Introduction
Liver cancer is the fifth most common malignant
tumor, and exhibits the second highest mortality rate among
malignant tumors (1–3). Liver cancer has a high incidence
rate, and accounts for ~55% of the cancer incidence worldwide
(4). Additionally, the majority of
patients with liver cancer at an advanced stage have a poor
prognosis (5). Therefore, it is
necessary to identify novel drugs to overcome the chemotherapeutic
resistance of liver cancer cells and to develop novel treatments
for patients with liver cancer.
Tumor necrosis factor (TNF) superfamily member 10
(TNFSF10) belongs to the TNF family, and is able to induce cell
apoptosis in various types of tumor cells; however, its
pro-apoptotic activity has not been detected in normal healthy
cells (6–9). Cell apoptosis may be initiated by the
interaction between TNFSF10 and TNF receptor superfamily member 10a
(TNFRSF10A)/TNFRSF10B (10,11).
Subsequently, caspase-8 (CASP8) is recruited to the death-inducing
signaling complex and may cause the activation of CASP3, inducing
cell apoptosis (12–14). TNFSF10 resistance may be acquired
via multiple molecular mechanisms, including the differential
expression of death receptors and the increased expression of
anti-apoptotic factors (15).
Notably, treatments combining TNFSF10 with various anticancer drugs
have been demonstrated to improve the anticancerogenic effects of a
number of compounds (16).
Due to the antiproliferative activity exhibited by
ent-3α-formylabieta-8(14),13(15)-dien-16,12β-olide (EFLDO) in MCF-7
cells and NCI-H460 cells (17),
EFLDO has attracted the interest of various research studies
(18). The aim of the present
study was to investigate the antitumor activity of EFLDO and the
potential molecular mechanisms underlying EFLDO function in
promoting the inhibitory effects of TNFSF10 in liver cancer
cells.
Materials and methods
Cell culture
HepG2 cells (19)
and non-cancerous normal liver LO2 cells were purchased from The
American Type Culture Collection (Manassas, VA, USA). The cells
were cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone;
GE Healthcare Life Sciences, Logan, UT, USA), supplemented with 10%
(v/v) fetal bovine serum (HyClone; GE Healthcare Life Sciences) and
1% penicillin-streptomycin (HyClone; GE Healthcare Life Sciences)
in an incubator with 5% CO2 at 37°C.
Drug treatments
Non-cancerous normal liver LO2 cells were treated
with EFLDO (3 µM) and/or TNFSF10 (25 ng/ml, Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and cultured in an incubator
with 5% CO2 at 37°C for 24 h. EFLDO was extracted from
Euphorbia lunulata Bge as previously described (17). Hepatoblastoma cells were treated
with EFLDO (3 µM), TNFSF10 (25 ng/ml), caspase inhibitor z-DEVD-FMK
(working concentration, 40 µM; R&D Systems, Inc., Minneapolis,
MN, USA; cat. no. FMK004) and/or the p53 inhibitor pifithrin-α
(PFT-α; working concentrations, 20 and 40 µM; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) and cultured in an incubator with 5%
CO2 at 37°C for 24 h prior to further
experimentation.
MTT assay
Hepatoblastoma HepG2 cells and non-cancerous normal
liver LO2 cells were seeded into a 96-well plate at a density of
3×103 cells/well. Following addition of the indicated
drug into the culture medium, the cells were incubated in an
incubator with 5% CO2 at 37°C for 24 h. Subsequently,
the cell viability was measured using the MTT assay at 570 nm. The
MTT assay was performed using DMSO (Sigma-Aldrich; Merck KGaA) as
the solvent to dissolve the formazan as previously described
(20).
Annexin V-fluorescein isothiocyanate
(FITC) assay
The Annexin V-FITC assay (Becton, Dickinson and
Company, Franklin Lakes, NJ, USA) was performed to quantify the
percentage of apoptotic cells, according to the manufacturer's
protocol. HepG2 cells were incubated for 24 h, collected, washed
twice with PBS and incubated in 1X binding buffer (provided in the
Annexin V-FITC kit). Subsequently, Annexin V-FITC and propidium
iodide (PI) were added to the cells and cells were incubated for 20
min at room temperature in the dark. Following incubation with
Annexin V-FITC and PI, the cells were analyzed using a flow
cytometer (Becton, Dickinson and Company) and the results were
analyzed using the CellQuest Pro software (version 5.1; Becton,
Dickinson and Company).
CASP3 Activity assay
CASP3 activity was measured using the CASP3 activity
assay kit (Chemicon International; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. HepG2 cells were lysed
using lysis buffer provided in the kit on ice for 10 min and
centrifuged for 5 min at 12,000 × g at 4°C. Caspase substrate
solution was added to the supernatant and incubated for 2 h at
37°C, subsequently, the activity was measured using an ELISA plate
reader at 405 nm.
Western blot
HepG2 cells were incubated with DMEM for 48 h. Cells
at a confluency of 70% were lysed using radioimmunoprecipitation
assay buffer (Beyotime Institute of Biotechnology, Haimen, China)
containing a protease inhibitor cocktail (Roche Applied Science,
Penzberg, Germany). The protein concentration was determined using
Bradford Protein Assay kit (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). Each lane was loaded with 0.05 µg total protein. Proteins
were separated by 10% SDS-PAGE and transferred onto polyvinylidene
fluoride (PVDF) membranes. The PVDF membranes were blocked with 5%
non-fat milk for 1 h at room temperature, and incubated with the
following primary antibodies: Anti-cleaved CASP3 (cat. no. 9661),
cleaved CASP8 (cat. no. 9496), p53 (cat. no. 2527) (all 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA), GAPDH (cat. no.
