Introduction
Colorectal cancer (CRC) is a widespread malignant
tumor, representing the third most common cancer in men and women
globally (1). It is unfortunate
that only a small fraction of patients with metastatic CRC can
undergo curative resection and experience disease-free survival
(2). Surgical treatment and
radiotherapy are currently used, but both have certain limitations.
It has been identified that radiotherapy and surgery can trigger
undesirable invasion or metastasis in certain cases (3–7).
Therefore, chemotherapy is frequently applied in cancer therapy.
The majority of anti-cancer drugs are investigated and used based
on their inhibitory effect on the growth and induction of apoptosis
in tumor cells. Understanding the mechanisms underlying cellular
responses is important to improve chemotherapeutic efficacy.
Butyrate, a short chain fatty acid, is produced by
fermenting dietary fiber using gut microbiota (8). Butyrate serves an important role in
inhibiting cell growth and inducing the apoptosis of tumors
(9,10) and it functions by regulating the
activity of histone deacetylase (HDAC), silent mating type
information regulation 2 homolog (SIRT)1, and caspase-3 (11). Butyrate has been identified as a
chemotherapeutic strategy based on its effects on cell growth,
proliferation and apoptosis in tumor cells (12). However, the role of butyrate in the
tumorigenesis and progression of CRC, and the underlying
anti-cancer mechanism of butyrate remains to be elucidated.
SIRT1, as a member of the HDAC family, is known to
be positively correlated with tumor growth (13). Knockdown of SIRT1 has been reported
to increase the chemosensitivity of hepatic cancer cells to
anti-tumor agent cisplatin and to inhibit cell metastasis (14). It has been identified that the
expression of SIRT1 is involved in transcription factor nanog
expression in CRC (15). SIRT1 has
been employed as the target of anti-tumor drugs to inhibit cell
growth in cancers (16). It has
also been demonstrated that butyrate can inhibit SIRT1 expression
to induce apoptosis in hepatic cancer cells (17). Whether butyrate, as a HDAC
inhibitor, can modulate cell apoptosis and proliferation via SIRT1
regulation in CRC remains to be investigated.
The mammalian target of rapamycin (mTOR) is a
serine/threonine kinase that affects cell metabolism and growth
(18), and regulates protein
synthesis and controls cell growth by mediating the phosphorylation
of p70S6 ribosomal kinase (S6K) and eukaryotic translation
initiation factor 4E binding protein, thus activating S6 via
phosphorylation (19).
Phosphorylation of S6K at T389 has been reported to be enhanced by
SIRT1-mediated deacetylation (20). However, the exact association
between activity of mTOR/S6K signaling and SIRT1 in CRC requires
investigation.
The present study confirmed the effect of butyrate
on cell proliferation and apoptosis in CRC cells in vitro,
and explored the underlying molecular mechanism in order to reveal
the potential therapeutic target for CRC cells.
Materials and methods
Cell culture
The human CRC cell line HCT116 was purchased from
the Cell Bank of the Chinese Academy of Sciences Institute
(Shanghai, China). Cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Lonza Group, Ltd., Basel, Switzerland), 5 mM L-glutamine, 5 mM
non-essential amino acids, 100 U/ml penicillin and streptomycin
(Invitrogen; Thermo Fisher Scientific, Inc.) in a humidified 5%
CO2 incubator at 37°C.
Proliferation assay
Cell proliferation was evaluated using MTT
(Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). A total of 2,000
cells were seeded into each well of a 96-well plate in 100 µl
medium and incubated with or without 0.5, 1, 2, 3, 4 and 5 mM
butyrate for 24, 48 and 72 h at 37°C in a 5% CO2
incubator. Subsequently, cells were incubated with 20 µl 5 mg/ml
MTT at 37°C for 4 h, and then lysed for 10 min with the addition of
200 µl DMSO (OriGen Biomedical, Inc., Austin, TX, USA). Absorbance
was measured at 490 nm using a Rainbow microplate reader (Tecan
Group, Ltd., Mannedorf, Switzerland).
