% non-denaturing polyacrylamide gels in 0.5X
Tris/Borate/EDTA buffer, and then the bands were transferred to a
nylon membrane. The nylon membrane was visualized using the
LightShift™ Chemiluminescent EMSA Kit (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions.
Cell viability assay
Cell viability was assessed by a Cell Counting Kit-8
(CCK-8; Beyotime Institute of Biotechnology, Haimen, China) assay
according to the manufacturer's instructions. Transfected cells
were seeded into a 96-well plate at 5,000 cells/well. After
transfection for 24, 48 and 72 h, 10 µl CCK-8 assay solution was
added to each well and incubated for 1 h. Absorbance was then
measured at 450 nm using a microplate reader.
Flow cytometry analysis of cell
apoptosis
Cells were collected after transfection for 48 h,
then an Annexin V-FITC Apoptosis Detection Kit (Beyotime Institute
of Biotechnology) was used to assess apoptosis according to the
manufacturer's instructions. The rate of apoptosis was analyzed by
flow cytometry with BD FACSDiva software version 8.0 (BD
Biosciences, San Jose, CA, USA).
Cell migration and invasion
assays
The migration and invasion assays were performed
using Transwell inserts. For the migration assay, at 48 h after
transfection the indicated cells were starved for 24 h,
1×105 cells resuspended in serum-free medium and then
added to the upper chamber of Transwell plates (Corning
Incorporated, Corning, NY, USA). The lower chamber was filled with
L15 medium and 10% FBS (Gibco; Thermo Fisher Scientific, Inc.).
After incubation for 24 h, the cells attached to the lower surface
of the membrane were fixed in 95% ethanol at 37°C for 30 min, and
stained with 0.5% crystal violet for 30 min at room temperature;
cells were then counted in five randomly selected fields under a
light microscope (Olympus Corporation, Tokyo, Japan). For the cell
invasion assay, the indicated cells were plated in Transwell
polycarbonate membrane inserts precoated with a layer of diluted
Matrigel (BD Biosciences). The remaining experimental procedures
were consistent with those described for cell migration
experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from MDA-MB-231 breast cancer
cells using RNAiso Plus (Takara Biotechnology Co., Ltd., Dalian,
China) according to manufacturer's instructions. cDNA was
synthesized using the PrimeScript™ RT reagent Kit with gDNA Eraser
(Takara Biotechnology Co., Ltd.), the temperature protocol for
reverse transcription was as follows: 37°C for 15 min; 85°C for 5
sec and 4°C for 5 min. RT-qPCR was conducted using SYBR®
Premix Ex Taq™ II (Takara Biotechnology Co., Ltd.). The expression
of target genes was normalized to GAPDH. The reaction thermocycling
conditions for qPCR were as follows: 95°C for 30 sec, followed by
40 cycles of 95°C for 5 sec, and 60°C for 31 sec. The sequences of
the primers used for RT-qPCR are presented in Table I. All of the qPCR data were
processed using the 2−ΔΔCq method (20).
 | Table I.Primers used for reverse
transcritpion-quantitative polymerase chain reaction. |
Table I.
Primers used for reverse
transcritpion-quantitative polymerase chain reaction.
| Primer | Forward (5′ to
3′) | Reverse (5′ to
3′) |
|---|
| EVI-1 |
TTATAGAGCGATACAAGGGGGAG |
CGCCGTCTGATTATCTTGATGAG |
| Calreticulin |
AAGGAGCAGTTTCTGGACGG |
GCCGACAGAGCATAAAAGCG |
| Snail 1 |
TGCTGTCCCCGGCGATATT |
GTAGCTGCCCTGGTAGGTT |
| Slug |
TCCCTCGCGTGAGGTGAAGCA |
TCTGTCTCTGGAGCCAGGTGC |
| E-cadherin |
TGCAGGGGCAGCCATCTCCT |
TTCCCCCAGCGTCCTCCACC |
| N-cadherin |
CGGCCGCTGCCACCACAGTT |
AGTCCCCACGCTGCTCTTCT |
| BAX |
GGTCGCTTGTGGCCTTTTTC |
TGCTGCATTGTTCCCATAGAG |
| Caspase-3 |
ATGGAGCGAATCAATGGACTC |
CTGTACCAGACCGAGATGTCA |
| BCL-2 |
GACTGGGGGAGGATTGTGG |
CCGGTTCAGGTACTCAGTCA |
| GAPDH |
CTCCTCCTGGCCTCGCTGT |
GCTGTCACCTTCACCGTTCC |
Western blot analysis
Total protein was isolated from MDA-MB-231 breast
cancer cells using radioimmunoprecipitation assay lysis buffer
(Beyotime Institute of Biotechnology) and protein concentration was
measured using a bicinchoninic acid assay. Equal amounts of protein
(20 µg/lane) separated by SDS-PAGE on 8% gels and then transferred
to polyvinylidene difluoride membranes. The membranes were blocked
with 5% nonfat milk for 2 h at room temperature and then incubated
with rabbit polyclonal anti-EVI-1 (1:1,000; cat. no. ab28457),
rabbit polyclonal anti-CRT (1:1,000; cat. no. ab2907), rabbit
polyclonal anti-E-cadherin (1:1,000; cat. no. ab15148), rabbit
polyclonal anti-N-cadherin (1:1,000; cat. no. ab18203), rabbit
polyclonal anti-apoptosis regulator BAX (BAX; 1:1,000; cat. no.
