Introduction
Programmed cell death domain 2 (PDCD2), is a highly
conserved zinc finger MYND domain-containing protein and is
expressed in a variety of tissues (1). The original PDCD2 clone (RP-8) was
isolated from a rat gene that was associated with programmed cell
death (2). Generally, PDCD2
contributes to stem cell activity and tissue remodeling by the
induction of apoptosis (3).
Accumulating data demonstrated that PDCD2 is involved in the
development of cancer. For example, the expression of PDCD2 is
decreased in gastric cancer tissue, and it may induce gastric
cancer cell growth arrest and apoptosis in a p53-dependent manner
(4,5). PDCD2 serves as a tumor suppresser
gene involved in the pathogenesis of osteosarcoma (3). However, its functions in
carcinogenesis are debatable. For example, in human acute leukemia
cells, PDCD2 was identified to be expressed at a high level, and
its knockdown impaired cancer cell proliferation, suggesting that
PDCD2 significantly facilitates leukemia progression (6). A previous study demonstrated that
PDCD2 is downregulated in drug-resistant breast cancer cells,
indicating that PDCD2 may be involved in the process of the
acquisition of multidrug resistance (MDR) (7). However, at present, the underlying
mechanism of the involvement of PDCD2 in drug resistance in liver
cancer cells remains to be elucidated.
Liver cancer is the fifth most common type of cancer
worldwide, and is the third most frequent cause of
cancer-associated mortality to the poor prognosis and rapid
progression (8). Chemotherapy
remains an optional treatment for liver cancer. However, drug
resistance in patients diagnosed with liver cancer frequently leads
to the failure of chemotherapeutic administration (9). At present, the molecular mechanisms
underlying drug resistance remain to be fully understood.
Elucidating the molecular mechanisms of MDR is urgently required
for the development of effective chemotherapeutic drugs. The
activation of epithelial-mesenchymal transition (EMT) serves a
principal role in the process of MDR (10). Cancer stem cell (CSC)-like cells
may facilitate tumor cell acquisition of chemotherapy and
radiotherapy resistance by the activation of EMT (11). The CSC-like cells are responsible
for drug resistance and tumor metastasis, and are the principal
reason for tumor treatment failure and cancer-associated mortality
(12). Clinically, sorafenib is
the first-line treatment drug to prolong the overall survival rate
of patients with advanced liver cancer (13). However, drug resistance of
sorafenib remains a primary challenge in improving the prognoses of
patients with liver cancer (14).
Generally, sorafenib exerts an inhibitory function against EMT via
the inhibition of mitogen-activated protein kinase (MAPK) signaling
and expression of Snail in liver cancer (15). However, sorafenib-resistant liver
cancer cells exhibit EMT and MDR phenotypes, indicating that EMT is
important in sorafenib-resistant liver cancer cells (16,17).
Therefore, identifying the molecular mechanism underlying sorafenib
resistance is indispensable for the development of effective
chemotherapeutic treatments.
In the present study, it was demonstrated that PDCD2
was decreased in the sorafenib-resistant HepG2 cell line and that
the overexpression of PDCD2 increased the sensitivity of
chemoresistant HepG2 cells to sorafenib. Following experiments
demonstrated that PDCD2 increased the expression of apoptotic
proteins, suppressed resistant HepG2 cell metastasis and led to an
elevated apoptotic rate when treated with sorafenib.
Mechanistically, PDCD2 inhibited EMT, possibly in a Snail-dependent
manner. Taken together, the present study preliminarily
demonstrated that PDCD2 serves as a pivotal molecule to overcome
therapy failure in the treatment of resistant liver cancer.
Materials and methods
Cell line and vectors
The HepG2 human liver cancer cell line was obtained
from the Shanghai Institute of Cell Biology, Chinese Academy of
Sciences (Shanghai, China). The related sorafenib-resistant cell
line (HepG2/SF) was generated by exposing cells to increasing
concentrations (≤2 µM) of sorafenib. The MDR phenotype was
evidenced by the half maximal inhibitory concentration (IC50; data
not shown). These cells were maintained in RPMI-1640 (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS, Gibco; Thermo Fisher Scientific, Inc.) at
37°C in a 5% CO2 humidified incubator. The PDCD2
expression vector was constructed in the Laboratory of
Hepatobiliary-Pancreatic Surgery (Jilin University, Jilin, China).
The PDCD2 lentiviral vector and Snail interfering vector (Snail-sh)
were constructed by Shanghai GeneChem Co., Ltd. (Shanghai, China),
and were transfected into HepG2 cells at a 10 multiplicity of
infection (MOI) value. After 48 h, the cells were used in
subsequent experiments.
Analysis of cell viability
Cell viability was detected using the Cell Counting
Kit-8 (Dojindo Molecular Technologies, Inc., Shanghai, China). Each
experiment was repeated three times. In detail, the
sorafenib-resistant cell line (HepG2/SF) was plated into 96-well
plates with a total of 2×103 cells/well and subsequently
treated with sorafenib (concentrations ranged between 0 and 15 µM)
at 37°C for 24 h following transfection with or without PDCD2. The
cytotoxicity index was calculated as (1-OD450 of drug-treated
cells/OD450 of untreated cells) ×100 according to a previous study
(18). The IC50 values of
sorafenib were calculated using GraphPad Prism 5 (GraphPad
Software, Inc., La Jolla, CA, USA).