Apoptosis assay
HCT116 cells (4×103) were seeded into each well of a
96-well plate and were cultured to 80% confluence at 37°C, and then
treated with 1 mM butyrate for 0, 24, 48 and 72 h. Cells were
collected and cell apoptosis was detected using a Cell Death
Detection ELISAPLUS kit (cat. no. 11774425001; Roche Applied
Science, Mannheim, Germany) according to the manufacturer's
protocols. Results are presented as the fold induction of DNA
fragmentation relative to untreated control. To verify the effect
of butyrate on apoptosis, activation of caspase-3 was also assessed
in butyrate-treated or untreated cells using human active caspase-3
ELISA (cat. no. AF835; R&D Systems, Inc., Minneapolis, MN,
USA). Cells (5×105/well) were cultured in 6-well plates to 80%
confluence at 37°C. Then the cells were treated with 1 mM butyrate
for 0, 24, 48 and 72 h. The cells were lysed and detected in
accordance with the manufacturer's protocol.
Western blot analysis
Cells were cultured to 80% confluence at 37°C. The
cells (5×106/well) were washed twice with PBS and proteins were
extracted using the M-PER Mammalian Protein Extraction Reagent
(Pierce; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Following centrifugation at 12,000 × g for
10 min at 4°C, the supernatant was collected and quantified using a
bicinchoninic acid quantification kit (Beyotime Institute of
Biotechnology, Haimen, China). The proteins (50 µg) were separated
by SDS-PAGE on a 12% gel (Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China) and transferred onto polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 5% non-fat dried milk in Tris-buffered
saline with 0.1% Tween-20 for 1 h at room temperature, and
incubated with specific primary antibodies overnight at 4°C. Mouse
monoclonal anti-B-cell lymphoma-2 (Bcl-2; 1:500; cat. no. sc7382;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA), rabbit polyclonal
anti-Bcl-2-associated X protein (Bax; 1:500, cat. no. sc493; Santa
Cruz Biotechnology, Inc.), mouse monoclonal anti-GAPDH antibody
(1:3,000; cat. no. sc-365062; Santa Cruz Biotechnology, Inc.),
rabbit polyclonal anti-mTOR (1:1,000; cat. no. 2972; Cell Signaling
Technology, Inc., Danvers, MA, USA), rabbit monoclonal
anti-phosphorylated (p)-mTOR (Ser2448; 1:1,000; cat. no. 5536; Cell
Signaling Technology, Inc.), rabbit polyclonal anti-S6K1 (1:1,000;
cat. no. 9202; Cell Signaling Technology, Inc.), rabbit monoclonal
anti-p-S6K1 (Thr389; 1:1,000; cat. no. 9205; Cell Signaling
Technology, Inc.), rabbit polyclonal anti-acetylated-lysine
(1:1,000; cat. no. 9441; Cell Signaling Technology, Inc.), rabbit
monoclonal anti-S6 (1:1,000; cat. no. 2217; Cell Signaling
Technology, Inc.), rabbit monoclonal anti-p-S6 (Ser235/236;
1:2,000; cat. no. 4858; Cell Signaling Technology, Inc.) and mouse
monoclonal anti-SIRT1 (1:1,000; cat. no. 8469; Cell Signaling
Technology, Inc.) antibodies were used, followed by the horseradish
peroxidase-conjugated secondary antibodies goat anti-mouse
(1:2,000; cat. no. sc-2005; Santa Cruz Biotechnology, Inc.) and
anti-rabbit immunoglobulin G (1:2,000; cat. no. sc-2004; Santa Cruz
Biotechnology, Inc.) for 2 h at room temperature. Development was
performed using an enhanced chemiluminescence detecting reagent (GE
Healthcare Life Sciences, Little Chalfont, UK). The protein blots
were quantified by densitometry using Quantity One software version
4.6.2 (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and the
amounts were expressed relative to the internal reference
GAPDH.
