Introduction
Acetyl-coenzyme A (CoA) carboxylases (ACCs) are the
most highly regulated enzymes in the fatty acid (FA) synthesis
pathway; they catalyse the carboxylation of acetyl-CoA into
malonyl-CoA, which represents the rate limiting step in de
novo FA synthesis (1–4). Additionally, two isoforms of ACC
encoded by two different genes in mammalian cells have been
described, ACC1 and ACC2; ACC1 is highly enriched in lipogenic
tissues (liver and adipose), while ACC2 is mainly expressed in
oxidative tissues (heart, skeletal muscle and liver) (5,6). As
they are located in a variety specialised tissues, ACC1 and ACC2
serve different metabolic roles. ACC1 generates malonyl-CoA for
de novo synthesis of long-chain FAs in the cytosol, while
ACC2 generates malonyl-CoA; thus carnitine palmitoyl transferase I
is inhibited, preventing FA degradation in the mitochondria
(3,5).
A previous study reported that ACC1 is overexpressed
in different human cancer cells, and is likely involved in
lipogenesis and the development and progression of tumours
(7). Knockdown or chemical
inhibition of ACC1 in prostate cancer cells has been successful in
inducing cell apoptosis (8).
Inhibition of ACC1 downregulates epidermal growth factor receptor
variant III (EGFRvIII) during human glioblastoma cell proliferation
and de novo lipogenesis (9). The interaction between ACC1 and
breast cancer 1 indicates the possible role of ACC1 in the
susceptibility to breast and ovarian cancers (10). A previous study reported that the
molecule is essential for breast cancer cell survival (11). Furthermore, ACC1 regulates
endothelial cell migration, and is associated with FA metabolism
and the migration of endothelial cells (7). ACCs have been used as targets for
treating metabolic diseases, including obesity and diabetes, and
its inhibitors have been developed in clinical trials (12–15).
In the present study, the mRNA expression profile of
ACC1 in certain types of cancer was investigated using the Oncomine
database, and the association between alterations in ACC1
expression and clinical outcomes in numerous types of cancers,
including liver, brain and kidney cancer, was analysed.
Furthermore, the effects of small interfering RNA (siRNA)-mediated
knockdown of ACC1 on the rat liver cell line BRL 3A and human
hepatoma Hep G2 cells were determined.
Materials and methods
Oncomine database analysis
The mRNA expression levels of ACC1 in various types
of cancers were analysed using the Oncomine database (https://www.oncomine.org/resource/login.html)
(16). Cancer tissues were
compared with normal tissues using t-tests, and the threshold was
set to a P<0.0001, fold change >2 and gene ranking in the top
10%. Roessler liver normal and cancer tissue samples were used in
the present study (datasets GSE1898 and GSE4024) (17).
Kaplan-Meier survival analysis
The association between ACC1 expression and survival
time of patients was determined using SurvExpress (http://bioinformatica.mty.itesm.mx/SurvExpress)
(18). The risk groups were
produced using an optimization algorithm from the ordered
prognostic index (PI), which is commonly used to generate risk
groups: A log-rank test was employed among all values of arranged
PI for two groups and the minimum P-value was selected as the
cut-off point.
Cell culture
The liver cell lines BRL 3A and Hep G2 were obtained
from the American Type Culture Collection (CRL-1442™ and HB-8065™,
Manassas, VA, USA). Cells were cultured in Dulbecco's Modified
Eagle's medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in an atmosphere containing 5%
CO2.
Knockdown of ACC1 with siRNA
treatment
BRL 3A and Hep G2 cells were plated with 10% FBS
medium at a density of 200,000 cells/well in six-well plates, and
incubated overnight at 37°C in a humidified incubator with 5%
CO2. The following day, the cells were treated with 50
nmol ACC1-targeting siRNA or an identical concentration of negative
control (NC) siRNA (siControl) formulated into lipid complexes
using Lipofectamine® RNAiMax (Thermo Fisher Scientific,
Inc.) transfection reagent. SiRNA-transfected cells were collected
24, 48 and 72 h post-transfection for quantitative polymerase chain
reaction (qPCR) and western blot analysis. human (h)ACC1 siRNAs
[sihACC1-(1–3) and sihACC1-2] and siControl were
synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). The
same NC siRNA was used for the transfection of BRL 3A and Hep G2
cells; the sequence was random and unrelated to the human or rat
genome. The sequences of siControl and siACC1 are presented in
Table I.
 | Table I.Sequences of ACC1 siRNAs. |
Table I.
Sequences of ACC1 siRNAs.
| siRNA | Sense | Antisense |
|---|
| hACC1-1 |
5′-GCUUCUACUUUCUGGAAUUTT-3′ |
5′-AAUUCCAGAAAGUAGAAGCTT-3′ |
| hACC1-2 |
5′-GCUCAUACACUUCUGAAUATT-3′ |
5′-UAUUCAGAAGUGUAUGAGCTT-3′ |
| hACC1-3 |
5′-GCAGCUAUGUUCAGAGAAUTT-3′ |
5′-AUUCUCUGAACAUAGCUGCTT-3′ |
| rACC1 |
5′-GCUGGAGACAGAAAGCUUUTT-3′ |
5′-AAGCUGGAGACAGAAAGCUTT-3′ |
| Control |
5′-UUCUCCGAACGUGUCACGUTT-3′ |
5′-ACGUGACACGUUCGGAGAATT-3′ |
RNA isolation and reverse
transcription-qPCR (RT-qPCR) analysis
Total RNA (from BRL 3A or HepG2 cells) was extracted
using TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocols. RNA (2 µg) was
used to synthesise the first strand of cDNA using the GoScript™
Reverse Transcription system (cat. no. A5001; Promega Corporation,
Madison, WI, USA). qPCR was performed on a Bio-Rad CFX96 PCR system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) with the SYBR Green
Master mix (Qiagen GmbH, Hilden, Germany) and the thermocycling
conditions used for all amplifications were: One cycle of 95°C for
2 min and 40 cycles of 95°C for 15 sec, 60°C for 15 sec, and 68°C
for 20 sec. mRNA values were normalised to the β-actin housekeeping
gene according to the 2−ΔΔCq method (19). Three replicates were performed for
each sample. The primers were synthesised by Shanghai Generay
Biotech Co., Ltd. (Shanghai, China) and are presented in Table II.
| Table II.Primer sequences used in reverse
transcription-quantitative polymerase chain reaction. |
Table II.