ab53154), rabbit polyclonal anti-caspase-3 (1:1,000; cat. no.
ab13847), rabbit polyclonal anti-zinc finger protein SNAI1 (Snail
1; 1:1,000; cat. no. ab110490), rabbit polyclonal anti-zinc finger
protein SNAI2 (Slug; 1:1,000; cat. no. ab27568) and rabbit
polyclonal anti-apoptosis regulator Bcl-2 (BCL-2; 1:1,000; cat. no.
ab59348) primary antibodies at 4°C overnight. GAPDH (1:3,000; cat.
no. ab9485) was used as the internal control to ensure equal
protein loading. All antibodies were purchased from Abcam
(Cambridge, UK). The signal was developed with Pierce™ ECL Western
Blotting Substrate (Thermo Fisher Scientific, Inc.) following
incubation with the corresponding goat anti-rabbit horseradish
peroxidase-conjugated (IgG H&L) secondary antibody at room
temperature for 1 h (1:2,000; cat. no. ab205718; Abcam).
Statistical analysis
All experiments were repeated three times
independently, and the data are presented as the mean ± standard
deviation. One-way analysis of variance followed by Bonferroni's
multiple comparisons procedure was used to compare multiple groups
and paired t-test was used to compare two groups. Analysis was
performed using SPSS 19.0 statistical software (IBM Corp., Armonk,
NY, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
EVI-1 may target the CRT promoter
region in vitro
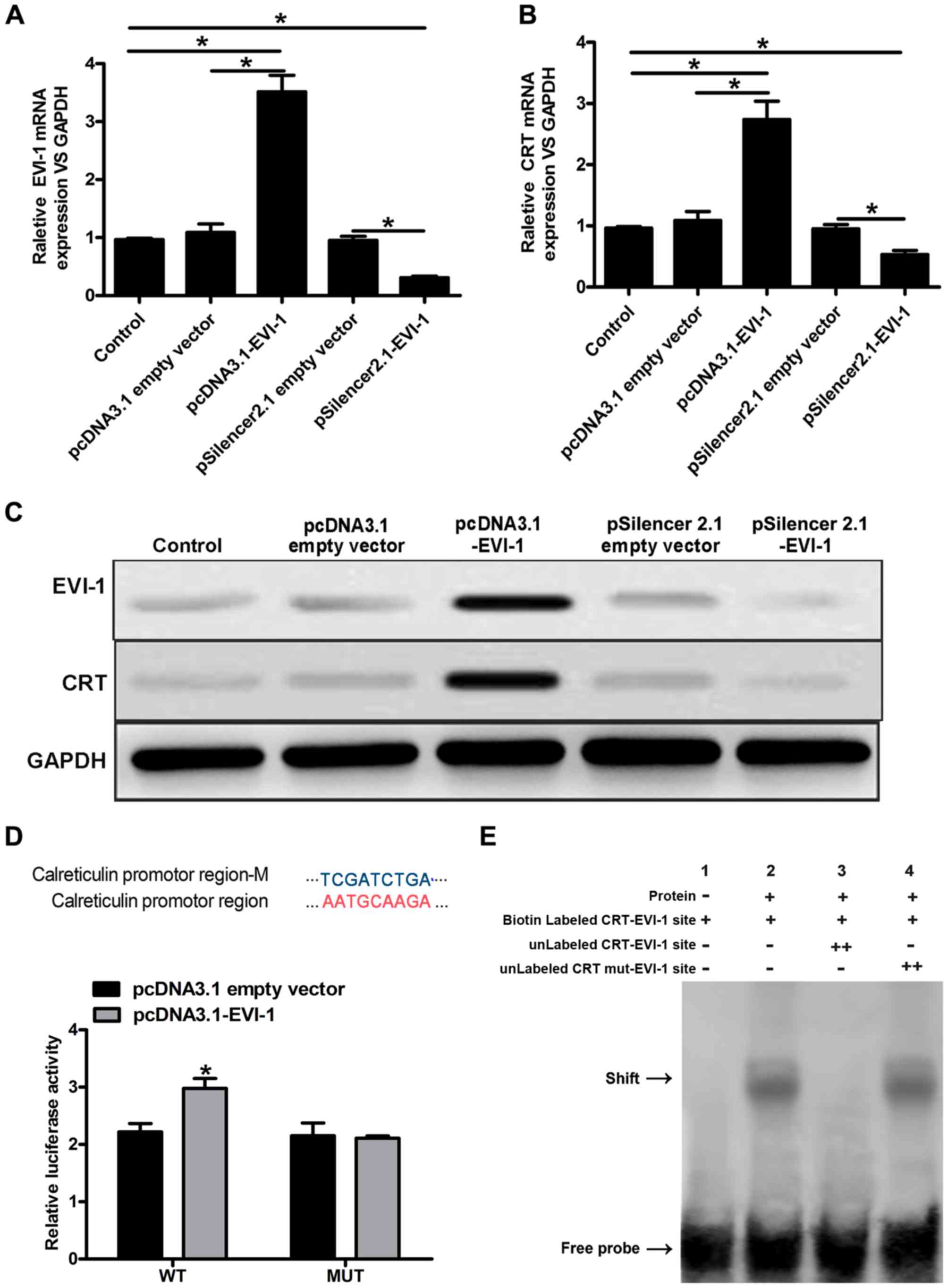
The expression of EVI-1 was markedly upregulated
following transfection with pcDNA3.1-EVI-1, whereas EVI-1 was
downregulated after transfection with pSilencer-2.1-EVI-1 (Fig. 1A). Subsequently, the expression of