RNA interference and overexpression of
SIRT1
Small interfering (si)-RNA against SIRT1 and the
negative control were designed and chemically synthesized by
Shanghai GenePharma Co., Ltd. (Shanghai, China). The target
sequence of siRNA against SIRT1 was 5′-GAAGTGCCTCAGATATTAA-3′. The
siRNA sequences of the negative control (siCtrl) was: Sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. A total of 2×104 HCT116 cells were
seeded into each well of a 12-well plate and were cultured to 80%
confluence in incubator with 5% CO2 at 37°C in DMEM
medium supplemented with 10% FBS. Cell transfections were performed
using 100 nmol siRNA and 5 µl Lipofectamine® 2000™
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The pcDNA3.1-His-SIRT1 plasmid was
obtained from Dr Tony Kouzarides (University of Cambridge,
Cambridge, UK) (21). Cells were
further cultured at 37°C for 48 h following transfection, and cells
were subsequently lysed and analyzed for the protein expression of
SIRT1 by western blotting as aforementioned. In addition, cells
were treated with 1 mM butyrate for 48 h and then the cell
proliferation and protein levels of the mTOR/S6K1 signaling pathway
were detected.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using an RNA isolation kit
(A&A Biotechnology, Gdynia, Poland), according to the
manufacturer's protocol. cDNA was obtained by RT using the
RevertAid™ First Strand cDNA synthesis kit (Fermentas; Thermo
Fisher Scientific, Inc., Pittsburgh, PA, USA) according to the
manufacturer's protocol. The RT reaction was performed at 25°C for
10 min, 42°C for 60 min and 70°C for 10 min. cDNA was amplified via
qPCR using a TaqMan® Gene Expression Assay (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with fluorogenic
carboxyfluorescein-labeled probes using specific primers for target
proteins. The specific primers for PCR were: Forward,
5′-TCGGCAGGTCCCTTTGTCATCC-3′ and reverse,
5′-TGCAGGTCAACTGGTGTCGT-3′ for SIRT1; and forward,
5′-GATCCCTCCAAAATCAAGTG-3′ and reverse, 5′-GAGTCCTTCCACGATACCAA-3′
for GAPDH. Real-time fluorescence detection was performed with the
ABI PRISM 7700 Sequence Detector (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions for RT-qPCR were as
follows: 40 amplification cycles of 94°C for 10 sec, 53°C for 30
sec and 72°C for 40 sec, followed by a final extension at 72°C for
10 min. mRNA expression of the target proteins was calculated using
the formula 2−ΔΔCq (22) and was normalized to the level of
GAPDH. The relative mRNA level was presented as a percentage of the
control.
Statistical analysis
Data were obtained from at least three experiments.
Values were expressed as the mean ± standard error of the mean.
Statistical analysis was performed using SPSS version 16.0 for
Windows (SPSS, Inc., Chicago, IL, USA). One-way analysis of
variance was used to assess differences between groups. The Duncan
method was employed for pairwise comparison followed by Bonferroni
correction. P<0.05 was considered to indicate a statistically
significant difference.
Results
Butyrate treatment inhibits
proliferation and induces apoptosis in HCT116 cells
HCT116 cells were treated with butyrate at 0.5, 1,
2, 3, 4 and 5 mM for 24, 48 and 72 h, respectively. Cell viability
was evaluated by MTT assay and the cellular growth inhibition rate
was calculated. As presented in Fig.
1A, butyrate treatment inhibited the proliferation of HCT116
cells in a dose- and time-dependent manner; 0.5 mM butyrate
treatment for 24 and 48 h and 1 mM butyrate treatment for 24 h did
not demonstrate significant inhibition of proliferation in HCT116
cells, while 1 mM butyrate treatment for 48 h and butyrate
treatment at all concentrations for 72 h demonstrated significant
inhibition of proliferation activity (Fig. 1A). In addition, cells were treated
with 1 mM butyrate for 24, 48 and 72 h, and DNA fragmentation and
caspase-3 activity were evaluated by ELISA assay. The results
demonstrated that butyrate treatment induced the apoptosis of
HCT116 cells in a time-dependent manner (Fig. 1B and C). To further confirm the
role of butyrate in the apoptosis of HCT116 cells, the
pro-apoptosis protein Bax and anti-apoptosis protein Bcl-2 were
evaluated. Western blot analysis indicated that butyrate-treated
HCT116 cells demonstrated higher expression of Bax and lower
expression of Bcl-2 in comparison with control cells, with
increased Bax/Bcl-2 ratios (Fig.