Primer sequences used in reverse
transcription-quantitative polymerase chain reaction.
| Genes | Forward primer | Reverse primer |
|---|
| hACC1 |
5′-GCTCCTTGTCACCTGCTTCT-3′ |
5′-CAAGGCCAAGCCATCCTGTA-3′ |
| rACC1 |
5′-TTCTTCTACTGGCGACTGAG-3′ |
5′-TCCCTGCTGATGTATTTGAT-3′ |
| CCND1 |
5′-AAAATGCCAGAGGCGGATGA-3′ |
5′-GAAAGTGCGTTGTGCGGTAG-3′ |
| JUN |
5′-GGCTGTTCATCTGTTTGTCTTCAT-3′ |
5′-CCCTTTTCTTTACGGTCTCGGT-3′ |
| MYC |
5′-ACCCAACATCAGCGGTCG-3′ |
5′-CGTGACTGTCGGGTTTTCCA-3′ |
| CCNA2 |
5′-CTTTTAGTGCCGCTGTCTCTTT-3′ |
5′-GCCCGCATACTGTTAGTGATGT-3′ |
| hβ-actin |
5′-AAATCTGGCACCACACCTTC-3′ |
5′-GGGGTGTTGAAGGTCTCAAA-3′ |
| rβ-actin |
5′-ACATCCGTAAAGACCTCTATGCCAACA-3′ |
5′-GTGCTAGGAGCCAGGGCAGTAATCT-3′ |
Cell viability assay
Cells were plated at a density of 10,000 cells/well
in 96-well plates. On the second day, cells were transfected with
50 nmol siACC1 or siControl. An MTT assay (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) was used to measure the viability of BRL
3A and Hep G2 cells 24, 48 and 72 h post-treatment with siRNA.
Briefly, 10% v/v of 5 mg/ml MTT was added to each well, after which
the cells were incubated at 37°C for 4 h. The supernatant was
discarded, and 0.2 ml of dimethyl sulfoxide (Sigma-Aldrich; Merck
KGaA) was added to each of the wells. The wells were gently
agitated for 10 min at room temperature, and the absorbance of each
well was measured at 570 nm by an ELx808 absorbance reader (BioTek
Instruments, Inc., Winooski, VT, USA) (20).
Cell migration assay
Transfected cells (100,000 cells/well) were seeded
in 24-well plates. The cell layer was scratched with the tip of a
10-µl pipette. The healing process was observed for 48 and 96 h.
The width of the wound was measured 48 and 96 h after scratching in
order to evaluate the wound healing ability of the cells. Images of
the migrated cells were taken using a microscope (magnification,
×10; Nikon Eclipse 80i; Nikon Corporation, Tokyo, Japan) and the
number of cells permeating the septum were counted in five random
fields.
Cell cycle analysis
Nuclear DNA content can be quantitatively measured
at high speed by flow cytometry. Cells were seeded into six-well
plates and transfected with siACC1 or siControl at a final
concentration of 50 nmol. After 48 h, the cells were harvested,
washed with cold PBS, and then fixed with 70% alcohol at ~20°C
overnight. The fixed cells were washed with cold PBS 2–3 times; PBS
was discarded and the cells were incubated in 1 ml of PBS
containing 50 µg propidium iodide (Sigma-Aldrich; Merck KGaA) and
100 µg RNase A (Sigma-Aldrich; Merck KGaA) for 30 min at 37°C.
Samples were then analysed for DNA content by flow cytometry with a
FACScan instrument with BD FACStation software version 5.2 (BD
Biosciences, Franklin Lakes, NJ, USA).
Western blot analysis
Cells were washed with ice-cold PBS and lysed with
radioimmunoprecipitation assay lysis buffer (50 mmol Tris, 150 mmol
NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing
protease inhibitors (Roche Diagnostics, Basel, Switzerland).
Protein concentrations were determined by a bicinchoninic acid
protein assay kit (Tiangen Biotech Co., Ltd., Beijing, China). SDS
loading sample buffer was applied to the proteins, which was
subsequently heated at 95°C for 5 min. Proteins (50 µg/lane) were
separated by 12% SDS-PAGE and transferred to nitrocellulose
membranes (GE Healthcare Life Sciences, Little Chalfont, UK). The
membranes were first blocked with 5% non-fat milk in Tris-buffered
saline containing 0.1% Tween-20 for 2 h at room temperature and
subsequently incubated overnight at 4°C with the following primary
antibodies: Rabbit anti-ACC1 (cat. no. BM4414; 1:1,000), rabbit
anti-cyclin A2 (CCNA2; cat. no. PB0402; 1:1,000), rabbit
anti-n-MYCN (cat. no. PB0769; 1:1,000), rabbit anti-Cyclin D1
(CCND1) (BM0771; 1:1,000) and rabbit anti-JUN (BA0208-2; 1:1,000;
all from Boster Biological Technology, Pleasanton, CA, USA). The
membrane was further incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG (cat. no. 12-348;
1:5,000; Sigma-Aldrich; Merck KGaA) as a secondary antibody for 1 h
at room temperature. Bands were visualized with BeyoECL plus
reagent (Beyotime Institute of Biotechnology, Haimen, China) and
the band density was measured using ImageQuant TL version 1.1 (GE
Healthcare Life Sciences) with β-actin (A1978; 1:1,000;
Sigma-Aldrich; Merck KGaA) as the internal reference.
Statistical analysis
All data are presented as the mean ± standard
deviation of three independent experiments. SPSS 19.0 (IBM Corp.,
Armonk, NY, USA) was used for statistical analyses. Student's
t-tests were used to compare the means of two groups. One-way
analyses of variance with Bonferroni's correction was used to
compare the means of three or more groups. Survival curves were
generated using a Kaplan-Meier analysis and the log-rank test was
used to determine P-values. P<0.05 was considered to indicate a
statistically significant difference.