CRT was analyzed at the mRNA and protein levels. CRT mRNA
expression was increased following transfection with
pcDNA3.1-EVI-1; however, pSilencer-2.1-EVI-1 reduced CRT mRNA
expression (Fig. 1B). In addition,
a similar effect on CRT protein expression was observed (Fig. 1C). To further demonstrate that CRT
was a target of EVI-1, dual luciferase reporter and EMSA assays
were performed. Luciferase activity was increased in cells
co-transfected with pcDNA3.1-EVI-1 and the psiCHECK2-CRT-WT
reporter vector. However, pcDNA3.1-EVI-1 had no effect on the
luciferase activity of cells co-transfected with the
psiCHECK2-CRT-MUT reporter vector (Fig. 1D). Based on the EMSA gel shifts, a
protein bound to the CRT promoter region, and this protein may be
EVI-1. No specific gel shift was observed in lane 1; however, a
marked shift was observed in lane 2. In addition, no shift was
observed with the 100-fold excess of the unlabeled EVI-1 probe
(cold probe) in lane 3, and there was no obvious interaction
between the nuclear extract and the mutated EVI-1 probe in lane 4
(Fig. 1E). In summary, these
results indicated that EVI-1 positively regulated CRT expression,
and EVI-1 is likely to regulate its expression by binding to the
promoter region of CRT in vitro experiment.
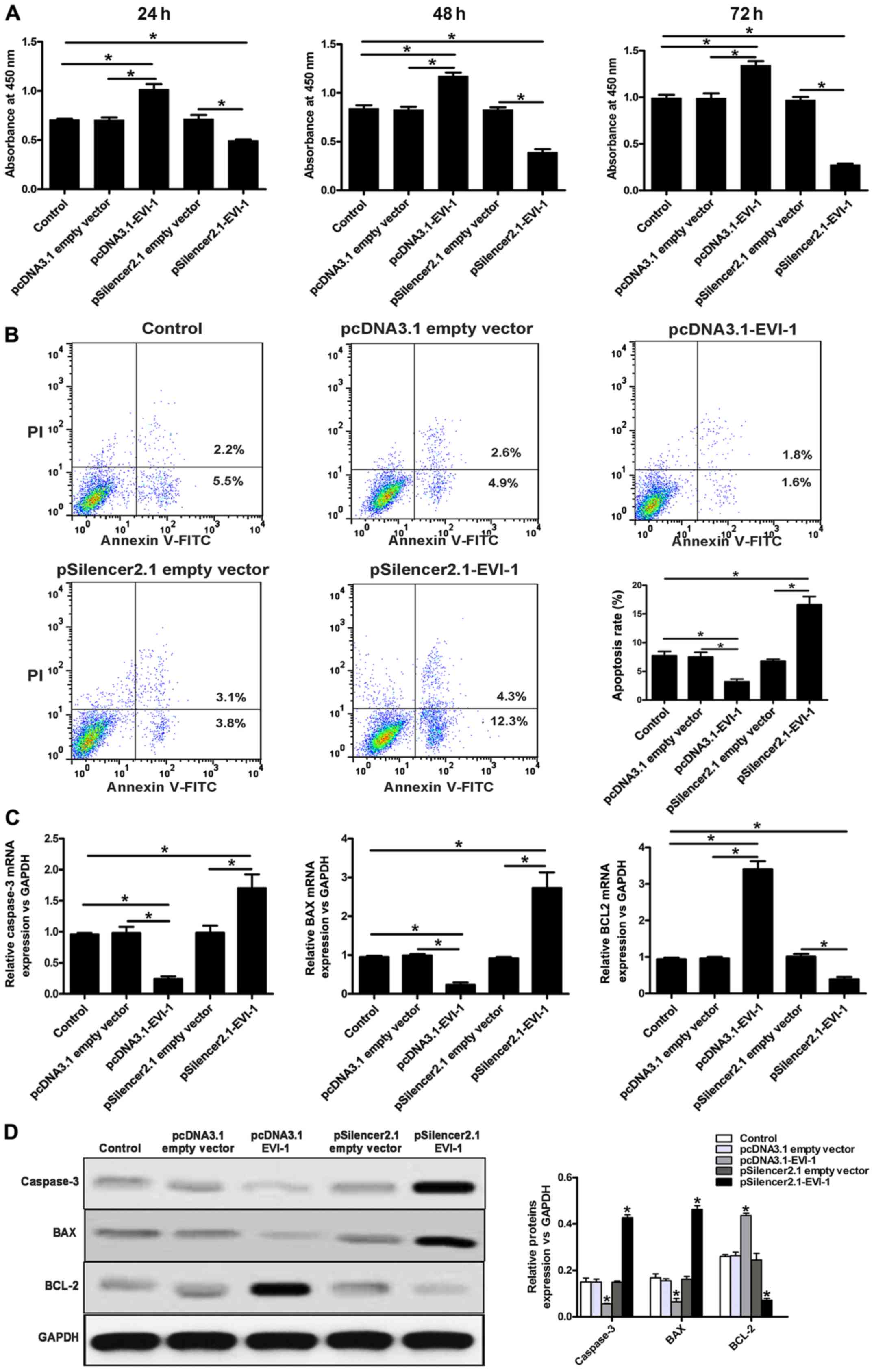
EVI-1 promotes cell proliferation and
inhibits cell apoptosis in human breast cancer
The overexpression of EVI-1 markedly increased cell
proliferation, while downregulation of EVI-1 inhibited cell
proliferation (Fig. 2A).