Transfection and cell migration
assay
PDCD2 lentiviral vector was transfected into HepG2
cells at a 10 MOI value, according to the manufacturer's protocol
(Shanghai GeneChem Co., Ltd.). After 48 h, the cells were assessed
to detect cell migration using polycarbonate membranes with an 8-µm
pore size (Corning, Inc., Corning, NY, USA). The cells
(6×104) were seeded into the upper chamber with 200 µl
serum-free medium, and the upper chambers were subsequently placed
onto the lower chambers of 24-well culture dishes containing 500 µl
RPMI-1640 containing 10% FBS. After 48 h, cells that had migrated
to the outer side of the membranes were fixed with 4%
paraformaldehyde for 30 min at room temperature and stained with
0.1% crystal violet for 20 min at room temperature. The number of
migrated cells was counted under a light microscope (magnification,
×100; Olympus CKX31; Olympus Corporation, Tokyo, Japan).
Consistently, the snail interfering vector (Snail-sh) was
additionally transfected into HepG2 cells at a 10 MOI value, and
after 48 h, the cells were used in the following experiments.
Western blot analysis
The total proteins from cells were extracted and
prepared using radioimmunoprecipitation assay buffer containing
phenylmethanesulfonyl fluoride (Thermo Fisher Scientific, Inc.).
Lysates were subsequently centrifuged at 11,000 × g for 15 min at
4°C. The protein concentration was determined using a Bicinchoninic
Acid Protein Assay kit (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). A total of 15 µg proteins in the supernatant were
separated on 10% SDS-PAGE and subsequently transferred to
polyvinylidene difluoride membranes. TBS with Tween 20 containing
5% nonfat milk powder (w/v) was used to block the membranes for 2 h
at room temperature. The membranes were incubated with primary
antibodies against PDCD2 (1:2,000; cat. no. ab133324; Abcam,
Cambridge, MA, USA), MDR1 (1:1,000; cat. no. 901401; BioLegend,
Inc., San Diego, CA, USA), matrix metalloproteinase (MMP)2
(1:2,000; cat. no. 87809; Cell Signaling Technology, Inc., Danvers,
MA, USA), MMP9 (1:2,000; cat. no. 2270; Cell Signaling Technology,
Inc.), Caspase 3 (1:1,000; cat. no. 9665; Cell Signaling
Technology, Inc.), Vimentin (1:1,000; cat. no. 5741; Cell Signaling
Technology, Inc.), E-cadherin (1:1,000; cat. no. 3195; Cell
Signaling Technology, Inc.) and Snail (1:1,000; cat. no. 3895; Cell
Signaling Technology, Inc.) for 12 h at 4°C, respectively. GAPDH
was used as the internal control (1:1,000; cat. no. 5174; Cell
Signaling Technology, Inc.). The membranes were subsequently
incubated with horseradish peroxidase-conjugated secondary
antibodies (1:5,000; cat. nos. A27022 and A16169; Thermo Fisher
Scientific, Inc.) for 2 h at room temperature. An Enhanced
Chemiluminescent Substrate Reagent kit (Thermo Fisher Scientific,
Inc.) was used to detect the bound antibodies. Finally, protein
expression was quantified using the Carestream IS4000MM Pro
Molecular Imaging System (cat. no. 8642985; Carestream Health,
Inc., Rochester, NY, USA) and analyzed with ImageJ software
(version no. 1.4.3.67; National Institutes of Health, Bethesda, MD,
USA). All experiments were performed in triplicate
independently.
Evaluation of apoptosis
The cells were transfected with PDCD2 and were
subsequently treated with sorafenib for 24 h. The apoptotic cells
were assessed using an Annexin V/fluorescein isothiocyanate and
propidium iodide apoptosis detection kit (Dojindo Molecular
Technologies, Inc.). Flow cytometry was used to measure apoptosis
using a Beckman Coulter flow cytometer (Beckman Coulter, Inc.,
Brea, CA, USA) and the data were analyzed with Kaluza software
(version. no 2.0; Beckman Coulter, Inc.).
Statistical analysis
GraphPad Prism 7.0 (GraphPad Software, Inc.) was
used to perform data analysis. All data are presented as the mean ±
standard deviation of at least three independent experiments.