Results
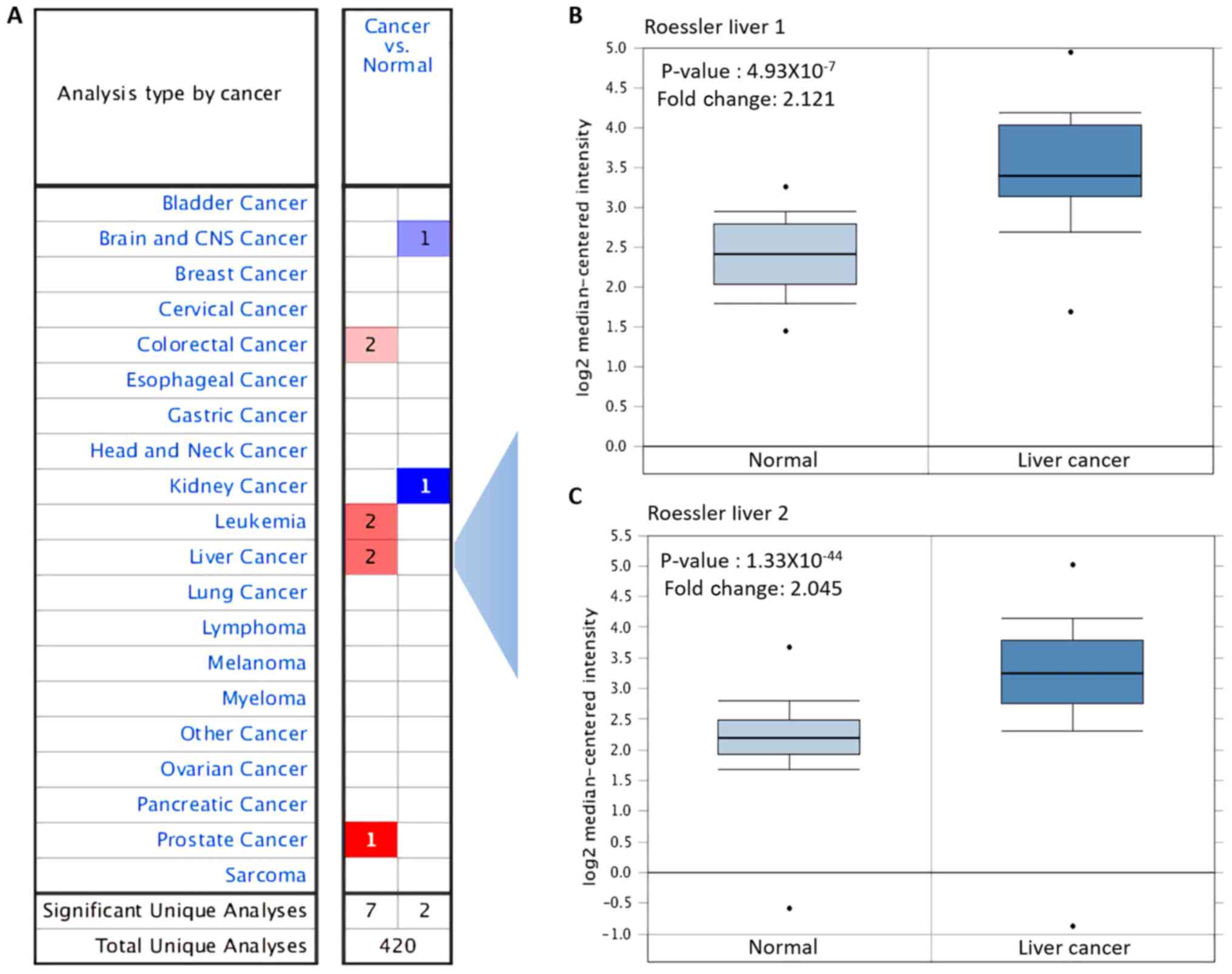
Oncomine database analysis
The expression levels of ACC1 mRNA in various types
of cancer and normal tissue were investigated using the Oncomine
database. Compared with the levels in the corresponding normal
tissues, ACC1 was overexpressed in colorectal, leukaemia, liver and
prostate cancer, but was downregulated in brain, central nervous
system and kidney cancer (Fig.
1A). Overexpression of ACC1 in liver cancer was analysed, with
P=4.93×10−7 and fold change, 2.121 reported in Roessler
liver; P=1.33×10−44 and fold change, 2.045 in Roessler
liver 2 (Table III; Fig. 1B and C).
| Table III.Overexpression of ACC1 in various
types of cancers. |
Table III.
Overexpression of ACC1 in various
types of cancers.
| Cancer type | P-value | Fold change |
|---|
| Roessler liver
1 |
4.93×10−07 | 2.121 |
| Roessler liver
2 |
1.33×10−44 | 2.045 |
| Colorectal
carcinoma |
1.95×10−09 | 2.297 |
| Sabates-Bellver
colon |
2.24×10−05 | 2.487 |
| Coustan-Smith
leukemia |
1.36×10−05 | 4.909 |
| Andersson
leukemia |
5.08×10−10 | 3.418 |
| Vanaja
prostate |
1.86×10−06 | 2.389 |
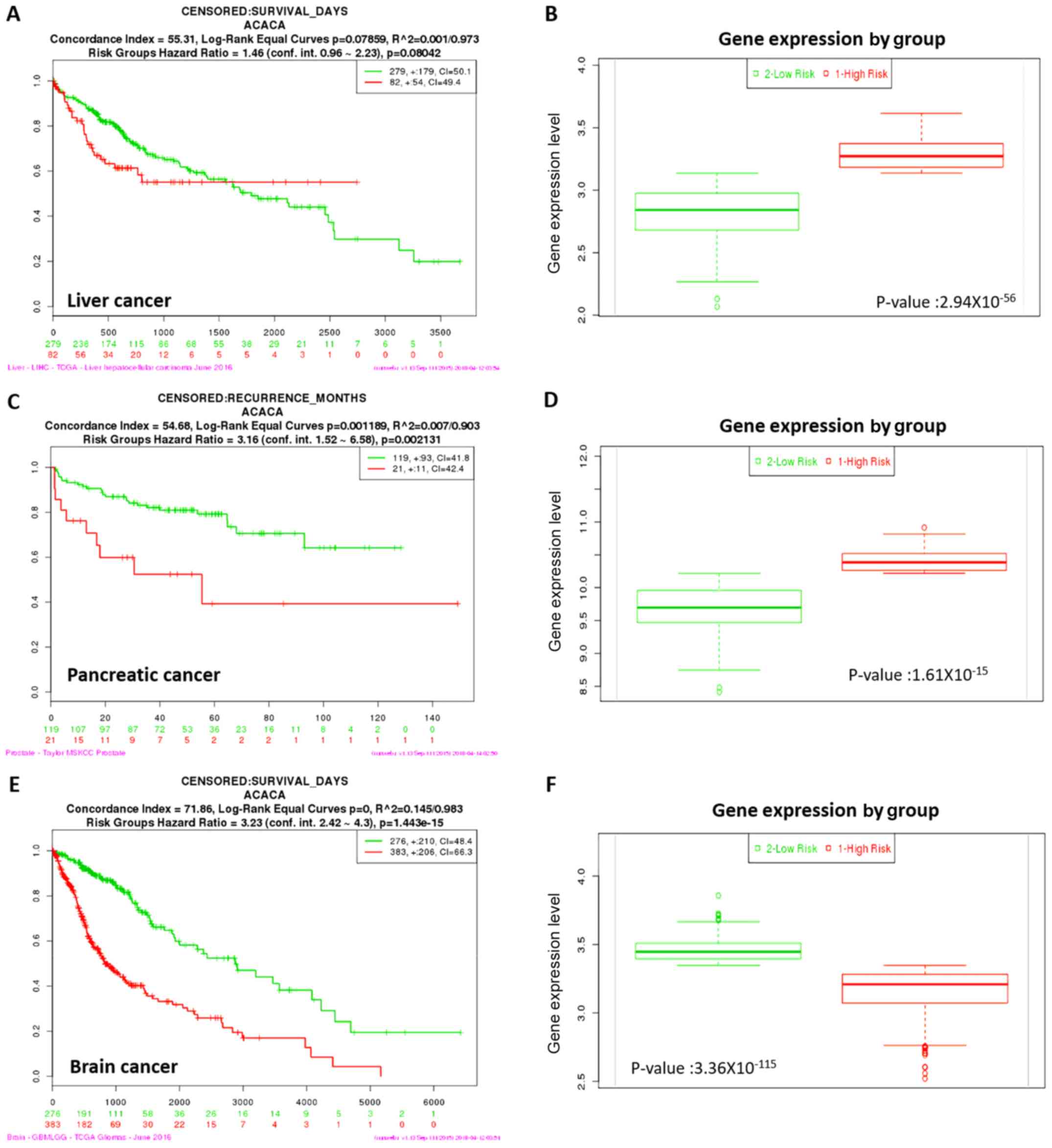
Kaplan-Meier survival analysis
Kaplan-Meier survival analysis was performed to
evaluate the prognostic value of ACC1 expression in liver,
pancreatic and brain cancer. Overexpression of ACC1 was associated
with increased risk, poorer prognosis and shorter overall survival
times for liver and pancreatic cancer (Fig. 2). Conversely, downregulation of
ACC1 was associated with lower risk, better prognosis and longer
overall survival times in brain cancer. Thus, ACC1 may be
considered to act as a tumour suppressor gene in brain cancer, and
as an oncogene in liver and pancreatic cancer.