59540), TNFRSF10A (cat. no. 32255), TNFRSF10B (cat. no. 166624),
BCL2 associated agonist of cell death (BAD; cat. no. 8044), BCL2
associated X, apoptosis regulator apoptosis regulator (BAX; cat.
no. 20067), BCL2 apoptosis regulator (BCL2; cat. no. 509) (all
1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) overnight
at 4°C. Horseradish peroxidase-conjugated goat anti-rabbit (cat.
no. 7407) and goat anti-mouse (cat. no. 7076) secondary antibodies
(all 1:10,000; Cell Signaling Technology, Inc.) were incubated with
the membranes at room temperature for 2 h. Bands were developed
with the electrochemiluminescence detection reagent (GE Healthcare,
Chicago, IL, USA). GAPDH was used as the loading control and Image
J software (version 1.8.0, National Institutes of Health, Bethesda,
MD, USA) was used to compare the relative band intensities.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
HepG2 cells were incubated with DMEM for 48 h, and
total RNA was extracted using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. mRNA was reverse-transcribed to cDNA using
the PrimeScript™ RT reagent kit (Takara Bio, Inc., Otsu, Japan),
according to the manufacturer's protocol. For cDNA synthesis, the
RT reactions were incubated at 37°C for 1 h and at 70°C for 15 min.
The sequences of the PCR primers used were as follows: TNFRSF10A
forward: 5′-ACCTTCAAGTTTGTCGTCGTC-3′; TNFRSF10A reverse:
5′-CCAAAGGGCTATGTTCCCATT-3′; TNFRSF10B forward:
5′-GCCCCACAACAAAAGAGGTC-3′; TNFRSF10B reverse:
5′-AGGTCATTCCAGTGAGTGCTA-3′; GAPDH forward:
5′-ACAACTTTGGTATCGTGGAAGG-3′; GAPDH reverse:
5′-GCCATCACGCCACAGTTTC-3′. GAPDH gene was used as the reference
gene. QuantiTect SYBR® Green RT-PCR kit (cat. no.
204245; Qiagen GmbH, Hilden, Germany) was used to analyze relative
gene expression. The thermocycling conditions were as follows:
Initial denaturation at 95°C for 10 min, followed by 40 cycles of
95°C for 15 sec and 60°C for 60 sec, and a final elongation at 72°C
for 15 sec.
Small interfering (siRNA)
transfection
p53 siRNA (cat. no. sc-29435) and control siRNA
(cat. no. sc-44238) were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). For the siRNA transfection, HepG2 cells
were seeded in 6-well plates at 3×105 cells/well and
cultured in an incubator at 37°C overnight. On the following day,
0.67 µg siRNAs were mixed with Lipofectamine® RNAiMAX
(Invitrogen; Thermo Fisher Scientific, Inc.) in Opti-MEM™ (Gibco;
Thermo Fisher Scientific, Inc.) and incubated at room temperature
for 15 min; subsequently, the mixtures were added into the culture
medium and incubated at 37°C for 24 h prior to further
experimentation.
Statistical analysis
All data were analyzed using GraphPad Prism 6.0
(GraphPad Software, Inc., La Jolla, CA, USA). Data are presented as
the mean ± standard deviation. Comparisons between two groups and
multiple groups were performed using Student's t-test and one-way
analysis of variance followed by Newman-Keuls test, respectively.
P<0.05 was considered to indicate a statistically significant
difference. All the experiments were performed ≥3 times.
Results
Synergistic effects of EFLDO and
TNFSF10 on HepG2 cell viability
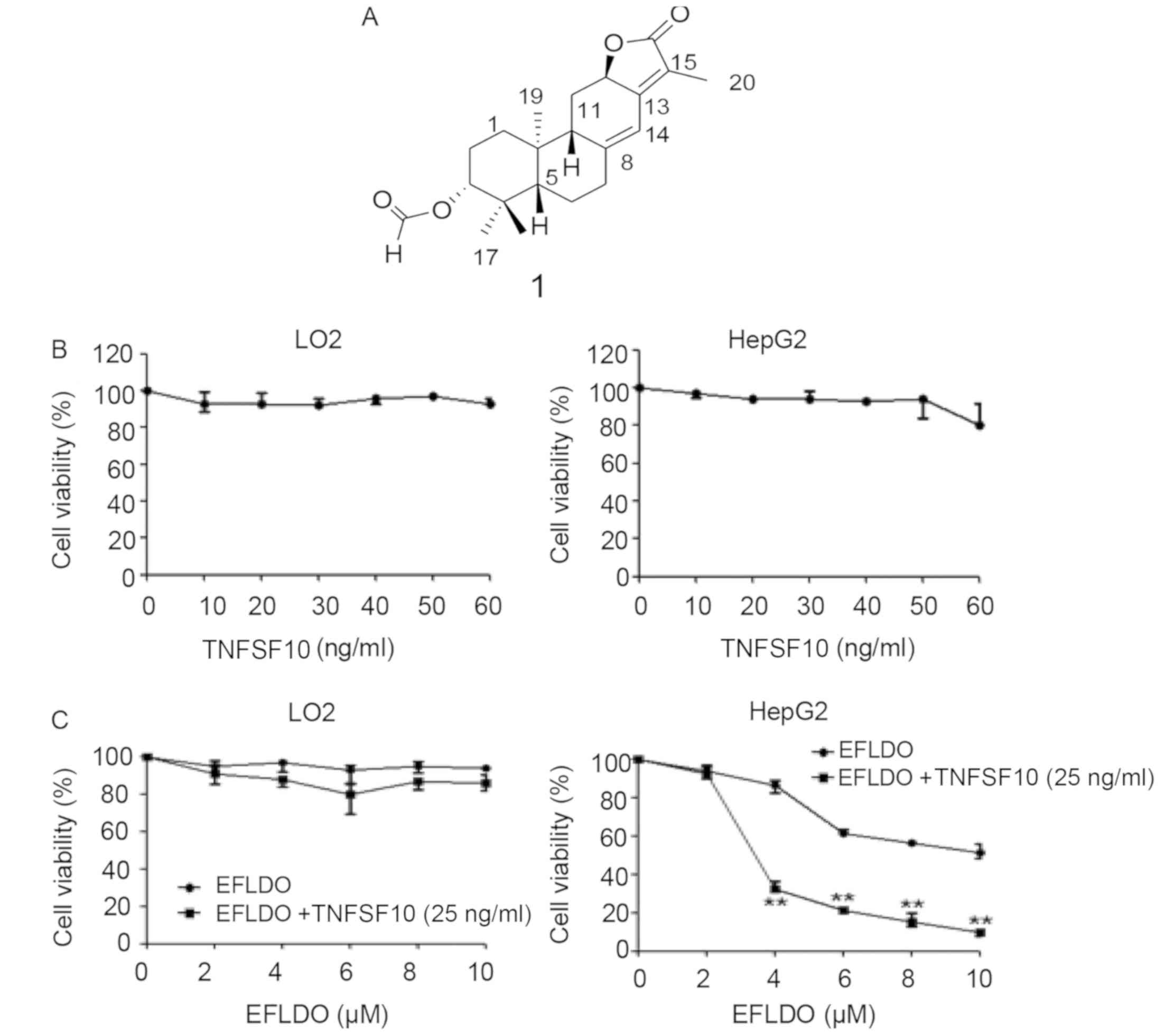
The chemical structure of EFLDO is presented in
Fig. 1A. The cytotoxicity of
TNFSF10 at various concentrations (10, 20, 30, 40, 50 and 60 ng/ml)
on LO2 and HepG2 cells was assessed using the MTT assay. The
present MTT results suggested that TNFSF10 exhibited no obvious
cytotoxicity in cells at concentrations ≤60 ng/ml (Fig. 1B).