Student's t-test was used to determine significant differences
between groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
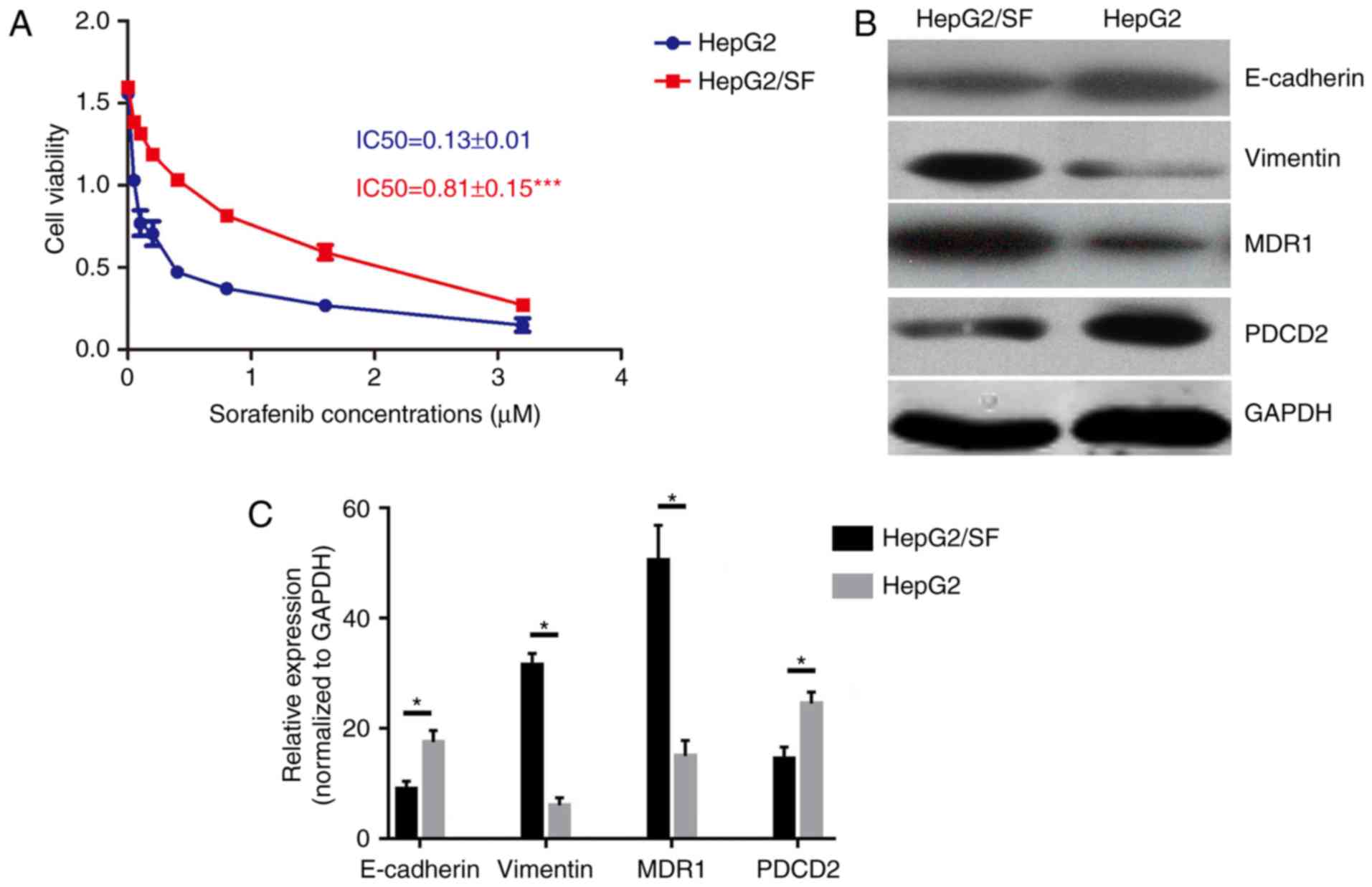
Sorafenib-resistant HepG2 cells
exhibit EMT and MDR phenotypes, and decreased expression of
PDCD2
The sorafenib-resistant HepG2 cell line (HepG2/SF)
was successfully established, determined by the IC50 for sorafenib
(Fig. 1A). The MDR1 level, which
represents the MDR phenotype, was significantly increased in the
sorafenib-resistant HepG2 cell line compared with that in its
matched sensitive cell line (Fig. 1B
and C). Secondly, the expression of PDCD2 was analyzed, and the
results demonstrated that the sorafenib-resistant HepG2 cells had a
lower expression level of PDCD2. Furthermore, the expression of
epithelial marker E-cadherin was decreased and that of the
mesenchymal marker Vimentin was increased (Fig. 1B and C). The reduced expression of
PDCD2 and the elevated EMT and MDR in the sorafenib-resistant HepG2
cell line preliminarily indicated that PDCD2 may be involved in the
formation of MDR by regulating EMT.