Overexpression of EVI-1 also decreased the apoptosis of breast
cancer cells, while EVI-1 silencing increased the rate of apoptosis
in breast cancer cells (Fig. 2B).
Subsequently, expression of apoptotic pathway-associated genes,
including caspase-3, Bax and Bcl-2, was investigated. As indicated
in Fig. 2C and D, a marked
decrease in caspase-3 and Bax expression, and a marked increase in
Bcl-2 expression was observed when EVI-1 was overexpressed. By
contrast, knockdown of EVI-1 increased the expression of caspase-3
and Bax, and reduced the expression of Bcl-2.
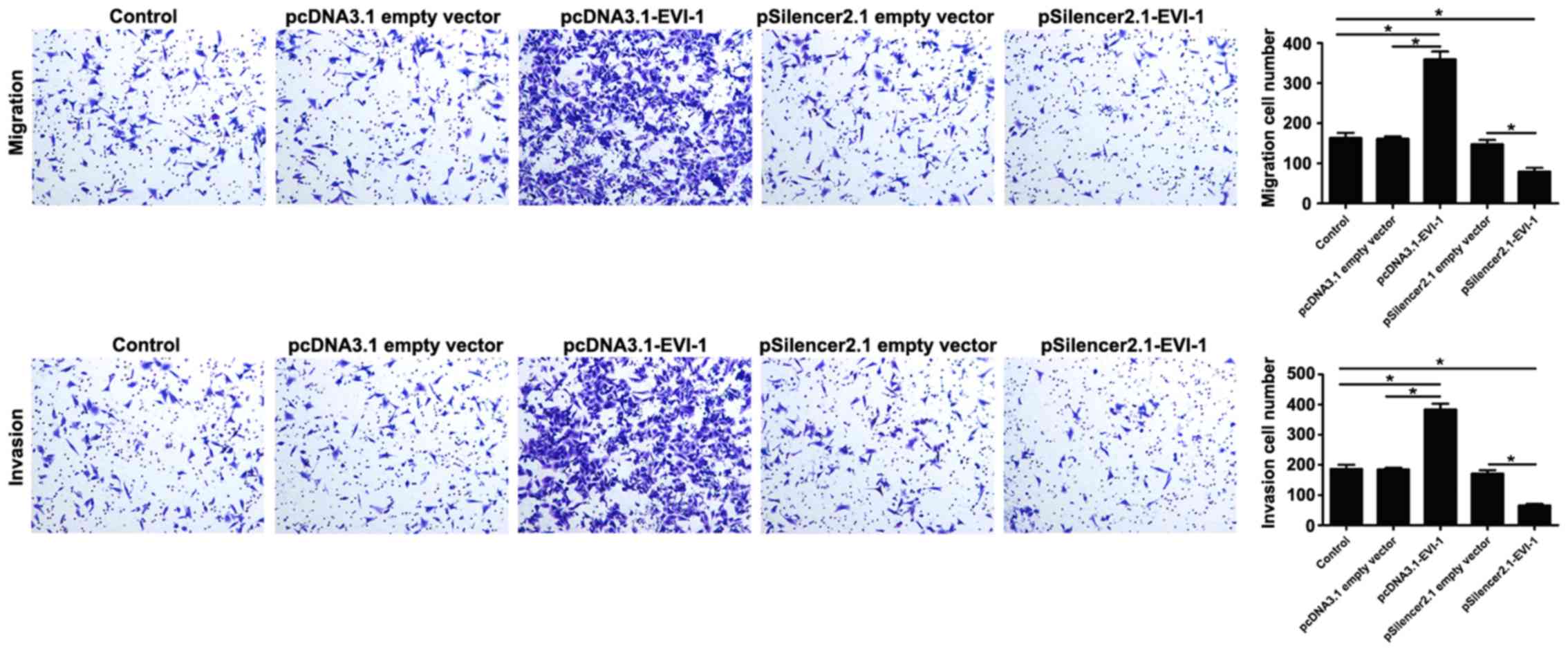
EVI-1 promotes cell migration and
invasion in human breast cancer
The present study further investigated the effect of
EVI-1 on cell migration and invasion. The results revealed that
overexpression of EVI-1 effectively promoted cell migration and
invasion, and silencing EVI-1 expression markedly suppressed cell
migration and invasion in human breast cancer cells compared with
the respective empty vector controls (Fig. 3).