Subsequently, the effects of various concentrations
of EFLDO (2, 4, 6, 8 and 10 µM) and EFLDO (2, 4, 6, 8 and 10 µM) +
TNFSF10 (25 ng/ml) on cell viability were detected. The present
results suggested that the cell viability of HepG2 cells
significantly decreased following treatment with EFLDO, in a
concentration-dependent manner. Notably, the cell viability of
normal liver LO2 cells was unaffected by treatment with EFLDO
(Fig. 1C). Additionally, the cell
viability of HepG2 cells was significantly decreased following the
administration of EFLDO + TNFSF10 compared with administration of
EFLDO alone. Furthermore, treatment with EFLDO (3 µM) + TNFSF10 (25
ng/ml) exhibited a significant antiproliferative activity (data not
shown). Therefore, 25 ng/ml was selected at the working
concentration of TNFSF10 and 3 µM was selected as the working
concentration of EFLDO for further experiments.
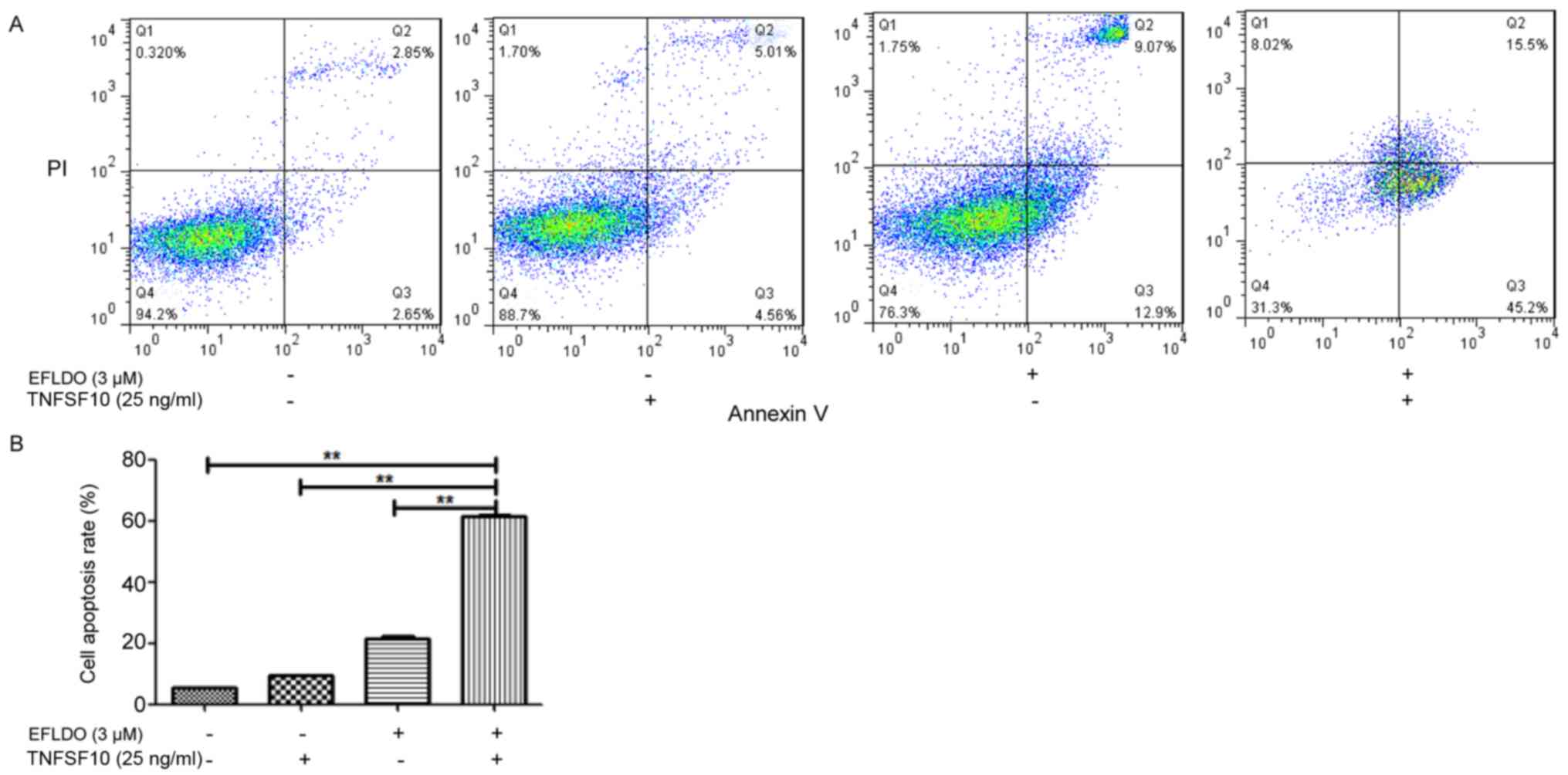
EFLDO increases TNFSF10-induced cell
apoptosis in HepG2 cells
The potential of EFLDO in sensitizing HepG2 cells to
TNFSF10-induced apoptosis was investigated. Flow cytometry results
suggested that the apoptotic rate of HepG2 cells following
treatment with EFLDO + TNFSF10 increased compared with the control,
TNFSF10 and EFLDO groups (Fig.