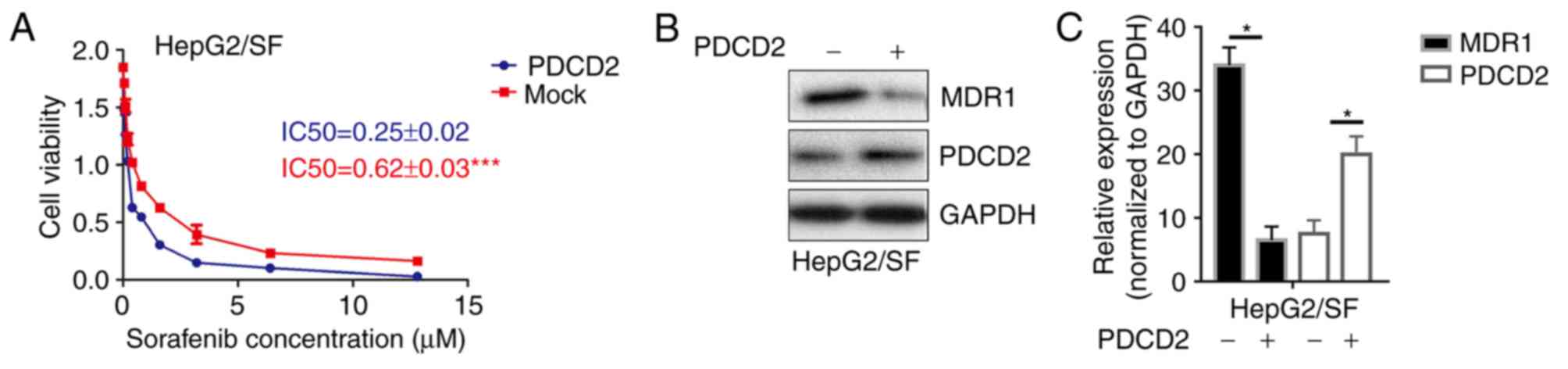
PDCD2 reverses the drug resistance of
sorafenib-resistant HepG2 cells
To further examine the function of PDCD2 in the
process of MDR, the IC50 was detected following transfection of the
sorafenib-resistant HepG2 cells with PDCD2. The results showed that
the overexpression of PDCD2 significantly increased the
sorafenib-induced cytotoxicity (Fig.
2A). Furthermore, the expression of MDR1 was decreased
following transfection with PDCD2 (Fig. 2B and C). The results preliminarily
demonstrated that PDCD2 may reverse the MDR of sorafenib-resistant
liver cancer cells.
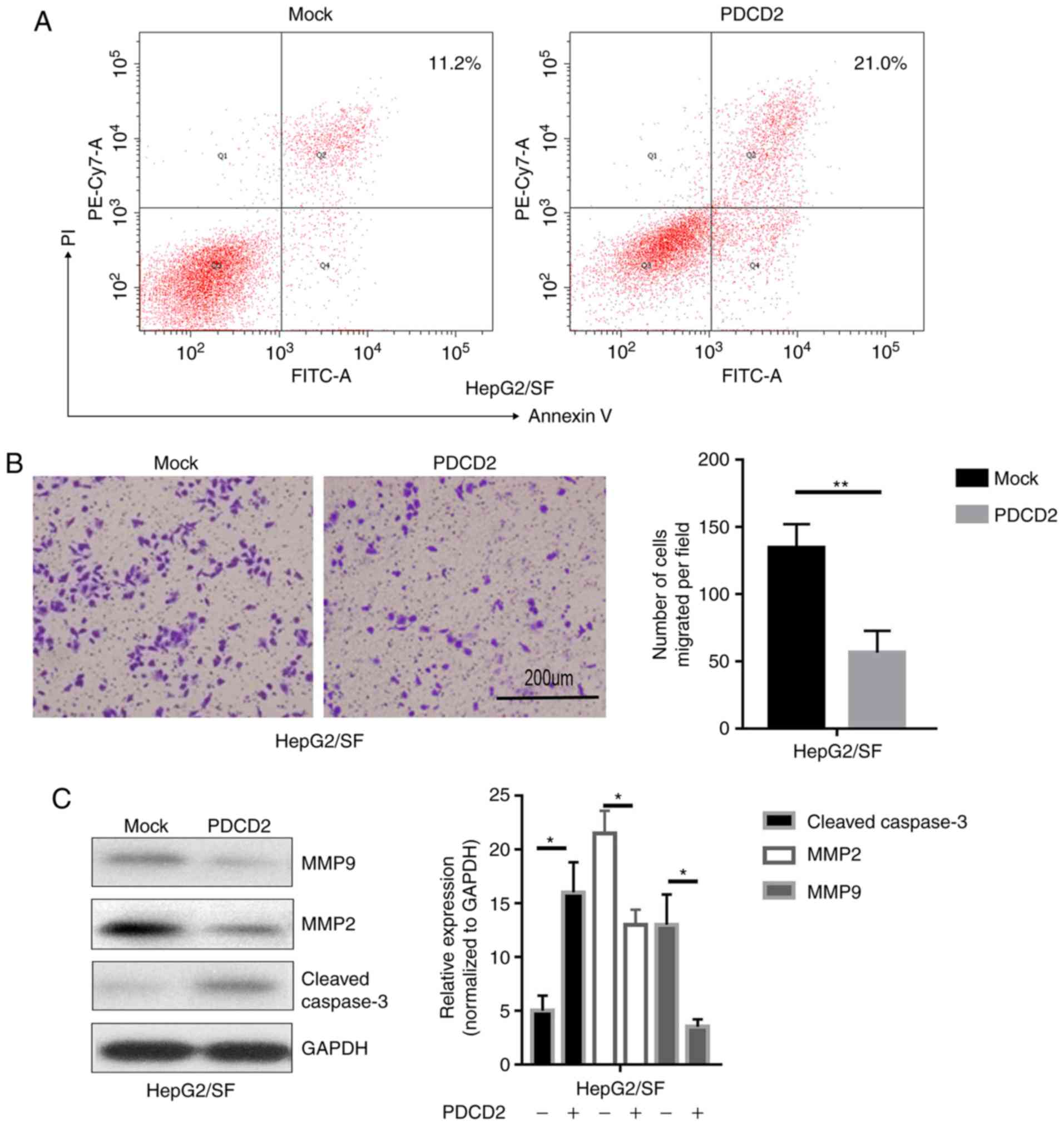
PDCD2 facilitates cell apoptosis and
suppresses cell migration in sorafenib-resistant HepG2 cells
To further detect the function of PDCD2 involved in
MDR, the apoptotic rates of sorafenib-resistant HepG2 cells
transfected with PDCD2 were measured following treatment with
sorafenib. As expected, the overexpression of PDCD2 enhanced
sorafenib-induced apoptosis in the drug-resistant cells as revealed
by flow cytometry (Fig. 3A).