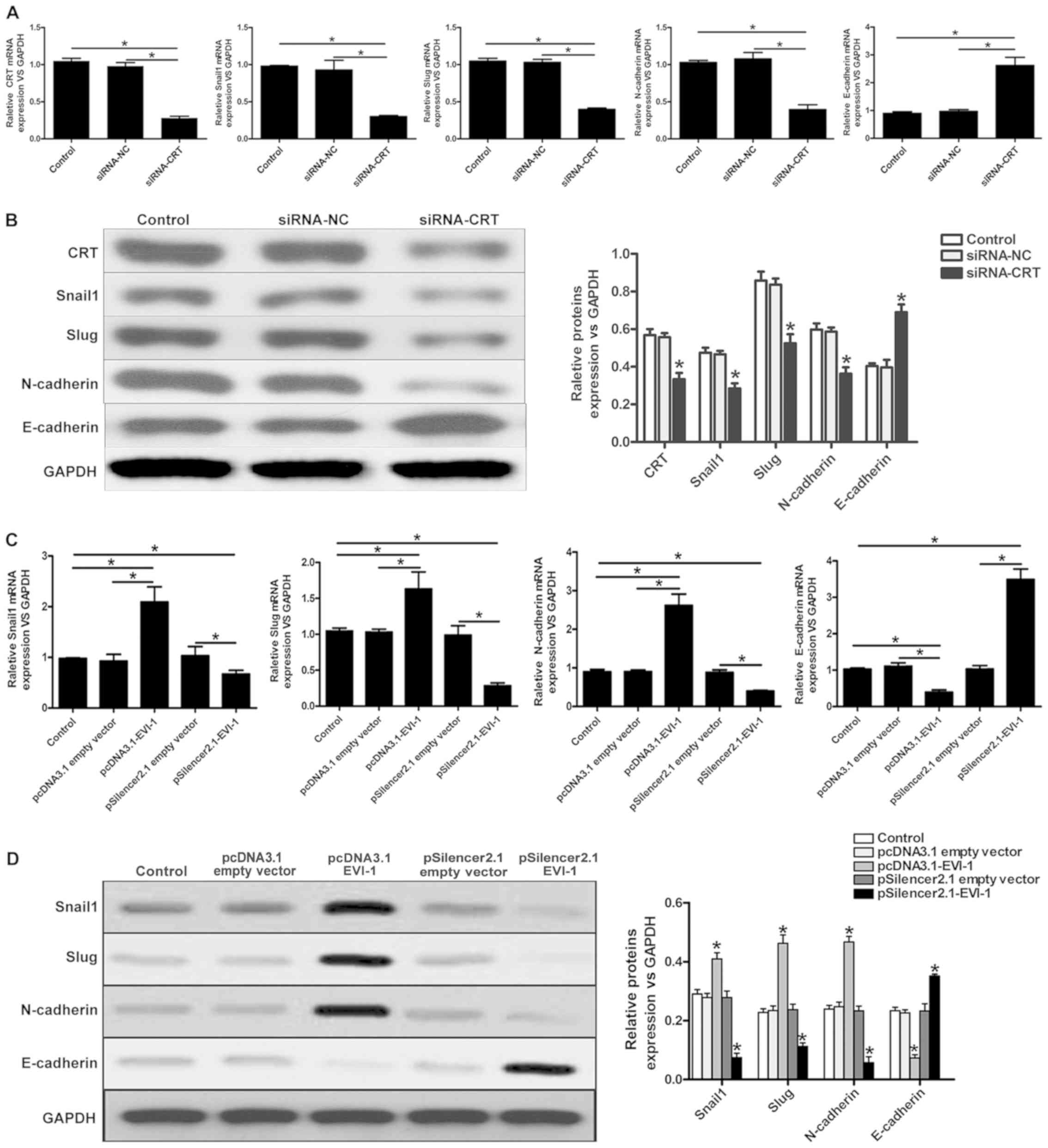
EVI-1 regulates EMT-related gene
expression in breast cancer
Previous studies have indicated that the abnormal
expression of CRT regulates the expression of EMT-associated genes
(8,9), and the present study revealed that
EVI-1 positively regulated CRT expression. As indicated in Fig. 4A and B, knockdown of CRT using
siRNA-CRT decreased the expression of Snail 1, Slug and N-cadherin,
and increase the expression of E-cadherin.
To determine whether EVI-1 can regulate
EMT-associated gene expression, RT-qPCR and western blot analyses
were performed to investigate the expression of EMT-associated
genes. As demonstrated in Fig. 4C and
D, a marked increase in Snail 1, Slug, and N-cadherin
expression, and a marked decrease in E-cadherin expression were
observed when EVI-1 was upregulated. By contrast, silencing EVI-1
decreased the expression of Snail 1, Slug and N-cadherin, and
increased the expression of E-cadherin. These results suggested
that EVI-1 may influence the process of EMT in breast cancer
cells.
Discussion
EVI-1, an oncogene located on chromosome 3q26,
specifically binds to promoter DNA sequences and has a role in
transcriptional regulation (10).