2A). The apoptotic rates for HepG2 cells in the TNFSF10 and
EFLDO + TNFSF10 group were 9.57 and 60.7%, respectively (Fig. 2B). The present results suggested
that the combination of EFLDO and TNFSF10 increased the effect of
TNFSF10 in inducing the apoptosis of HepG2 cells.
EFLDO promotes TNFSF10-induced
apoptosis of HepG2 cells in a caspase-dependent manner
Previous studies demonstrated that activation of
CASP8 induced by TNFSF10 may activate CASP3 and CASP9 (12–14).
To investigate the molecular mechanisms underlying cell apoptosis
induced by treatment with EFLDO + TNFSF10, the activity levels of
CASP8 and 3 in HepG2 cells treated with TNFSF10 and/or EFLDO were
detected. The present results suggested that proteolytic cleavage
of proCASP8 and proCASP3 increased following treatment with EFLDO +
TNFSF10 compared with the control, TNFSF10 and EFLDO groups
(Fig. 3A). CASP3 activity in the
EFLDO + TNFSF10 group increased to 7.0-fold compared with the
control group, which was increased compared with TNFSF10 (0.7-fold)
and EFLDO (1.4-fold) groups, and was significantly decreased
following treatment with the caspase inhibitor z-DEVD-FMK (Fig. 3B). Subsequently, treatment with
EFLDO + TNFSF10 significantly increased the protein expression
levels of BCL2 associated agonist of cell death (BAD) and BAX, and
decreased the protein expression level of BCL2 in HepG2 cells
compared with the control, TNFSF10 and EFLDO groups (Fig. 3C). The present results suggested
that EFLDO significantly sensitized HepG2 cells to TNFSF10-induced
apoptosis in a caspase-dependent manner.
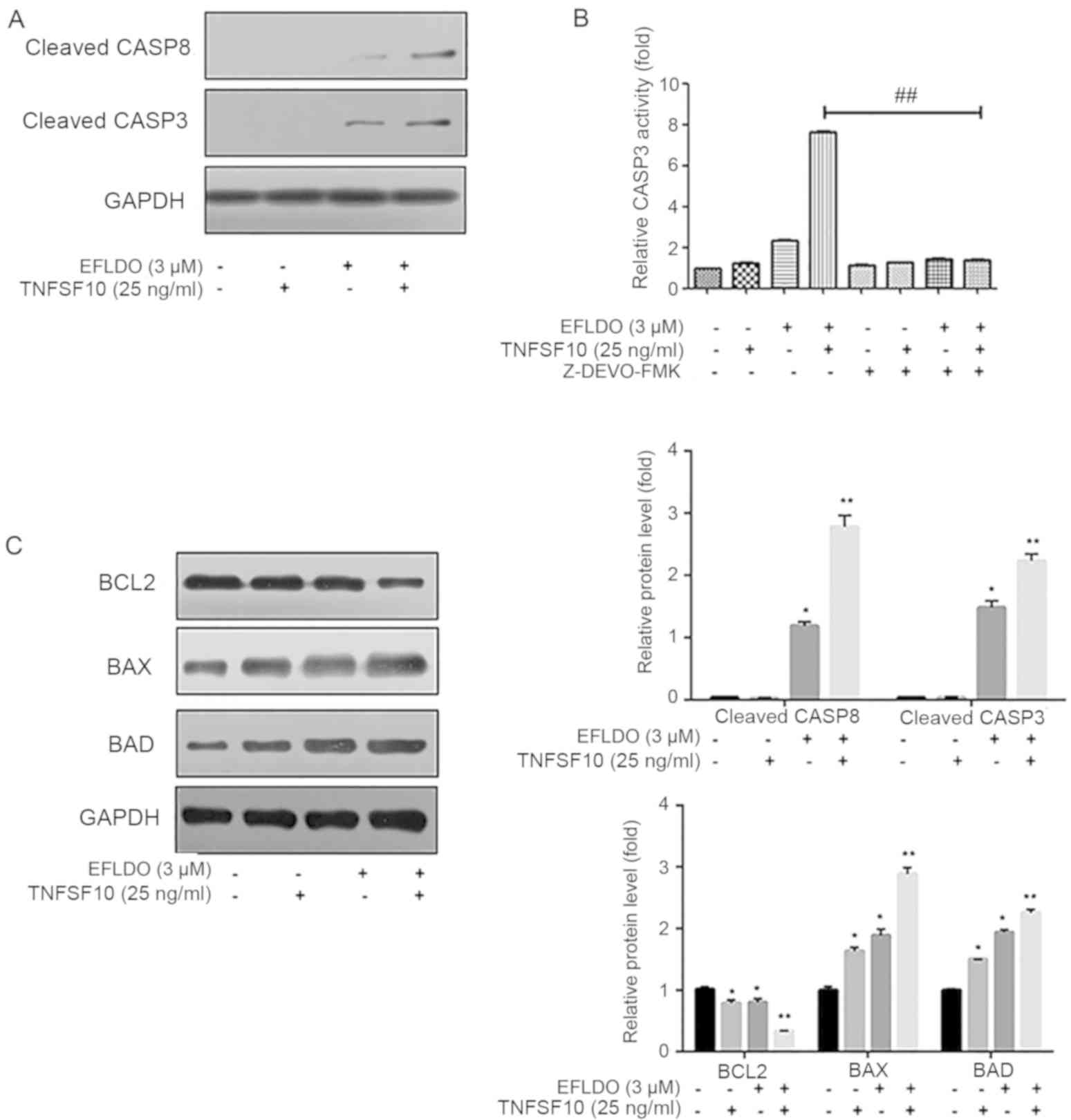 | Figure 3.Treatment with EFLDO sensitizes HepG2
cells to TNFSF10-induced apoptosis via the caspase pathway. (A)
Protein expression levels of cleaved CASP8 and CASP3 in HepG2 cells
following treatment with EFLDO and/or TNFSF10. (B) Activity of
CASP3 in liver cancer cells treated with EFLDO, TNFSF10 and/or
caspase inhibitor z-DEVD-FMK. Data are presented as fold change
relative to the mean CASP3 activity of the control group.