Furthermore, PDCD2 reduced the migration ability of
sorafenib-resistant HepG2 cells (Fig.
3B). Consistent with the proposed function of PDCD2, the
results of the western blotting demonstrated that PDCD2 upregulated
the apoptotic-associated proteins and downregulated
migration-associated proteins (Fig.
3C).
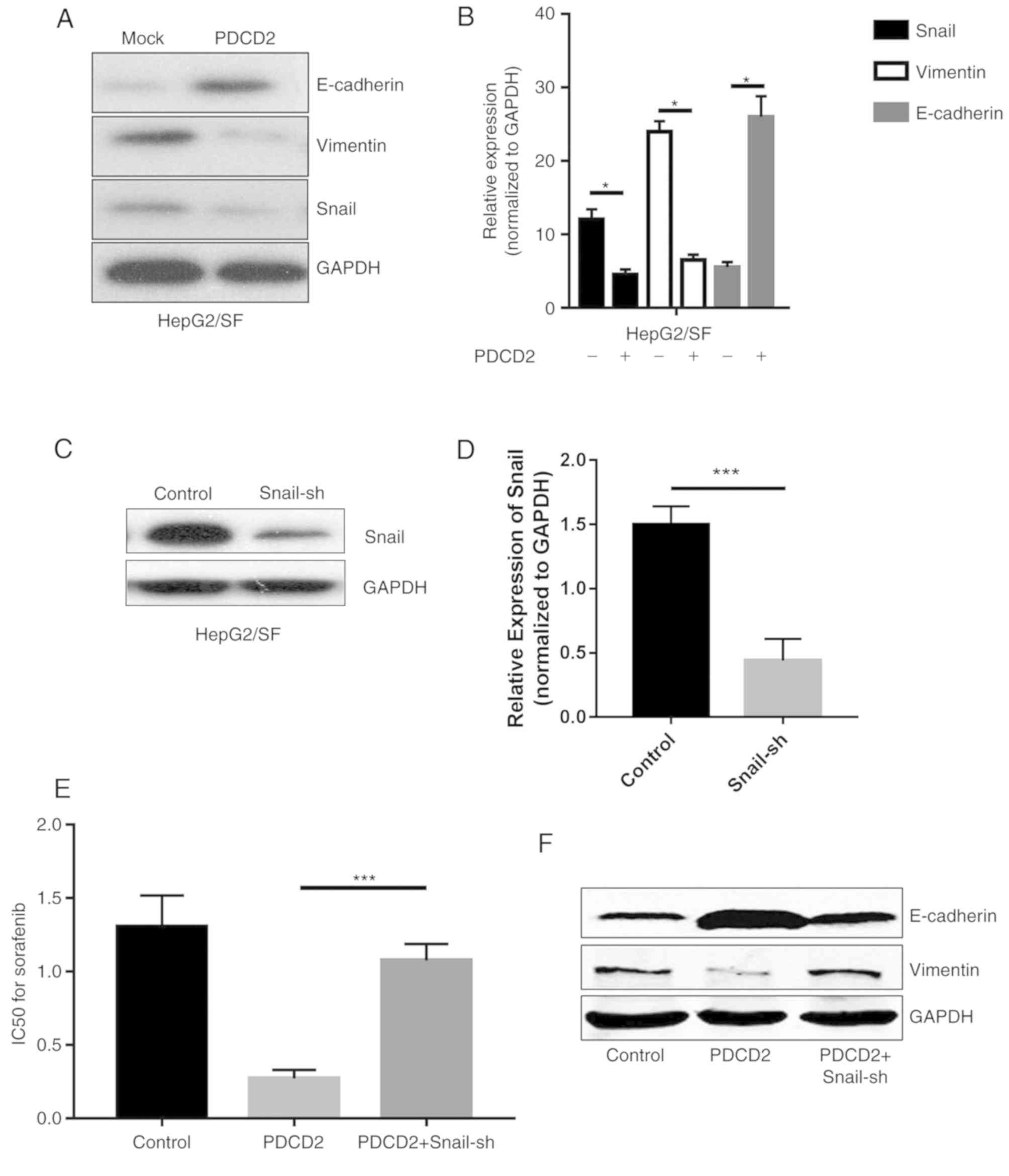
PDCD2 reduces EMT and MDR phenotypes
in a Snail-dependent manner in sorafenib-resistant HepG2 cells
Given the fact that PDCD2 sensitizes
sorafenib-resistant HepG2 cells to sorafenib, and the higher EMT
phenotype is responsible for MDR, it was hypothesized that PDCD2
may affect EMT and be involved in the process of reversing MDR. The
results of the western blotting demonstrated that the expression of
E-cadherin was increased whereas the expression of Vimentin was
decreased following transfection of the sorafenib-resistant HepG2
cells with PDCD2. Snail, a zinc-finger transcriptional repressor,
is critical in EMT-mediated tumor metastasis. Its expression was
additionally downregulated in the sorafenib-resistant HepG2 cells
transfected with PDCD2 (Fig. 4A and
B). A Snail interfering vector (Snail-sh) was transfected into
HepG2/SF cells, and the results demonstrated a significant
downregulation of Snail expression as presented in Fig. 4C and D. Cotransfection of the
HepG2/SF cells with PDCD2 and Snail-sh plasmids partially blunted
the MDR and EMT phenotypes, indicating that PDCD2 likely reverses
MDR and EMT in a Snail-dependent manner (Fig. 4E and F). From these results, it was
ascertained that PDCD2 controls EMT via the downregulation of
Snail.