Multiple studies have revealed that EVI-1 is upregulated in
leukemia, ependymoma, ovarian cancer, breast cancer and colon
cancer, and the downregulation of EVI-1 expression inhibited the
proliferation of tumor cells (11–13).
In addition, increased EVI-1 expression has also been revealed to
be a risk factor for poor prognosis in patients with leukemia
(21–23). However, whether EVI-1 exerts
specific regulatory effects on breast cancer has not been reported
thoroughly. In the present study, overexpression of EVI-1 increased
the proliferative ability of breast cancer.
c-Jun N-terminal protein kinase (JNK) has a critical
role in the process of cell apoptosis and the mechanism of
JNK-mediated apoptosis is associated with the regulated expression
of apoptosis-associated proteins, including Bax and Bcl-2 (24–26).
However, EVI-1 inhibits the activity of JNK and the subsequent cell
apoptosis by interfering with the interactions between JNK and its
physiological substrates (27). It
was hypothesized that EVI-1 may regulate breast cancer apoptosis
and the present study demonstrated that cell apoptosis was
inhibited by the overexpression of EVI-1 and enhanced by the
downregulation of EVI-1. Bcl-2 has a major role in the signal
transduction pathways of cell apoptosis (28). Bcl-2 and Bax are the most
representative genes for inhibiting and promoting apoptosis, and
Bax is the main regulator of Bcl-2 (29). Bcl-2 can bind to Bax to form a
heterodimer, and the increased expression of Bax can antagonize the
effect of Bcl-2 and promote cell apoptosis (30–32).
Caspase-3 is the key mediator of cell apoptosis and has a role in a
variety of apoptotic signaling pathways (33–35).
Previous studies have reported that overexpression of Bcl-2 can
effectively inhibit the activation of caspase-3, thus inhibiting
the occurrence of apoptosis (36).
In the present study, RT-qPCR and western blotting revealed that
EVI-1 overexpression/silencing altered the expression of the
aforementioned proteins associated with apoptosis, and subsequently
the process of apoptosis, in breast cancer. When EVI-1 was highly
expressed, the expression of Bax and caspase-3 was reduced, and
Bcl-2 was increased.
A number of previous studies investigating CRT
demonstrated that the abnormal expression of CRT leads to changes
in certain biological processes, including cell invasion and
proliferation, and may be associated with the cancer occurrence,
development and prognosis (3,6,37–39).
However, research on the association between EVI-1 and CRT is
rarely reported. EMSA is an invaluable tool to study interaction of
proteins with DNA. The experiment can simulate the specific binding
of protein and DNA in vitro. EMSA has some limitations in
the process of reconstructing the binding between proteins and DNA
in vivo. However, EMSA is still a good method to predict the
binding between EVI-1 and CRT in vitro in this study. By
using EMSA and dual luciferase assays, the present study revealed
that EVI-1 may bind to the promoter region of CRT, and positively
regulate its expression. Thus, it is reasonable to suggest that
EVI-1 may affect some of the biological functions of breast cancer
by regulating the expression of CRT.
EMT is a process by which epithelial cells gain the
phenotype of mesenchymal cells, and the close connection between
cells with the extracellular matrix and neighboring cells weakens
or is completely abolished; consequently, cell migration and
invasion is enhanced (40). In the
process of EMT, the expression of several proteins, including
E-cadherin, N-cadherin, Snail 1 and Slug, is altered (41–44).
Previous studies have revealed that the overexpression of CRT can
inhibit the expression of E-cadherin and enhance cell migration
abilities (8,45). Liu et al (9) reported that the overexpression of CRT
promoted cell invasion and metastasis in gastric cancer by
regulating the expression of Snail 1 and E-cadherin. In the present
study, the results demonstrated that increased expression of EVI-1
significantly reduced the expression of E-cadherin and enhanced the
expression of N-cadherin, Snail 1 and Slug. When the expression of
EVI-1 was suppressed, the expression of E-cadherin was increased,
and N-cadherin, Snail 1 and Slug were reduced. It was speculated
that EVI-1 may regulate EMT-associated genes via the CRT pathway in
breast cancer.
In conclusion, the results of the present study
revealed that EVI-1 overexpression promotes cell proliferation,
migration and invasion, and inhibits apoptosis in breast cancer
cells. EVI-1 is likely to bind to CRT and positively regulate the
expression of CRT in vitro environment; it was concluded
that EVI-1 may affect EMT-associated genes by regulating the
expression of CRT in breast cancer.