##P<0.01. (C) Protein expression levels of BLC2, BAX and BAD in
HepG2 cells treated with EFLDO and/or TNFSF10. *P<0.05,
**P<0.01 vs. respective control group. EFLDO,
Ent-3α-formylabieta-8(14),13(15)-dien-16,12β-olide; TNFSF10, tumor
necrosis factor superfamily member 10; CASP, caspase; BCL2, BCL2
apoptosis regulator; BAD, BCL2 associated agonist of cell death;
BAX, BCL2 associated X, apoptosis regulator. |
EFLDO sensitizes HepG2 cells to
TNFSF10-induced cell apoptosis by increasing the expression levels
of TNFRSF10A and TNFRSF10B
TNFRSF10A and TNFRSF10B (TNFSF10 receptors) were
identified to be able to initiate the apoptotic cascade by binding
to TNFSF10 (21). The present
RT-qPCR and western blotting results suggested that treatment with
EFLDO or EFLDO + TNFSF10 increased the mRNA (Fig. 4A) and protein expression levels
(Fig. 4B) of TNFRSF10A and
TNFRSF10B compared with the control group. A previous study
observed that p53 may serve a role in regulating the functions of
TNFRSF10A and TNFRSF10B (22). The
present western blotting results suggested that, compared with the
control group, the protein expression level of p53 was
significantly increased following treatment with EFLDO or EFLDO +
TNFSF10 (Fig. 4C). The effects of
EFLDO on the protein expression levels of p53 and TNFRSF10B were
inhibited by treatment with PFT-α (Fig. 4D). The present results suggested
that treatment with EFLDO + TNFSF10 increased the protein
expression levels of TNFRSF10A and TNFRSF10B by increasing the
protein expression level of p53 in HepG2 cells.
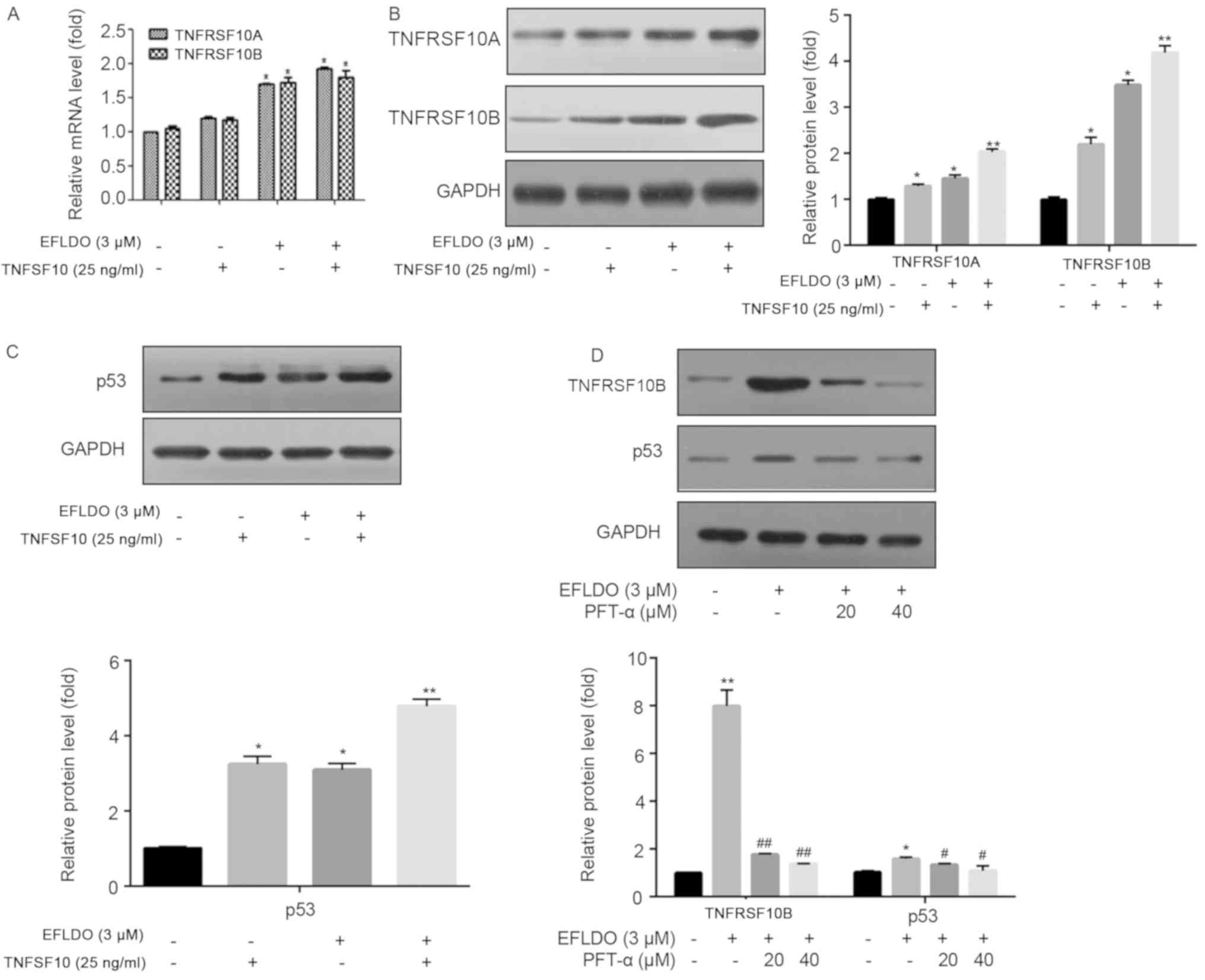 | Figure 4.EFLDO treatment sensitizes liver
cancer cells to TNFSF10-induced apoptosis by upregulating the
protein expression levels of TNFRSF10A and TNFRSF10B in a
p53-dependent manner. (A) HepG2 cells were treated with EFLDO
and/or TNFSF10 for 24 h. Reverse transcription-quantitative
polymerase chain reaction analysis was performed to detect the mRNA
expression levels of TNFRSF10A and TNFRSF10B. In total, three
independent experiments were performed. (B) Western blotting was
performed to detect the protein expression levels of TNFRSF10A and
TNFRSF10B. (C) Protein expression levels of p53 in HepG2 cells
treated with EFLDO and/or TNFSF10. (D) Protein expression levels of
TNFRSF10B and p53 in HepG2 cells treated with EFLDO, TNFSF10 and/or
PFT-α. *P<0.05, **P<0.01 vs. respective control group.