Discussion
Sorafenib, with anti-angiogenic and
antiproliferative effects, is a multi-kinase inhibitor that
suppresses the MAPK/extracellular signal-regulated kinase, vascular
endothelial growth factor receptor and platelet-derived growth
factor receptor signaling pathways (15). Generally, treatment with sorafenib
leads to tumor angiogenesis suppression, cell cycle arrest and
elevated apoptosis (19,20). However, resistance to sorafenib is
a principal cause of antineoplastic treatment failure, particularly
in certain patients with advanced liver cancer under long-term
treatment, which causes oncogenic relapse or distant metastasis
(21). Accumulating evidence has
demonstrated that EMT is the principal cause of sorafenib
resistance in liver cancer cells (10,22).
In the present study, a sorafenib-resistant HepG2 cell line was
established from long-term exposure to high-dose sorafenib.
Therefore, the sorafenib-resistant HepG2 cells exhibited MDR, EMT
phenotypes and decreased expression of PDCD2, preliminarily
indicating that PDCD2 may be involved in the process of drug
resistance by modulating EMT.
To further investigate the function of PDCD2 in
sorafenib-resistant HepG2 cell lines in the present study, the IC50
value for sorafenib and the expression levels of MDR1 following
transfection with PDCD2 were detected. The results suggested that
the overexpression of PDCD2 enhanced the inhibitory effect of
sorafenib, as determined by the decreased IC50 value and decreased
expression of MDR1. Notably, the overexpression of PDCD2
significantly increased sorafenib-induced cytotoxicity and
apoptosis, and decreased the migration rate, demonstrating that
PDCD2 may reverse the MDR and EMT phenotypes in sorafenib-resistant
HepG2 cells. Generally, EMT is a normal developmental program that
promotes cancer cells to trigger abnormal cell migration, invasion
and drug resistance (12,23). Therefore, EMT, as a critical
regulator, is closely associated with the CSC phenotype and is a
prerequisite for metastasis. The induction of EMT in epithelial
cells leads to CSC characteristics, including increased stem-cell
marker expression, enhanced ability to metastasize and drug
resistance (24). A number of
previous studies have identified the association between EMT and
drug resistance (10–12,25).
In the present study, the increased expression of mesenchymal
markers, including vimentin, and the inhibition of epithelial
markers, including E-cadherin, were observed in the
sorafenib-resistant liver cancer cell lines. Mechanistically,
octamer binding transcription factor 4 and Nanog are essential for
the maintenance of the stem cell phenotype that hijacks liver
cancer cells with CSC and EMT phenotypes via activation of the
signal transducer and activator of transcription 3/Snail pathway
(26). Oncogenes, including
epidermal growth factor receptor, Akt and nuclear factor-κB
additionally contribute to EMT (27). However, the molecular mechanisms of
how PDCD2 influences sorafenib resistance by regulating EMT in
cancer cells require further elucidation.
Numerous mechanisms involved in MDR are important in
the drug resistance of liver cancer, including the drug efflux pump
(e.g. MDR1), EMT and DNA damage repair (28–30).
EMT is more associated with the acquisition of the MDR phenotypes
in liver cancer. For example, liver cancer cells with MDR have been
shown to exhibit enhanced metastatic activity, and upregulated
expression of N-cadherin and Vimentin in a calcium-dependent manner
(31). In addition, a previous
study demonstrated that liver cancer cells underwent EMT and
exhibited increased invasiveness and MDR phenotypes when exposed to
hypoxia (32). A number of
transcriptional repressors, including the Snail/Slug family,
function as a molecular switch of EMT (33). The present study examined the
crosstalk between EMT and MDR involved in the acquired drug
resistance to sorafenib in liver cancer, and demonstrated that the
expression of Snail was decreased when the cells overexpressed
PDCD2. The Snail transcription factor is pivotal in the expression
of mesenchymal markers, including Vimentin, MMP2 and MMP9 in liver
cancer cells (34).
Mechanistically, Snail is involved in EMT via the downregulation of
cell metastasis by binding several E-boxes located in the
E-cadherin promoter region (35).
The overexpression of Snail facilitates the acquisition of
P-glycoprotein-mediated MDR (36).