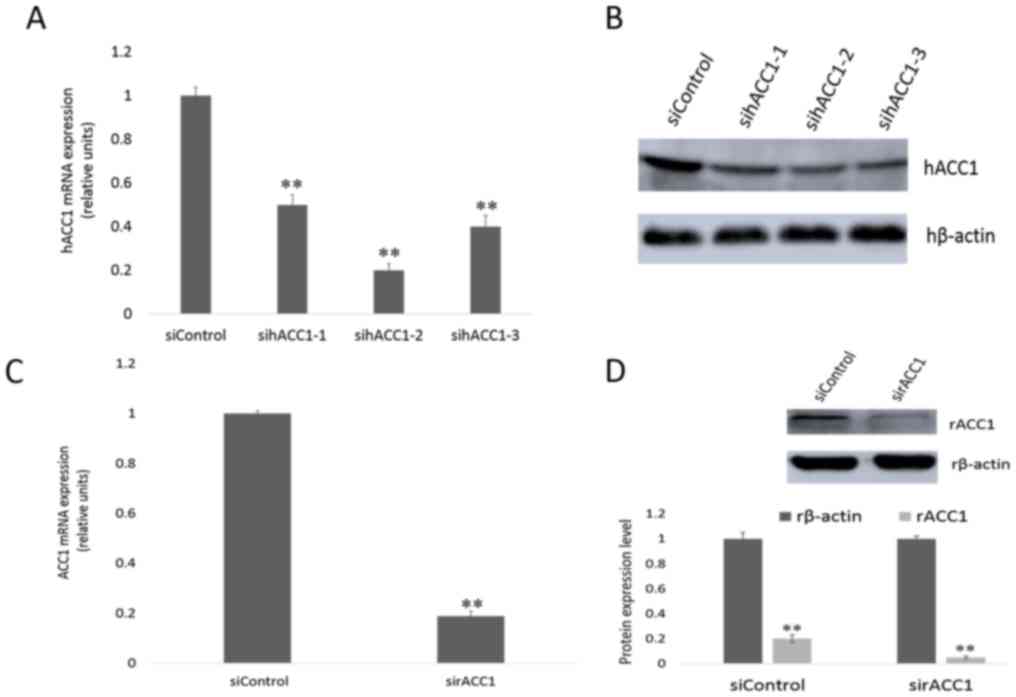
Effects of siRNA-targeting ACC1 on
mRNA and protein expression in Hep G2 and BRL 3A cells
To specifically silence ACC1 gene expression in Hep
G2 and BRL 3A cells, cells were transfected with siRNA targeting
ACC1 mRNA. RT-qPCR and western blotting were performed, and the
expression of ACC1 in transfected and control cells after 48 h was
detected. As presented in Fig. 3A,
human (h)ACC1 mRNA levels in Hep G2 cells were reduced to 0.50±0.04
(sihACC1-1), 0.20±0.03 (sihACC1-2) and 0.40±0.05 (sihACC1-3)
relative to those the expression of control cells. The protein
levels of hACC1 were determined (Fig.
3B); sihACC1-2 was selected for use in later experiments.
Furthermore, rat (r)ACC1 mRNA levels from the rat cell line BRL 3A
were significantly reduced to 0.19±0.01 compared with in control
cells (Fig. 3C); the protein
expression levels of ACC1 were notably suppressed (Fig. 3D).