Acknowledgements
The authors would like to thank Professor Ping Ma
(Molecular Oncology Laboratory of Cancer Research Institute, The
First Affiliated Hospital of China Medical University, Shenyang,
China) for designing the study and for their excellent technical
assistance.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW designed the study, performed the experiments and
drafted the manuscript. TW, DH and XL analyzed and interpreted the
experimental data. YJ conceived the study and participated in its
design and coordination.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ghislain I, Zikos E, Coens C, Quinten C,
Balta V, Tryfonidis K, Piccart M, Zardavas D, Nagele E,
Bjelic-Radisic V, et al: Health-related quality of life in locally
advanced and metastatic breast cancer: Methodological and clinical
issues in randomised controlled trials. Lancet Oncol. 17:e294–e304.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goitea VE and Hallak ME: Calreticulin and
arginylated calreticulin have different susceptibilities to
proteasomal degradation. J Biol Chem. 290:16403–16414. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holmström MO, Ocias LF, Kallenbach K, Kjær
L, Kristensen TK, Pallisgaard N, Petersen BL, Skov V, de Stricker
K, Larsen TS, et al: New disease markers within the chronic
myeloproliferative neoplasms. Ugeskr Laeger. 1772015.(In
Danish).
|
|
5
|
Wang WA, Groenendyk J and Michalak M:
Calreticulin signaling in health and disease. Int J Biochem Cell
Biol. 44:842–846. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zamanian M, Veerakumarasivam A, Abdullah S
and Rosli R: Calreticulin and cancer. Pathol Oncol Res. 19:149–154.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lwin ZM, Guo C, Salim A, Yip GW, Chew FT,
Nan J, Thike AA, Tan PH and Bay BH: Clinicopathological
significance of calreticulin in breast invasive ductal carcinoma.
Mod Pathol. 23:1559–1566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hayashida Y, Urata Y, Muroi E, Kono T,
Miyata Y, Nomata K, Kanetake H, Kondo T and Ihara Y: Calreticulin
represses E-cadherin gene expression in Madin-Darby canine kidney
cells via Slug. J Biol Chem. 281:32469–32484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu SH, Lee WJ, Lai DW, Wu SM, Liu CY,
Tien HR, Chiu CS, Peng YC, Jan YJ, Chao TH, et al: Honokiol confers
immunogenicity by dictating calreticulin exposure, activating ER
stress and inhibiting epithelial-to-mesenchymal transition. Mol
Oncol. 9:834–849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wieser R: The oncogene and developmental
regulator EVI1: Expression, biochemical properties, and biological
functions. Gene. 396:346–357. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Daghistani M, Marin D, Khorashad JS, Wang
L, May PC, Paliompeis C, Milojkovic D, De Melo VA, Gerrard G,
Goldman JM, et al: EVI-1 oncogene expression predicts survival in
chronic-phase CML patients resistant to imatinib treated with
second-generation tyrosine kinase inhibitors. Blood. 116:6014–6017.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gröschel S, Lugthart S, Schlenk RF, Valk
PJ, Eiwen K, Goudswaard C, van Putten WJ, Kayser S, Verdonck LF,
Lübbert M, et al: High EVI1 expression predicts outcome in younger
adult patients with acute myeloid leukemia and is associated with
distinct cytogenetic abnormalities. J Clin Oncol. 28:2101–2107.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vázquez I, Maicas M, Cervera J, Agirre X,
Marin-Béjar O, Marcotegui N, Vicente C, Lahortiga I, Gomez-Benito
M, Carranza C, et al: Down-regulation of EVI1 is associated with
epigenetic alterations and good prognosis in patients with acute
myeloid leukemia. Haematologica. 96:1448–1456. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patel JB, Appaiah HN, Burnett RM,
Bhat-Nakshatri P, Wang G, Mehta R, Badve S, Thomson MJ, Hammond S,
Steeg P, et al: Control of EVI-1 oncogene expression in metastatic
breast cancer cells through microRNA miR-22. Oncogene.
30:1290–1301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koos B, Bender S, Witt H, Mertsch S,
Felsberg J, Beschorner R, Korshunov A, Riesmeier B, Pfister S,
Paulus W and Hasselblatt M: The transcription factor evi-1 is
overexpressed, promotes proliferation, and is prognostically
unfavorable in infratentorial ependymomas. Clin Cancer Res.
17:3631–3637. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deng X, Cao Y, Liu Y, Li F, Sambandam K,
Rajaraman S, Perkins AS, Fields AP, Hellmich MR, Townsend CM Jr, et
al: Overexpression of Evi-1 oncoprotein represses TGF-β signaling
in colorectal cancer. Mol Carcinog. 52:255–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Venkov CD, Link AJ, Jennings JL, Plieth D,
Inoue T, Nagai K, Xu C, Dimitrova YN, Rauscher FJ and Neilson EG: A
proximal activator of transcription in epithelial-mesenchymal
transition. J Clin Invest. 117:482–491. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Venkov C, Plieth D, Ni T, Karmaker A, Bian
A, George AL Jr and Neilson EG: Transcriptional networks in
epithelial-mesenchymal transition. PLoS One. 6:e253542011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nayak KB, Sajitha IS, Kumar TRS and
Chakraborty S: Ecotropic viral integration site 1 promotes
metastasis independent of epithelial mesenchymal transition in
colon cancer cells. Cell Death Dis. 9:182018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goyama S and Kurokawa M: Evi-1 as a