Co-transfection of PDCD2 and Snail-sh plasmids into HepG2/SF cells
partially blunted the MDR and EMT phenotypes, indicating that PDCD2
likely reversed MDR and EMT in a Snail-dependent manner. Therefore,
the results indicated that PDCD2 modulates EMT by the suppression
of Snail in drug-resistant liver cancer cells.
In conclusion, the present study demonstrated that
sorafenib-resistant HepG2 cells exhibit EMT, MDR phenotypes and
downregulated expression of PDCD2. The overexpression of PDCD2
suppressed sorafenib-resistant HepG2 cells from undergoing EMT and
metastasis, and promoted cell apoptosis. Mechanistically, PDCD2
modulated EMT by the suppression of Snail in drug-resistant HepG2
cells. The results additionally identified that PDCD2, as a pivotal
regulator of EMT, may serve as a potential therapeutic target in
the treatment of sorafenib-resistant liver cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LG and HL designed and conducted the experiments; MW
and NL contributed to the statistical analysis. The manuscript was
drafted by LG. All authors read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
MDR
|
multidrug resistance
|
|
PDCD2
|
programmed cell death domain 2
|
References
|
1
|
Baron BW, Anastasi J, Thirman MJ, Furukawa
Y, Fears S, Kim DC, Simone F, Birkenbach M, Montag A, Sadhu A, et
al: The human programmed cell death-2 (PDCD2) gene is a target of
BCL6 repression: Implications for a role of BCL6 in the
down-regulation of apoptosis. Proc Natl Acad Sci USA. 99:2860–2865.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vaux DL and Häcker G: Cloning of mouse
RP-8 cDNA and its expression during apoptosis of lymphoid and
myeloid cells. DNA Cell Biol. 14:189–193. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang Y, Jin Y and Du W: Programmed cell
death 2 functions as a tumor suppressor in osteosarcoma. Int J Clin
Exp Pathol. 8:10894–10900. 2015.PubMed/NCBI
|
|
4
|
Zhang J, Wei W, Jin HC, Ying RC, Zhu AK
and Zhang FJ: Programmed cell death 2 protein induces gastric
cancer cell growth arrest at the early S phase of the cell cycle
and apoptosis in a p53-dependent manner. Oncol Rep. 33:103–110.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang W, Song XW, Bu XM, Zhang N and Zhao
CH: PDCD2 and NCoR1 as putative tumor suppressors in gastric
gastrointestinal stromal tumors. Cell Oncol (Dordr). 39:129–137.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barboza N, Minakhina S, Medina DJ, Balsara
B, Greenwood S, Huzzy L, Rabson AB, Steward R and Schaar DG: PDCD2
functions in cancer cell proliferation and predicts relapsed
leukemia. Cancer Biol Ther. 14:546–555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kars MD, Iseri OD and Gündüz U: A
microarray based expression profiling of paclitaxel and vincristine
resistant MCF-7 cells. Eur J Pharmacol. 657:4–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rani B, Malfettone A, Dituri F, Soukupova
J, Lupo L, Mancarella S, Fabregat I and Giannelli G: Galunisertib
suppresses the staminal phenotype in hepatocellular carcinoma by
modulating CD44 expression. Cell Death Dis. 9:3732018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mir N, Jayachandran A, Dhungel B, Shrestha
R and Steel JC: Epithelial-to-mesenchymal transition: A mediator of
sorafenib resistance in advanced hepatocellular carcinoma. Curr
Cancer Drug Targets. 17:698–706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyazaki H, Takahashi RU, Prieto-Vila M,
Kawamura Y, Kondo S, Shirota T and Ochiya T: CD44 exerts a
functional role during EMT induction in cisplatin-resistant head
and neck cancer cells. Oncotarget. 9:10029–10041. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park JH, Shin JE and Park HW: The role of
hippo pathway in cancer stem cell biology. Mol Cells. 41:83–92.
2018.PubMed/NCBI
|
|
13
|
El-Khoueiry AB, O'Donnell R, Semrad TJ,
Mack P, Blanchard S, Bahary N, Jiang Y, Yen Y, Wright J, Chen H, et
al: A phase I trial of escalating doses of cixutumumab (IMC-A12)
and sorafenib in the treatment of advanced hepatocellular
carcinoma. Cancer Chemother Pharmacol. 81:957–963. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang K, Chen J, Zhou H, Chen Y, Zhi Y,
Zhang B, Chen L, Chu X, Wang R and Zhang C: PU.1/microRNA-142-3p
targets ATG5/ATG16L1 to inactivate autophagy and sensitize
hepatocellular carcinoma cells to sorafenib. Cell Death Dis.