1D). These results indicated that butyrate treatment inhibited
cell proliferation by inducing apoptosis signaling via the
modulation of Bax and Bcl-2 expression.
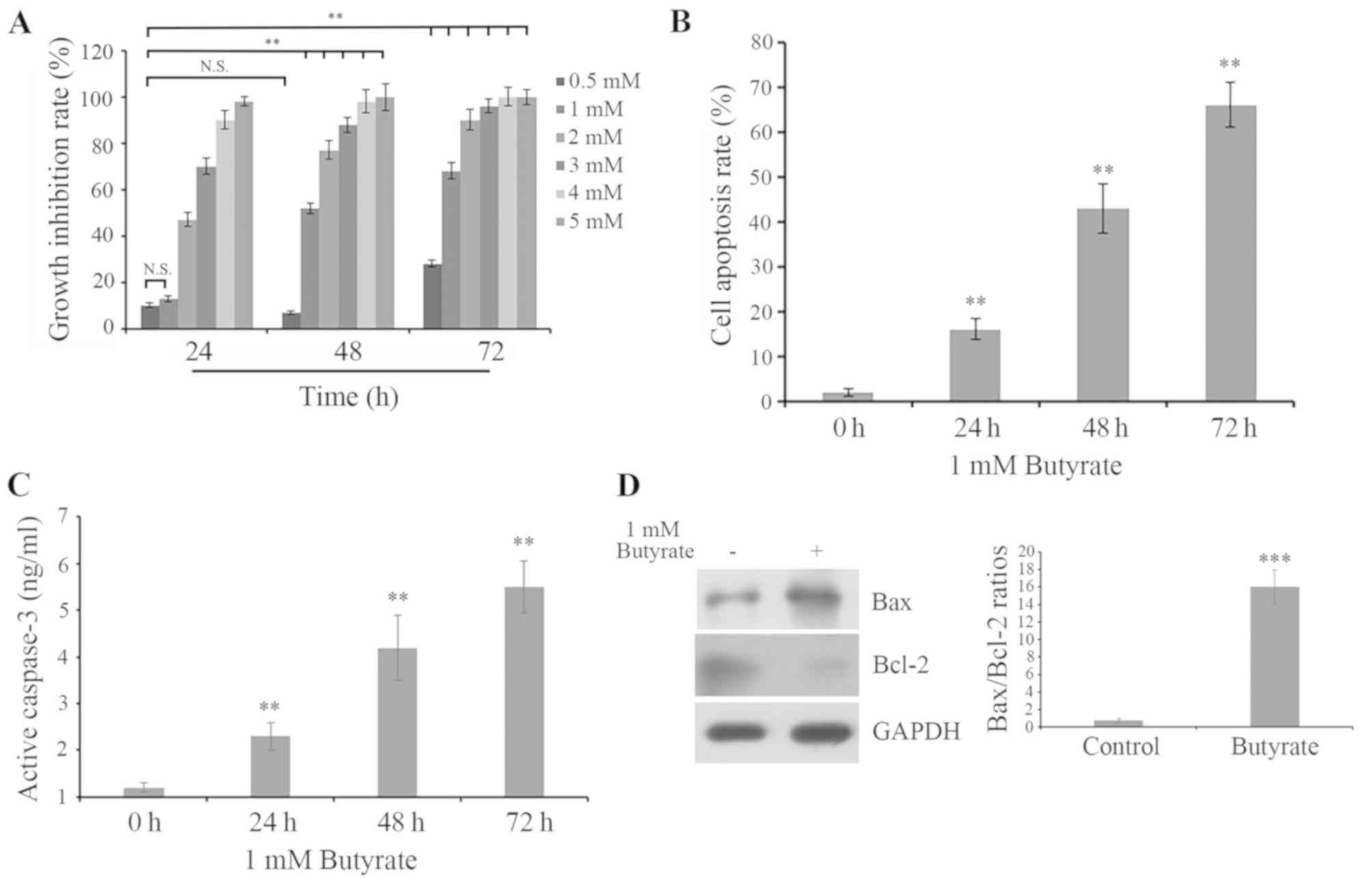 | Figure 1.Butyrate treatment inhibits
proliferation and promotes apoptosis in HCT116 cells. (A) Cells
were treated with 0.5, 1, 2, 3, 4 and 5 mM butyrate for 24, 48 and
72 h, respectively. Cell growth was evaluated by MTT assay and the
cell growth inhibition rate was calculated. N.S., no significance;
**P<0.01 vs. 0.5 mM butyrate for 24. (B) Cells were treated with
1 mM butyrate for 0, 24, 48 and 72 h. DNA fragmentation was
determined by histone release. Histone release was measured by
ELISA assay, indicating cell apoptosis. The fold induction of DNA
fragmentation in HCT116 cells treated with butyrate was presented
relative to untreated control cells. (C) Caspase-3 activation was
determined by ELISA assay. **P<0.01 vs. 0 h. (D) Alterations in
Bax and Bcl-2 protein expression in HCT116 cells with or without 1
mM butyrate for 48 h were assessed by western blot analysis with
anti-Bax or anti-Bcl-2 antibodies, respectively. Bax/Bcl-2 ratios
were calculated by protein expression/Bcl-2 protein expression.