#P<0.05, ##P<0.01 vs. respective EFLDO group. EFLDO,
Ent-3α-formylabieta-8(14),13(15)-dien-16,12β-olide; TNFSF10, tumor
necrosis factor superfamily member 10; TNFRSF10A, TNF receptor
superfamily member 10a; TNFRSF10B, TNF receptor superfamily member
10b. |
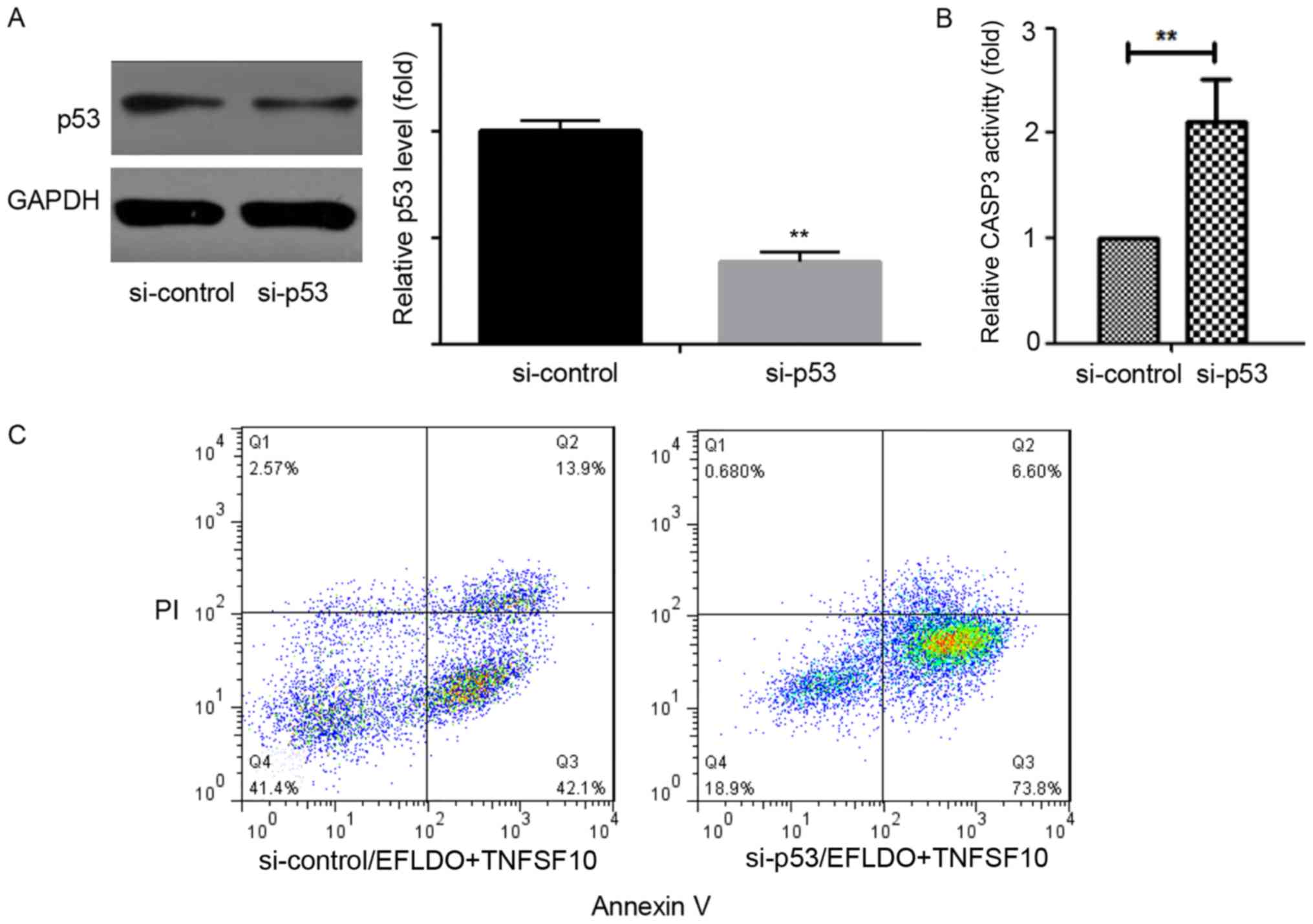
EFLDO increases TNFSF10-induced cell
apoptosis in a p53-dependent manner in HepG2 cells
To investigate the role of p53 on the effects of
EFLDO in increasing TNFSF10-induced cell apoptosis, p53 was
silenced by transfecting a siRNA targeting p53 (si-p53) in HepG2
cells (Fig. 5A). Knockdown of p53
significantly increased the activity of CASP3 in liver cancer cells
(Fig. 5B). si-p53 significantly
increased the apoptosis of HepG2 cells following treatment of
TNFSF10 + EFLDO (Fig. 5C). The
present results suggested that inhibition of p53 may increase
EFLDO-induced TNFSF10 sensitivity in HepG2 cells.