9:3122018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nagai T, Arao T, Furuta K, Sakai K, Kudo
K, Kaneda H, Tamura D, Aomatsu K, Kimura H, Fujita Y, et al:
Sorafenib inhibits the hepatocyte growth factor-mediated epithelial
mesenchymal transition in hepatocellular carcinoma. Mol Cancer
Ther. 10:169–177. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dong J, Zhai B, Sun W, Hu F, Cheng H and
Xu J: Activation of phosphatidylinositol 3-kinase/AKT/snail
signaling pathway contributes to epithelial-mesenchymal
transition-induced multi-drug resistance to sorafenib in
hepatocellular carcinoma cells. PLoS One. 12:e01850882017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Malenstein H, Dekervel J, Verslype C,
Van Cutsem E, Windmolders P, Nevens F and van Pelt J: Long-term
exposure to sorafenib of liver cancer cells induces resistance with
epithelial-to-mesenchymal transition, increased invasion and risk
of rebound growth. Cancer Lett. 329:74–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao P, Wang S, Jiang J, Liu H, Zhu X,
Zhao N, Li J, Yin Y, Pan X, Yang X, et al: TIPE2 sensitizes
osteosarcoma cells to cis-platin by down-regulating MDR1 via the
TAK1-NF-κB and -AP-1 pathways. Mol Immunol. 101:471–478. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marra M, Sordelli IM, Lombardi A, Lamberti
M, Tarantino L, Giudice A, Stiuso P, Abbruzzese A, Sperlongano R,
Accardo M, et al: Molecular targets and oxidative stress biomarkers
in hepatocellular carcinoma: An overview. J Transl Med. 9:1712011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao P, Li M, Wang Y, Chen Y, He C, Zhang
X, Yang T, Lu Y, You J, Lee RJ and Xiang G: Enhancing anti-tumor
efficiency in hepatocellular carcinoma through the autophagy
inhibition by miR-375/sorafenib in lipid-coated calcium carbonate
nanoparticles. Acta Biomater. 72:248–255. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Caraglia M, Giuberti G, Marra M, Addeo R,
Montella L, Murolo M, Sperlongano P, Vincenzi B, Naviglio S, Prete
SD, et al: Oxidative stress and ERK1/2 phosphorylation as
predictors of outcome in hepatocellular carcinoma patients treated
with sorafenib plus octreotide LAR. Cell Death Dis. 2:e1502011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen J, Jin R, Zhao J, Liu J, Ying H, Yan
H, Zhou S, Liang Y, Huang D, Liang X, et al: Potential molecular,
cellular and microenvironmental mechanism of sorafenib resistance
in hepatocellular carcinoma. Cancer Lett. 367:1–11. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Otsuki Y, Saya H and Arima Y: Prospects
for new lung cancer treatments that target EMT signaling. Dev Dyn.
247:462–472. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou L, Lv X, Yang J, Zhu Y, Wang Z and Xu
T: Overexpression of Napsin A resensitizes drug-resistant lung
cancer A549 cells to gefitinib by inhibiting EMT. Oncol Lett.
16:2533–2538. 2018.PubMed/NCBI
|
|
26
|
Yin X, Zhang BH, Zheng SS, Gao DM, Qiu SJ,
Wu WZ and Ren ZG: Coexpression of gene Oct4 and Nanog initiates
stem cell characteristics in hepatocellular carcinoma and promotes
epithelial-mesenchymal transition through activation of Stat3/Snail
signaling. J Hematol Oncol. 8:232015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng HC: The molecular mechanisms of
chemoresistance in cancers. Oncotarget. 8:59950–59964.
2017.PubMed/NCBI
|
|
28
|
Teicher BA: Acute and chronic in vivo
therapeutic resistance. Biochem Pharmacol. 77:1665–1673. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
van Zijl F, Zulehner G, Petz M, Schneller
D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H and Mikulits
W: Epithelial-mesenchymal transition in hepatocellular carcinoma.
Future Oncol. 5:1169–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang H, Mizumachi T, Carcel-Trullols J,
Li L, Naito A, Spencer HJ, Spring PM, Smoller BR, Watson AJ,
Margison GP, et al: Targeting human 8-oxoguanine DNA glycosylase
(hOGG1) to mitochondria enhances cisplatin cytotoxicity in hepatoma
cells. Carcinogenesis. 28:1629–1637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wen L, Liang C, Chen E, Chen W, Liang F,
Zhi X, Wei T, Xue F, Li G, Yang Q, et al: Regulation of Multi-drug
Resistance in hepatocellular carcinoma cells is TRPC6/Calcium
dependent. Sci Rep. 6:232692016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiao M and Nan KJ: Activation of PI3
kinase/Akt/HIF-1α pathway contributes to hypoxia-induced
epithelial-mesenchymal transition and chemoresistance in
hepatocellular carcinoma. Int J Oncol. 40:461–468. 2012.PubMed/NCBI
|
|
33
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen JS, Li HS, Huang JQ, Zhang LJ, Chen
XL, Wang Q, Lei J, Feng JT, Liu Q and Huang XH: Down-regulation of
Gli-1 inhibits hepatocellular carcinoma cell migration and
invasion. Mol Cell Biochem. 393:283–291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zucchini-Pascal N, Peyre L and Rahmani R:
Crosstalk between beta-catenin and snail in the induction of
epithelial to mesenchymal transition in hepatocarcinoma: Role of
the ERK1/2 pathway. Int J Mol Sci. 14:20768–20792. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li W, Liu C, Tang Y, Li H, Zhou F and Lv
S: Overexpression of Snail accelerates adriamycin induction of
multidrug resistance in breast cancer cells. Asian Pac J Cancer
Prev. 12:2575–2580. 2011.PubMed/NCBI
|