GAPDH was detected as the internal reference. ***P<0.001 vs.
control. Bcl-2, B-cell lymphoma-2; Bax, Bcl-2-associated X
protein. |
mTOR/S6K1 signaling is deactivated by
butyrate treatment
To investigate the underlying mechanism of butyrate
inhibiting proliferation and inducing apoptosis in HCT116 cells,
the cells were treated with 1 mM butyrate for 48 h. Then the
activity of mTOR/S6K1 signaling were investigated using western
blot analysis. The results demonstrated that the phosphorylation
levels of mTOR at Ser2448, S6K1 at Thr389 and S6 at Ser235/236 were
significantly downregulated, while the acetylation level of S6K1
was significantly upregulated and the expression of SIRT1 was
decreased following butyrate treatment (Fig. 2). Based on these data, it was
speculated that butyrate treatment inhibited cell proliferation,
possibly by deactivating mTOR/S6K1 signaling, which may be mediated
by the downregulation of SIRT1 and the enhancement of S6K1
acetylation.
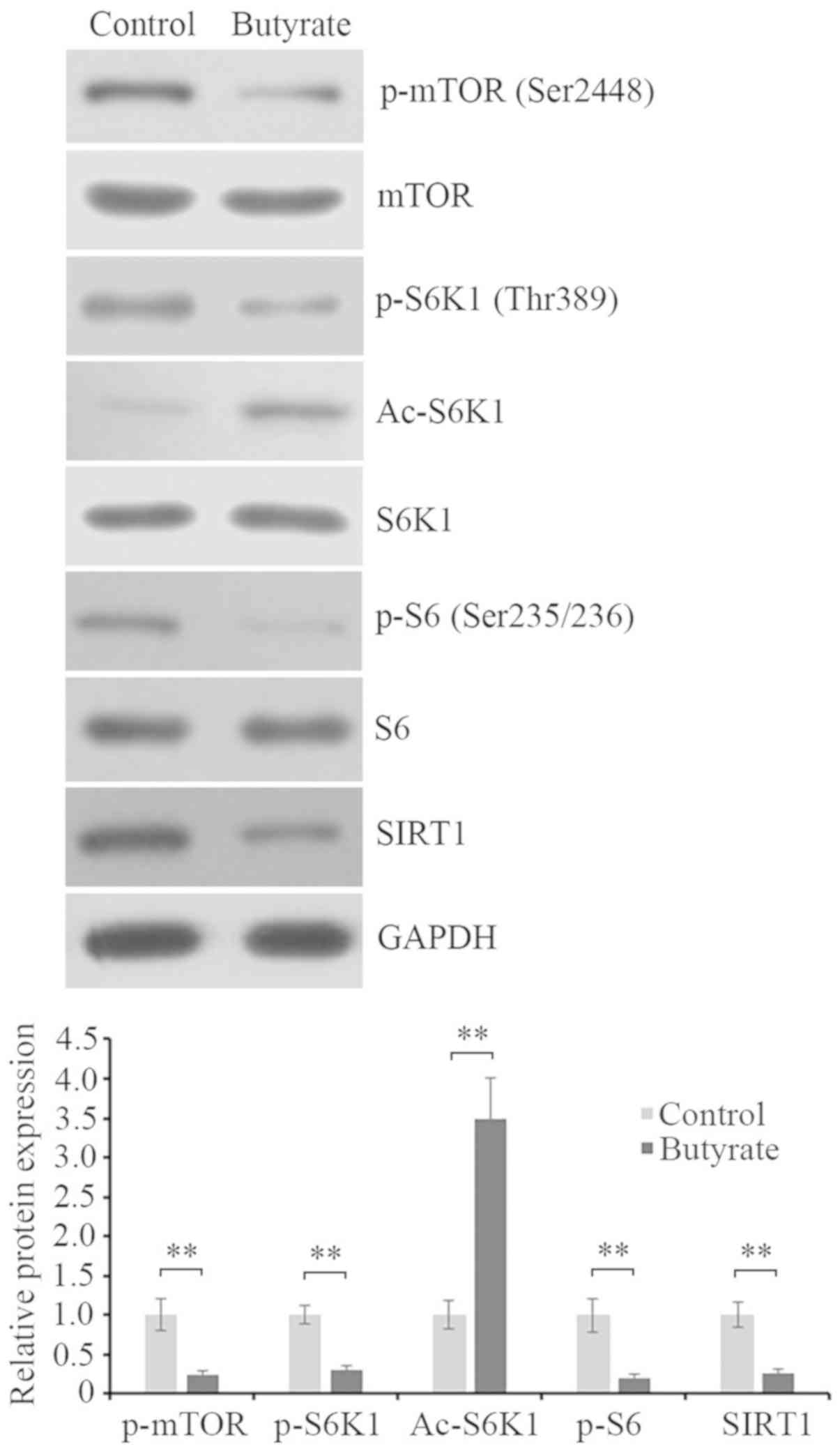 | Figure 2.mTOR/S6K1 signaling is inhibited and
the acetylation of S6K1 is increased by butyrate treatment. Protein
expression levels of the phosphorylation of mTOR at Ser2448
(p-mTOR), mTOR, phosphorylation of S6K1 at Thr389 (p-S6K1),