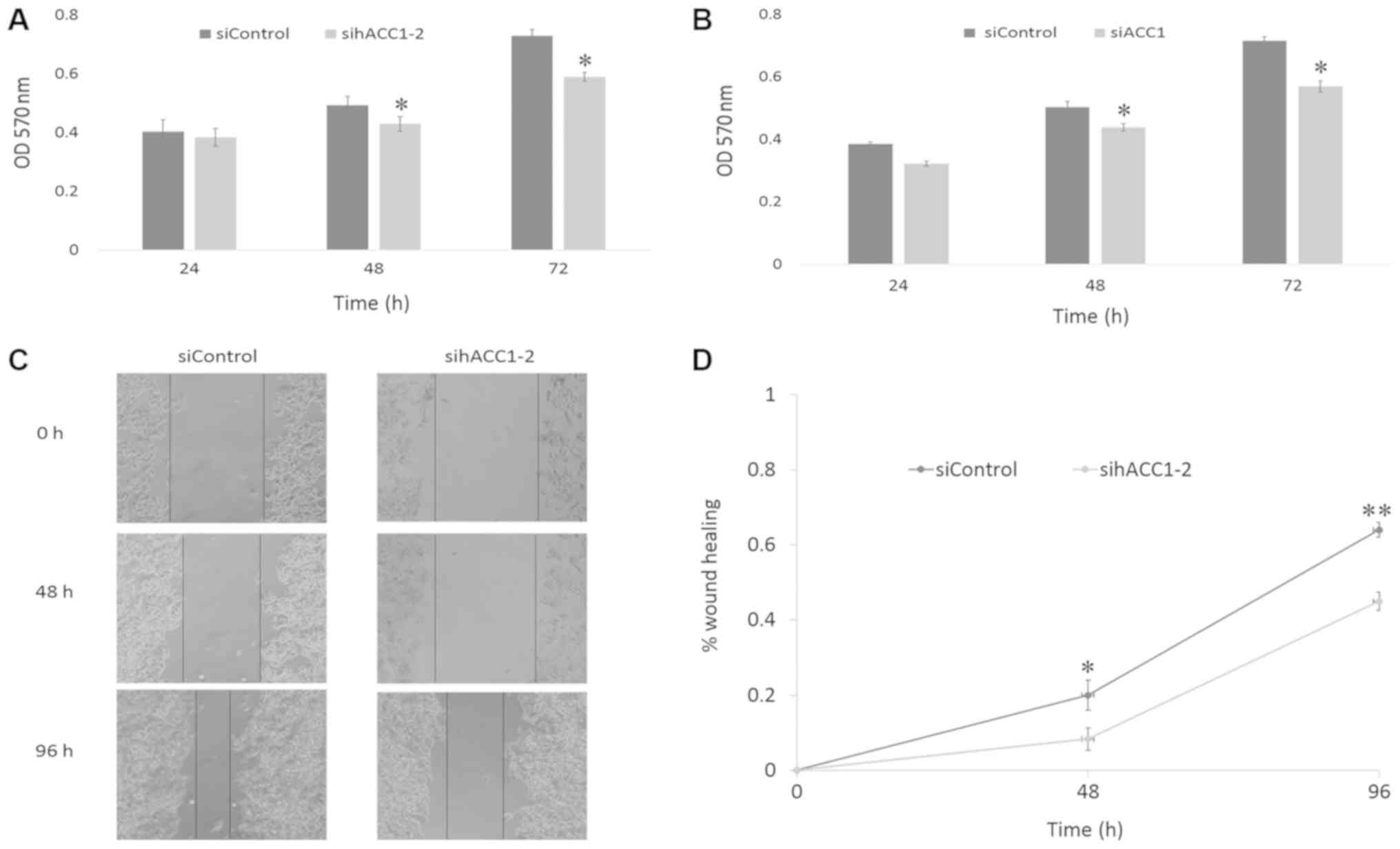
Effects of siRNA-targeting ACC1 on
cell viability and migration
To investigate the effects of ACC1 RNAi on cell
viability, an MTT assay was used to measure the viability of Hep G2
and BRL 3A cells 24, 48 and 72 h post-transfection with siRNA
targeting ACC1. As presented in Fig.
4A and B, cell viability began to decline after 24 h, and
significantly decreased at 48 and 72 h in Hep G2 and BRL 3A cells,
compared with the siControl. To further investigate the effects of
siACC1 on cell migration, Hep G2 cells were treated with sihACC1-2,
which significantly reduced cell migration at 48 and 96 h compared
with the siControl, respectively (Fig.
4C and D).
Effects of siRNA-targeting ACC1 on
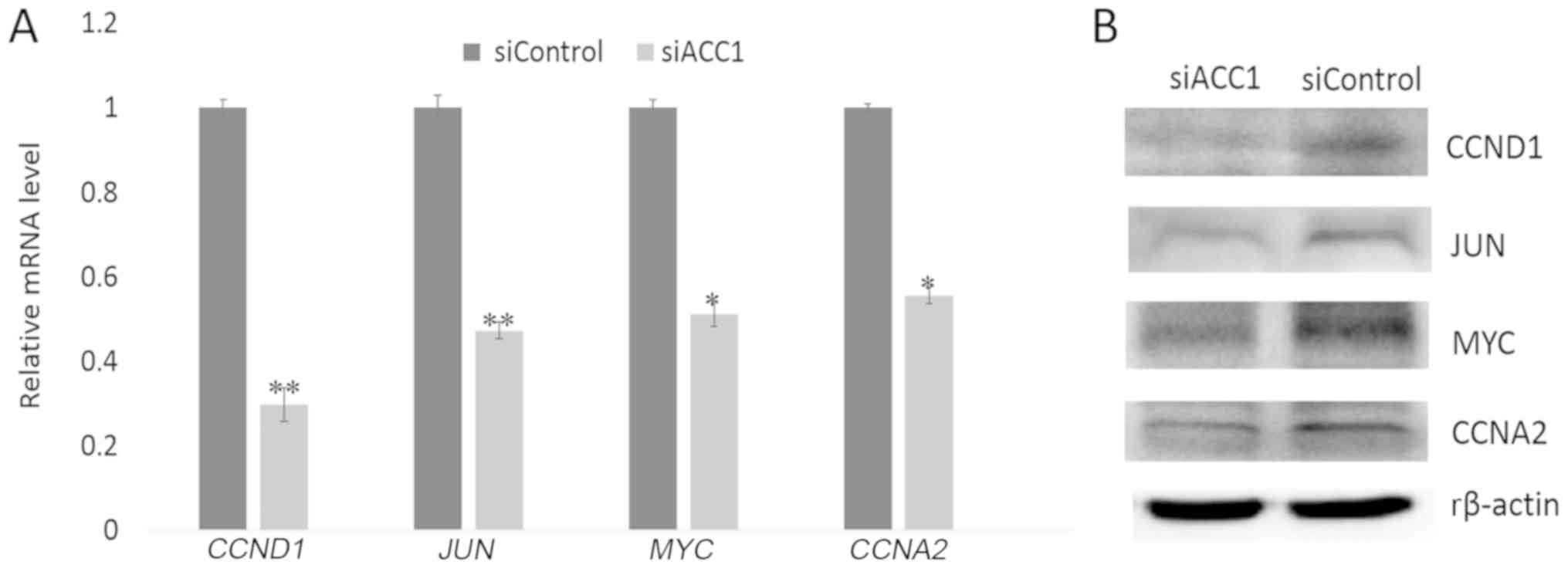
cell proliferation- associated genes
To further investigate the effects of ACC1 RNA
interference (RNAi) on cell proliferation, RT-qPCR and western blot
analyses were performed to determine the expression of
proliferation-associated genes 48 h following interference. The
results revealed that the expression of cell
proliferation-associated genes, including CCND1, JUN, MYCN
and CCNA2, were significantly decreased in
siACC1-transfected cells compared with the control group (Fig. 5A); a similar trend in protein was
observed (Fig. 5B).