Discussion
Previous studies in liver cancer demonstrated that
the upregulation of antiapoptotic genes, and the increase in the
protein expression levels of dysadherin and various cytokines, may
serve roles in the negative outcome of certain therapies and in
cancer metastasis (23–25). Combinatorial treatments were
identified to exhibit an increased effectiveness compared with
single-agent chemotherapy in various types of cancer, including
liver cancer (26). Nevertheless,
the efficacy of chemotherapy to treat cancer may be decreased by
the occurrence of drug resistance. Therefore, the identification of
novel therapies is required to improve the treatment of patients
with liver cancer.
Due to its strong antiproliferative activity in
MCF-7 and NCI-H460 cells (17),
EFLDO has attracted the interest of various research studies
(18). However, the effects of the
combination of EFLDO with additional anticancer drugs in liver
cancer remain unknown.
The present results suggested that TNFSF10 did not
induce cytotoxicity. By contrast, the combination of EFLDO and
TNFSF10 presented a synergistic effect in suppressing cell
viability of HepG2 cells; however, this combinatorial treatment did
not affect the viability of non-cancerous normal liver LO2 cells.
Subsequently, EFLDO was identified to sensitize HepG2 cells to
TNFSF10-induced apoptosis. The present results suggested that
apoptotic rates of HepG2 cells increased significantly following
the cotreatment with TNFSF10 and EFLDO compared with
single-treatment with TNSFS10 or EFLDO, indicating a potential
synergistic effect between TNFSF10 and EFLDO in inducing apoptosis
in liver cancer cells.
Furthermore, the activities of CASP8 and 3 following
treatment with TNFSF10 and/or EFLDO were detected to investigate
the molecular mechanisms underlying cell apoptosis induced by the
combined treatment with EFLDO and TNFSF10. The present results
suggested that liver cancer cell apoptosis was induced by EFLDO +
TNFSF10 via the activation of CASP3 and CASP8. Treatment with EFLDO
and TNSFS10 induced the upregulation of pro-apoptotic molecules,
including BAD and BAX, and the downregulation of the anti-apoptotic
factor BCL2. The present results suggested that EFLDO may promote
TNFSF10-induced liver cancer cell apoptosis in a caspase-dependent
manner.
p53 was previously identified to serve a role in
regulating the function of TNFRSF10A and TNFRSF10B (22). p53 is able to inhibit cell
proliferation by inducing cell apoptosis and cell cycle arrest in
response to cellular stresses (27–29).
The present results suggested that the synergistic effects between
EFLDO and TNFSF10 in increasing the protein expression level of
TNFRSF10B may be due to the upregulation of p53, since the protein
expression level of TNFRSF10B was decreased following treatment
with the p53 inhibitor PFT-α. In addition, knockdown of p53
increased the activity of CASP3 and increased cell apoptosis in
HepG2 cells, suggesting that p53 may regulate the synergistic
effects between EFLDO and TNFSF10 in liver cancer cells. However,
treatment with EFLDO and TNFSF10 led to an increase in the
expression level of p53, in contrast with the results of p53
knockdown. p53 may be involved in various pathways regulating HepG2
cells, and further studies are required to confirm the present
results.
In conclusion, combinatorial treatment with TNFSF10
and EFLDO induced cell apoptosis in HepG2 cells by promoting the
caspase pathway, suggesting a potential therapeutic effect of the
combination of EFLDO and TNFSF10 in treating liver cancer.
Acknowledgements
The authors would like to thank The Pharmaceutical
Engineering Department of Southeast University for technical
support of this study.
Funding
The project was funded by the priority academic
program development of Jiangsu Higher Education Institutions (grant
no. 1107047002), and the Key Laboratory for Chemistry and Molecular
Engineering of Medicinal Resources (Guangxi Normal University;
grant nos. CMEMR2016-B06 to ZXL).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YQ acquired the data, analyzed and interpreted the
data, drafted the manuscript and revised it critically. ZL designed
and conceived the study and revised the manuscript. XW, JZ and CL
performed the experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Takada Y, Tohyama T and Watanabe J:
Biological markers of hepatocellular carcinoma for use as selection
criteria in liver transplantation. J Hepatobiliary Pancreat Sci.
22:279–286. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ling D, Xia H, Park W, Hackett MJ, Song C,
Na K, Hui KM and Hyeon T: pH-sensitive nanoformulated triptolide as
a targeted therapeutic strategy for hepatocellular carcinoma. ACS
Nano. 8:8027–8039. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kassebaum NJ, Bertozzi-Villa A, Coggeshall
MS, Shackelford KA, Steiner C, Heuton KR, Gonzalez-Medina D, Barber
R, Huynh C, Dicker D, et al: Global, regional, and national levels
and causes of maternal mortality during 1990–2013: A systematic
analysis for the Global burden of disease study 2013. Lancet.
384:980–1004. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alsaied OA, Sangwan V, Banerjee S, Krosch
TC, Chugh R, Saluja A, Vickers SM and Jensen EH: Sorafenib and
triptolide as combination therapy for hepatocellular carcinoma.
Surgery. 156:270–279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen W, Zhang Y, Li Y, Zhang J, Zhang T,
Fu B, Zhang Q and Jiang N: Constitutive expression of Wnt/β-catenin
target genes promotes proliferation and invasion of liver cancer
stem cells. Mol Med Rep. 13:3466–3474. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He S, Chen Y, Chen XP, Zhang WG, Wang HP,
Zhao YZ and Wang SF: Antitumor effects of soluble TRAIL in human
hepatocellular carcinoma. J Huazhong Univ Sci Technolog Med Sci.