critical regulator of leukemic cells. Int J Hematol. 91:753–757.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lugthart S, van Drunen E, van Norden Y,
van Hoven A, Erpelinck CA, Valk PJ, Beverloo HB, Löwenberg B and
Delwel R: High EVI1 levels predict adverse outcome in acute myeloid
leukemia: Prevalence of EVI1 overexpression and chromosome 3q26
abnormalities underestimated. Blood. 111:4329–4337. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barjesteh van Waalwijk van
Doorn-Khosrovani S, Erpelinck C, van Putten WL, Valk PJ, van der
Poel-van de Luytgaarde S, Hack R, Slater R, Smit EM, et al: High
EVI1 expression predicts poor survival in acute myeloid leukemia: A
study of 319 de novo AML patients. Blood. 101:837–845. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aoki H, Kang PM, Hampe J, Yoshimura K,
Noma T, Matsuzaki M and Izumo S: Direct activation of mitochondrial
apoptosis machinery by c-Jun N-terminal kinase in adult cardiac
myocytes. J Biol Chem. 277:10244–10250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang CB, Pei WJ, Zhao J, Cheng YY, Zheng
XH and Rong JH: Bornyl caffeate induces apoptosis in human breast
cancer MCF-7 cells via the ROS- and JNK-mediated pathways. Acta
Pharmacol Sin. 35:113–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kurokawa M, Mitani K, Yamagata T,
Takahashi T, Izutsu K, Ogawa S, Moriguchi T, Nishida E, Yazaki Y
and Hirai H: The evi-1 oncoprotein inhibits c-Jun N-terminal kinase
and prevents stress-induced cell death. Embo J. 19:2958–2968. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Garrison SP, Phillips DC, Jeffers JR,
Chipuk JE, Parsons MJ, Rehg JE, Opferman JT, Green DR and Zambetti
GP: Genetically defining the mechanism of Puma- and Bim-induced
apoptosis. Cell Death Differ. 19:642–649. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rossé T, Olivier R, Monney L, Rager M,
Conus S, Fellay I, Jansen B and Borner C: Bcl-2 prolongs cell
survival after Bax-induced release of cytochrome c. Nature.
391:496–499. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brooks C and Dong Z: Regulation of
mitochondrial morphological dynamics during apoptosis by Bcl-2
family proteins: A key in Bak? Cell Cycle. 6:3043–3047. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng EH, Kirsch DG, Clem RJ, Ravi R,
Kastan MB, Bedi A, Ueno K and Hardwick JM: Conversion of Bcl-2 to a
Bax-like death effector by caspases. Science. 278:1966–1968. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Y, Goodyer C and LeBlanc A:
Selective and protracted apoptosis in human primary neurons
microinjected with active caspase-3, −6, −7, and −8. J Neurosci.
20:8384–8389. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cryns V and Yuan J: Proteases to die for.
Genes Dev. 12:1551–1570. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang Y, Duan W, Liang Z, Yi W, Yan J, Wang
N, Li Y, Chen W, Yu S, Jin Z and Yi D: Curcumin attenuates
endothelial cell oxidative stress injury through Notch signaling
inhibition. Cell Signal. 25:615–629. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lončarević-Vasiljković N, Milanović D,
Pešić V, Tešić V, Brkić M, Lazić D, Avramović V and Kanazir S:
Dietary restriction suppresses apoptotic cell death, promotes Bcl-2
and Bcl-xl mRNA expression and increases the Bcl-2/Bax protein
ratio in the rat cortex after cortical injury. Neurochem Int.
96:69–76. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sazawal S, Singh N, Mahapatra M and Saxena
R: Calreticulin mutation profile in Indian patients with primary
myelofibrosis. Hematology. 20:567–570. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chiang WF, Hwang TZ, Hour TC, Wang LH,
Chiu CC, Chen HR, Wu YJ, Wang CC, Wang LF, Chien CY, et al:
Calreticulin, an endoplasmic reticulum-resident protein, is highly
expressed and essential for cell proliferation and migration in
oral squamous cell carcinoma. Oral Oncol. 49:534–541. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Raghavan M, Wijeyesakere SJ, Peters LR and
Del Cid N: Calreticulin in the immune system: Ins and outs. Trends
Immunol. 34:13–21. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li X, Wang X, Tan Z, Chen S and Guan F:
Role of glycans in cancer cells undergoing epithelial-mesenchymal
transition. Front Oncol. 6:332016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Morra L and Moch H: Periostin expression
and epithelial-mesenchymal transition in cancer: A review and an
update. Virchows Arch. 459:465–475. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xiong H, Hong J, Du W, Lin YW, Ren LL,
Wang YC, Su WY, Wang JL, Cui Y, Wang ZH and Fang JY: Roles of STAT3
and ZEB1 proteins in E-cadherin down-regulation and human
colorectal cancer epithelial-mesenchymal transition. J Biol Chem.
287:5819–5832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang Y, Dey S and Matsunami H:
Calreticulin: Roles in cell-surface protein expression. Membranes
(Basel). 4:630–641. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lu Z, Wang J, Zheng T, Liang Y, Yin D,
Song R, Pei T, Pan S, Jiang H and Liu L: FTY720 inhibits
proliferation and epithelial-mesenchymal transition in
cholangiocarcinoma by inactivating STAT3 signaling. BMC Cancer.
14:7832014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ihara Y, Inai Y and Ikezaki M: Alteration
of integrin-dependent adhesion and signaling in EMT-like MDCK cells
established through overexpression of calreticulin. J Cell Biochem.
112:2518–2528. 2011. View Article : Google Scholar : PubMed/NCBI
|