Effects of siRNA-targeting ACC1 on the
cell cycle of BRL 3A cells
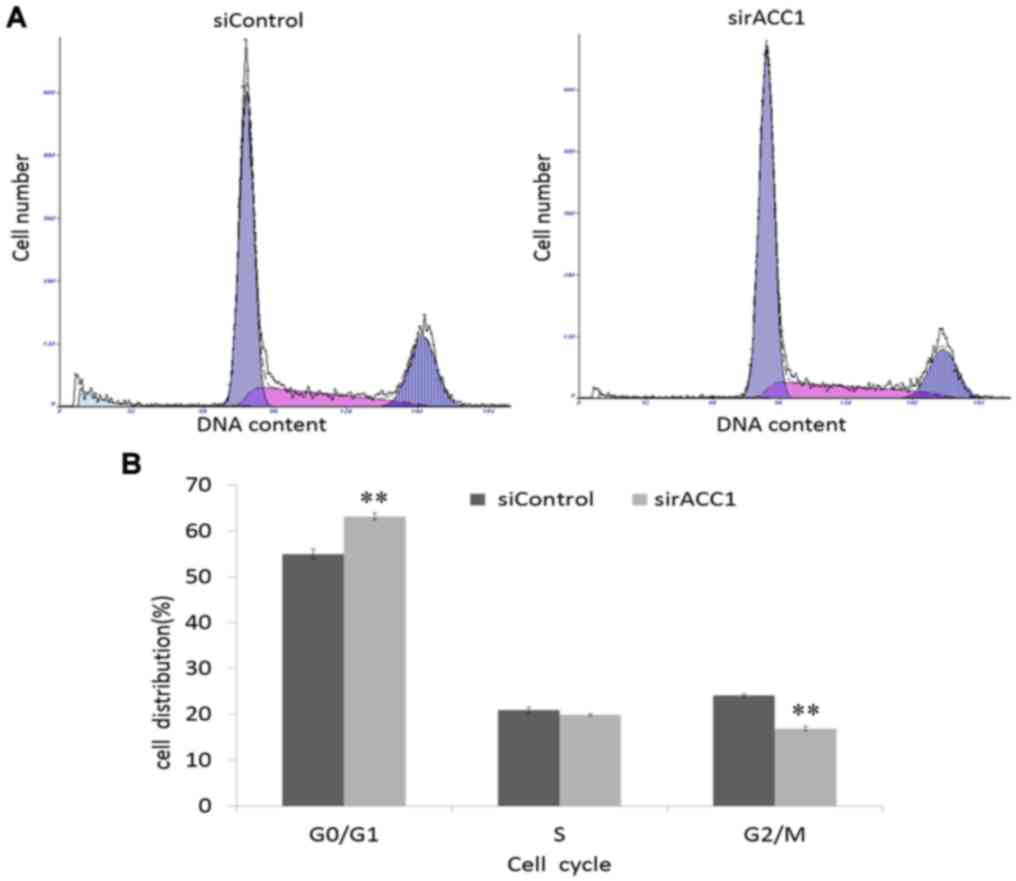
To investigate how ACC1 affects cell proliferation,
flow cytometry was used to analyse the cell cycle distribution of
BRL 3A cells. Knockdown of ACC1 expression significantly decreased
the percentage of G2/M phase cells (16.73±0.32%)
compared with that of control cells (24.10±0.30%; Fig. 6). Furthermore, the percentage of
G0/G1 phase cells significantly increased;
however, the percentage of S phase remained unchanged. Therefore,
ACC1 may modulate BRL 3A cell proliferation by controlling the cell
cycle.
Discussion
In the present study, the Oncomine database was
employed to analyse the association between the overexpression of
ACC1 and liver cancer, and determine whether ACC1 affects the
survival time of patients with liver cancer. The extent of ACC1
mRNA overexpression in liver cancer was closely associated with the
survival time of patients. To determine the role of ACC1 in cancer
and normal liver cells, ACC1 expression in Hep G2 and BRL 3A cells
was knocked down using RNAi. Downregulation of ACC1 significantly
suppressed Hep G2 and BRL 3A cell proliferation and decreased BRL
3A cells in G2/M phase (from 24.10±0.30 to 16.73±0.32%).
To further understand the mechanisms underlying
ACC1-mediated BRL 3A cell proliferation, the expression of cell
proliferation-associated genes, including MYCN, JUN, CCND1
and CCNA2, was analysed. MYCN is a key regulator of
mammalian cell proliferation and is required for oncogenesis
(21). Numerous studies have
demonstrated that MYCN can induce the transition of liver
cells from G0 phase to G1 phase (22,23).
CCND1 is another oncogene that drives cell cycle
progression, and its presence signifies that liver cells are
entering the G1 phase (24). Previous studies have reported that
CCND1 is significantly upregulated in liver cells following
partial hepatectomy; the protein regulates progression via the
G1-phase checkpoint and promotes cell proliferation
(25,26). MYCN can induce
CCND1-mediated cell cycle progression (27), whilst treatment of MCF7 cells with
antisense MYCN can inhibit cell proliferation by decreasing
CCND1 expression (28–30).
Thus, MYCN may induce CCND1 and enhance the activity
of CCND1-cyclin-dependent kinase complexes to drive cell cycle
progression and transformation (31). JUN is necessary for cells to
progress to G1 phase of the cell cycle and serves as an
important regulator for initiating liver regeneration (23,32).
Additionally, JUN regulates the transcriptional levels of
CCND1, which is required for the efficient proliferation of
mouse fibroblasts (33,34). Cyclin A2 (CCNA2), a core
component of the cell cycle, is involved in the G2/M
transition; thus, it may also affect cell proliferation (35). As a key member of the cyclin
family, CCNA2 significantly promotes hepatocyte cell cycle
progression (36). The present
study revealed that knockdown of ACC1 induced the downregulation of
CCNA2 and CCND1, and decreased BRL 3A cells in G2/M
phase. This suggested that ACC1 may bind and activate CCNA2,
CCND1, MYCN and JUN to promote BRL 3A proliferation.
Thus, downregulation of CCNA2 and CCND1 may affect
cells decreased in G2/M phase (from 24.10±0.30 to 16.73±0.32%);
however, further investigation is required to determine the exact
mechanism in detail.
As a master regulator of FA metabolism, ACC1
converts acetyl-CoA to malonyl-CoA, which is a critical substrate
for FA synthesis. Compared with normal cells, cancer cells
synthesise FAs at higher rates (9). The factors involved in lipid
synthesis are also observed in the proliferation, cell growth and
viability of certain cancers, including lung (37), colon (38), prostate (8) and breast cancer (11). In LNCaP cells, ACC1 RNAi-mediated
silencing induced cell growth inhibition and cytotoxicity (39). In non-small-cell lung cancer cells,
ACC inhibition reduces de novo lipid synthesis, and
decreases cell growth and viability (40). In human U87 EGFRvIII cells, ACC1/2
knockdown not only suppresses de novo lipogenesis, but
notably reduces U87 EGFRvIII cellular proliferation and viability
(9). In yeast, inactivation of
ACC1 completely inhibits vegetative growth and causes cell death
following treatment with FAs; further investigation has revealed
that siACC1 induces severe abnormalities in spindle formation,
arrests cells in the G2/M phase of the cell cycle, and
suppressed cell viability (41,42).