25:51–54. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He SQ, Rehman H, Gong MG, Zhao YZ, Huang
ZY, Li CH, Zhang WG and Chen XP: Inhibiting survivin expression
enhances TRAIL-induced tumoricidal activity in human hepatocellular
carcinoma via cell cycle arrest. Cancer Biol Ther. 6:1247–1257.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Ma H, Zhang J, Liu S, Liu Y and
Zheng D: AAV-mediated TRAIL gene expression driven by hTERT
promoter suppressed human hepatocellular carcinoma growth in mice.
Life Sci. 82:1154–1161. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y, Qu ZH, Cui M, Guo C, Zhang XM, Ma
CH and Sun WS: Combined endostatin and TRAIL gene transfer
suppresses human hepatocellular carcinoma growth and angiogenesis
in nude mice. Cancer Biol Ther. 8:466–473. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang B, Shan H, Li D, Zhu KS, Jiang ZB
and Huang MS: Cisplatin sensitizes human hepatocellular carcinoma
cells, but not hepatocytes and mesenchymal stem cells, to TRAIL
within a therapeutic window partially depending on the upregulation
of DR5. Oncol Rep. 25:461–468. 2011.PubMed/NCBI
|
|
11
|
Lee JY, Jung KH, Morgan MJ, Kang YR, Lee
HS, Koo GB, Hong SS, Kwon SW and Kim YS: Sensitization of
TRAIL-induced cell death by 20(S)-ginsenoside Rg3 via CHOP-mediated
DR5 upregulation in human hepatocellular carcinoma cells. Mol
Cancer Ther. 12:274–285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okano H, Shiraki K, Inoue H, Kawakita T,
Yamanaka T, Deguchi M, Sugimoto K, Sakai T, Ohmori S, Fujikawa K,
et al: Cellular FLICE/caspase-8-inhibitory protein as a principal
regulator of cell death and survival in human hepatocellular
carcinoma. Lab Invest. 83:1033–1043. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ganten TM, Haas TL, Sykoraet J, Stahl H,
Sprick MR, Fas SC, Krueger A, Weigand MA, Grosse-Wilde A, Stremmel
W, et al: Enhanced caspase-8 recruitment to and activation at the
DISC is critical for sensitisation of human hepatocellular
carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic
drugs. Cell Death Differ. 11 (Suppl 1):S86–S96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Su CL, Wu CJ, Chen FN, Wang BJ, Sheu SR
and Won SJ: Supernatant of bacterial fermented soybean induces
apoptosis of human hepatocellular carcinoma Hep 3B cells via
activation of caspase 8 and mitochondria. Food Chem Toxicol.
45:303–314. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Youness ER, El Nemr M, Oraby FS, Ahmed NM,
Moghni MA, Aly HF and Ahmed HH: Evaluation of apoptotic marker
Bcl2, CD4+, human hepatocyte growth factor and metalloproteinase-9
as tumor markers for patients with hepatocellular carcinoma. Indian
J Clin Biochem. 29:351–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang L and Fang B: Mechanisms of
resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther.
12:228–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu C, Liao ZX, Liu SJ, Qu YB and Wang HS:
Two new derivatives from Euphorbia lunulata bge and their
anti-proliferation activities. Fitoterapia. 96:33–38. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qu YB, Liao ZX, Liu C, Wang XZ and Zhang
J: EFLDO induces apoptosis in hepatic cancer cells by caspase
activation in vitro and suppresses tumor growth in
vivo. Biomed Pharmacother. 100:407–416. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: HepG2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|
|
20
|
Twentyman P and Luscombe M: A study of
some variables in a tetrazolium dye (MTT) based assay for cell
growth and chemosensitivity. Br J Cancer. 56:279–285. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fossati S, Ghiso J and Rostagno A: TRAIL
death receptors DR4 and DR5 mediate cerebral microvascular
endothelial cell apoptosis induced by oligomeric Alzheimer's Aβ.
Cell Death Dis. 3:e3212012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tomasetti M, Andera L, Alleva R, Borghi B,
Neuzil J and Procopio A: Alpha-tocopheryl succinate induces DR4 and
DR5 expression by a p53-dependent route: Implication for
sensitisation of resistant cancer cells to TRAIL apoptosis. FEBS
Lett. 580:1925–1931. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dalerba P, Dylla SJ, Park IK, Liu R, Wang
X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al:
Phenotypic character-ization of human colorectal cancer stem cells.
Proc Natl Acad Sci USA. 104:10158–10163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ino Y, Gotoh M, Sakamoto M, Tsukagoshi K
and Hirohashi S: Dysadherin, a cancer-associated cell membrane
glycoprotein, down-regulates E-cadherin and promotes metastasis.
Proc Natl Acad Sci USA. 99:365–370. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nam JS, Hirohashi S and Wakefield LM:
Dysadherin: A new player in cancer progression. Cancer Lett.
255:161–169. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
El-Shemi AG, Ashshi AM, Na Y, Li YM,
Al-Allaf FA, Oh E, Jung BK and Yun CO: Combined therapy with
oncolytic adenoviruses encoding TRAIL and IL-12 genes markedly
suppressed human hepatocellular carcinoma both in vitro and
in an orthotopic transplanted mouse model. J Exp Clin Cancer Res.
35:742016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chiu CT, Yeh TS, Hsu JC and Chen M:
Expression of Bcl-2 family modulated through p53-dependent pathway
in human hepatocellular carcinoma. Dig Dis Sci. 48:670–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Franklin DA, He Y, Leslie PL, Tikunov AP,
Fenger N, Macdonal JM and Zhang Y: p53 coordinates DNA repair with
nucleotide synthesis by suppressing PFKFB3 expression and promoting
the pentose phosphate pathway. Sci Rep. 6:380672016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Romano FJ, Rossetti S, Conteduca V,
Schepisi G, Cavaliere C, Di Franco R, La Mantia E, Castaldo L,
Nocerino F, Ametrano G, et al: Role of DNA repair machinery and p53
in the testicular germ cell cancer: A review. Oncotarget.
7:85641–85649. 2016. View Article : Google Scholar : PubMed/NCBI
|