However, the importance of increased lipogenesis in tumour cells,
and the mechanisms by which interference with this increased
lipogenesis promotes tumour cell proliferation, growth and
viability require further investigation.
To the best of our knowledge, the present study is
the first to determine the association between ACC1 and cell
proliferation in the rat liver cell line BRL 3A. ACC1 is not only
important for the proliferation of Hep G2 cancer cells, but may
also be necessary for the proliferation of BRL 3A cells. ACC1 is
highly expressed in liver tissue and hepatocytes, and is the sole
regulator of FA synthesis (43).
Mutant mice lacking ACC1 exhibit embryonic lethality, which
suggests that de novo FA synthesis is essential for
embryonic development (44). The
present study suggests that ACC1 is a valuable drug target for
treating various metabolic pathologies, including hepatic
steatosis, non-alcoholic fatty liver disease, metabolic syndrome,
obesity and hepatic insulin resistance (7). Thus, ACC1 may be a potential target
for the development of novel approaches to liver cancer prevention
and therapy. ACC1 can upregulate the cell proliferation-related
genes MYCN, JUN, CCND1 and CCNA2 in the rat liver
cell line BRL 3A. In future studies, the association between FA
synthesis and cell proliferation-related genes should be
investigated to improve understanding of the function and targets
of ACC1. Additionally, whether ACC1 performs the same functions
in vivo merits further investigation by utilising a model of
rat partial hepatectomy and knockout of ACC1 via CRISPR-Cas9
technology.
Acknowledgements
The authors' thank members of our laboratory for
critical discussions.
Funding
The present study was supported by the Research Fund
for the Postdoctoral Program of He'nan, China, Natural Science
Foundation of China (grant no. 31572270), the National Fostering
Science Foundation Project of Henan Normal University (grant no.
2016PL21) and the Key Scientific Research Projects of Henan Higher
Education (grant no. 15A180007).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CX made substantial contributions to the conception
of the present study. BY and LY performed the experiments and wrote
the manuscript; QW made substantial contributions to the design of
the present study, and interpreted the data. All authors read and
approved the final version of the manuscript for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Harwood HJ Jr: Treating the metabolic
syndrome: Acetyl-CoA carboxylase inhibition. Expert Opin Ther
Targets. 9:267–281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim KH: Regulation of mammalian
acetyl-coenzyme A carboxylase. Annu Rev Nutr. 17:77–99. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McGarry JD, Mannaerts GP and Foster DW: A
possible role for malonyl-CoA in the regulation of hepatic fatty
acid oxidation and ketogenesis. J Clin Invest. 60:265–270. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nugteren DH: The enzymic chain elongation
of fatty acids by rat-liver microsomes. Biochim Biophys Acta.
106:280–290. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Munday MR: Regulation of mammalian
acetyl-CoA carboxylase. Biochem Soc Trans. 30:1059–1064. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saggerson D: Malonyl-CoA, a key signaling
molecule in mammalian cells. Annu Rev Nutr. 28:253–272. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Glatzel DK, Koeberle A, Pein H, Löser K,
Stark A, Keksel N, Werz O, Muller R, Bischoff I and Fürst R:
Acetyl-CoA carboxylase 1 regulates endothelial cell migration by
shifting the phospholipid composition. J Lipid Res. 59:298–311.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brusselmans K, De Schrijver E, Verhoeven G
and Swinnen JV: RNA interference-mediated silencing of the
acetyl-CoA-carboxylase-alpha gene induces growth inhibition and
apoptosis of prostate cancer cells. Cancer Res. 65:6719–6725. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jones JE, Esler WP, Patel R, Lanba A, Vera
NB, Pfefferkorn JA and Vernochet C: Inhibition of Acetyl-CoA
Carboxylase 1 (ACC1) and 2 (ACC2) reduces proliferation and de novo
lipogenesis of EGFRvIII human glioblastoma cells. PLoS One.
12:e01695662017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Magnard C, Bachelier R, Vincent A,
Jaquinod M, Kieffer S, Lenoir GM and Venezia ND: BRCA1 interacts
with acetyl-CoA carboxylase through its tandem of BRCT domains.
Oncogene. 21:6729–6739. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chajes V, Cambot M, Moreau K, Lenoir GM
and Joulin V: Acetyl-CoA carboxylase alpha is essential to breast
cancer cell survival. Cancer Res. 66:5287–5294. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abu-Elheiga L, Matzuk MM, Abo-Hashema KA
and Wakil SJ: Continuous fatty acid oxidation and reduced fat
storage in mice lacking acetyl-CoA carboxylase 2. Science.
291:2613–2616. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harriman G, Greenwood J, Bhat S, Huang X,
Wang R, Paul D, Tong L, Saha AK, Westlin WF, Kapeller R and Harwood
HJ Jr: Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic
steatosis, improves insulin sensitivity, and modulates dyslipidemia
in rats. Proc Natl Acad Sci USA. 113:E1796–E1805. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harwood HJ Jr: Acetyl-CoA carboxylase
inhibition for the treatment of metabolic syndrome. Curr Opin
Investig Drugs. 5:283–289. 2004.PubMed/NCBI
|
|
15
|
Schreurs M, van Dijk TH, Gerding A,
Havinga R, Reijngoud DJ and Kuipers F: Soraphen, an inhibitor of
the acetyl-CoA carboxylase system, improves peripheral insulin
sensitivity in mice fed a high-fat diet. Diabetes Obes Metab.
11:987–991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rhodes DR, Kalyana-Sundaram S, Mahavisno
V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ,
Kincead-Beal C, Kulkarni P, et al: Oncomine 3.0: Genes, pathways,
and networks in a collection of 18,000 cancer gene expression
profiles. Neoplasia. 9:166–180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roessler S, Jia HL, Budhu A, Forgues M, Ye
QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX and Wang XW: A
unique metastasis gene signature enables prediction of tumor
relapse in early-stage hepatocellular carcinoma patients. Cancer
Res. 70:10202–10212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aguirre-Gamboa R, Gomez-Rueda H,
Martínez-Ledesma E, Martínez-Torteya A, Chacolla-Huaringa R,
Rodriguez- Barrientos A, Tamez-Peña JG and Treviño V: SurvExpress:
An online biomarker validation tool and database for cancer gene
expression data using survival analysis. PLoS One. 8:e742502013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boncler M, Różalski M, Krajewska U,
Podsędek A and Watala C: Comparison of PrestoBlue and MTT assays of
cellular viability in the assessment of anti-proliferative effects
of plant extracts on human endothelial cells. J Pharmacol Toxicol
Methods. 69:9–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bouchard C, Staller P and Eilers M:
Control of cell proliferation by Myc. Trends Cell Biol. 8:202–206.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miquet JG, Freund T, Martinez CS, Gonzalez
L, Diaz ME, Micucci GP, Zotta E, Boparai RK, Bartke A, Turyn D and
Sotelo AI: Hepatocellular alterations and dysregulation of
oncogenic pathways in the liver of transgenic mice overexpressing
growth hormone. Cell Cycle. 12:1042–1057. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morello D, Lavenu A and Babinet C:
Differential regulation and expression of jun, c-fos and c-myc
proto-oncogenes during mouse liver regeneration and after
inhibition of protein synthesis. Oncogene. 5:1511–1519.
1990.PubMed/NCBI
|
|
24
|
Pauklin S and Vallier L: The cell-cycle
state of stem cells determines cell fate propensity. Cell.
155:135–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nelsen CJ, Rickheim DG, Timchenko NA,
Stanley MW and Albrecht JH: Transient expression of cyclin D1 is
sufficient to promote hepatocyte replication and liver growth in
vivo. Cancer Res. 61:8564–8568. 2001.PubMed/NCBI
|
|
26
|
Rickheim DG, Nelsen CJ, Fassett JT,
Timchenko NA, Hansen LK and Albrecht JH: Differential regulation of
cyclins D1 and D3 in hepatocyte proliferation. Hepatology.
36:30–38. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mateyak MK, Obaya AJ and Sedivy JM: c-Myc
regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle
progression at multiple independent points. Mol Cell Biol.
19:4672–4683. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carroll JS, Swarbrick A, Musgrove EA and
Sutherland RL: Mechanisms of growth arrest by c-myc antisense
oligonucleotides in MCF-7 breast cancer cells: Implications for the
antiproliferative effects of antiestrogens. Cancer Res.
62:3126–3131. 2002.PubMed/NCBI
|
|
29
|
Daksis JI, Lu RY, Facchini LM, Marhin WW
and Penn LJ: Myc induces cyclin D1 expression in the absence of de
novo protein synthesis and links mitogen-stimulated signal
transduction to the cell cycle. Oncogene. 9:3635–3645.
1994.PubMed/NCBI
|
|
30
|
Perez-Roger I, Kim SH, Griffiths B, Sewing
A and Land H: Cyclins D1 and D2 mediate myc-induced proliferation
via sequestration of p27(Kip1) and p21(Cip1). EMBO J. 18:5310–5320.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oswald F, Lovec H, Möröy T and Lipp M:
E2F-dependent regulation of human MYC: Trans-activation by cyclins
D1 and A overrides tumour suppressor protein functions. Oncogene.
9:2029–2036. 1994.PubMed/NCBI
|
|
32
|
Riehle KJ, Campbell JS, McMahan RS,
Johnson MM, Beyer RP, Bammler TK and Fausto N: Regulation of liver
regeneration and hepatocarcinogenesis by suppressor of cytokine
signaling 3. J Exp Med. 205:91–103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Herber B, Truss M, Beato M and Müller R:
Inducible regulatory elements in the human cyclin D1 promoter.
Oncogene. 9:1295–1304. 1994.PubMed/NCBI
|
|
34
|
Musgrove EA, Lee CS, Buckley MF and
Sutherland RL: Cyclin D1 induction in breast cancer cells shortens
G1 and is sufficient for cells arrested in G1 to complete the cell
cycle. Proc Natl Acad Sci USA. 91:8022–8026. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gong D, Pomerening JR, Myers JW,
Gustavsson C, Jones JT, Hahn AT, Meyer T, Meyer T and Ferrell JE
Jr: Cyclin A2 regulates nuclear-envelope breakdown and the nuclear
accumulation of cyclin B1. Curr Biol. 17:85–91. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Garnier D, Loyer P, Ribault C,
Guguen-Guillouzo C and Corlu A: Cyclin-dependent kinase 1 plays a
critical role in DNA replication control during rat liver
regeneration. Hepatology. 50:1946–1956. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hatzivassiliou G, Zhao F, Bauer DE,
Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA and
Thompson CB: ATP citrate lyase inhibition can suppress tumor cell
growth. Cancer Cell. 8:311–321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhan Y, Ginanni N, Tota MR, Wu M, Bays NW,
Richon VM, Kohl NE, Bachman ES, Strack PR and Krauss S: Control of
cell growth and survival by enzymes of the fatty acid synthesis
pathway in HCT-116 colon cancer cells. Clin Cancer Res.
14:5735–5742. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rysman E, Brusselmans K, Scheys K,
Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D,
Daniëls VW, Machiels J, et al: De novo lipogenesis protects cancer
cells from free radicals and chemotherapeutics by promoting
membrane lipid saturation. Cancer Res. 70:8117–8126. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Svensson RU, Parker SJ, Eichner LJ, Kolar
MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A,
Vera L, et al: Inhibition of acetyl-CoA carboxylase suppresses
fatty acid synthesis and tumor growth of non-small-cell lung cancer
in preclinical models. Nat Med. 22:1108–1119. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hasslacher M, Ivessa AS, Paltauf F and
Kohlwein SD: Acetyl-CoA carboxylase from yeast is an essential
enzyme and is regulated by factors that control phospholipid
metabolism. J Biol Chem. 268:10946–10952. 1993.PubMed/NCBI
|
|
42
|
Al-Feel W, DeMar JC and Wakil SJ: A
Saccharomyces cerevisiae mutant strain defective in acetyl-CoA
carboxylase arrests at the G2/M phase of the cell cycle. Proc Natl
Acad Sci USA. 100:3095–3100. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Savage DB, Choi CS, Samuel VT, Liu ZX,
Zhang D, Wang A, Zhang XM, Cline GW, Yu XX, Geisler JG, et al:
Reversal of diet-induced hepatic steatosis and hepatic insulin
resistance by antisense oligonucleotide inhibitors of acetyl-CoA
carboxylases 1 and 2. J Clin Invest. 116:817–824. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Abu-Elheiga L, Matzuk MM, Kordari P, Oh W,
Shaikenov T, Gu Z and Wakil SJ: Mutant mice lacking acetyl-CoA
carboxylase 1 are embryonically lethal. Proc Natl Acad Sci USA.
102:12011–12016. 2005. View Article : Google Scholar : PubMed/NCBI
|