acetylation of S6K1 (Ac-S6K1), S6K1, phosphorylation of S6 at
Ser235/236 (p-S6), S6 and SIRT1 in HCT116 cells with or without 1
mM butyrate for 48 h were assessed by western blot analysis using
their corresponding antibodies. GAPDH was detected as the internal
reference. The blots were quantified by densitometry, and data were
presented as the ratios of p-mTOR/mTOR, p-S6K1/S6K1, Ac-S6K1/S6K1,
p-S6/S6 and SIRT1/GAPDH. The results were expressed as a ratio
relative to the value obtained in untreated control cells.
**P<0.01, as indicated. mTOR, mammalian target of rapamycin;
S6K1, S6 kinase β-1; p-, phosphorylated; Ac-, acetylated; SIRT,
silent mating type information regulation 2 homolog. |
SIRT1 silencing enhances the
inhibition of butyrate treatment on mTOR/S6K1 signaling and cell
growth
To verify the above hypothesis, SIRT1 was knocked
down using siRNA technology. The mRNA and protein expression of
SIRT1 were evaluated by RT-qPCR analysis and western blotting. The
results demonstrated that SIRT1 expression in SIRT1-silenced cells
was significantly decreased compared with non-silenced control
cells (Fig. 3A and B).
Subsequently, SIRT1-silenced and non-silenced HCT116 cells were
treated with or without 1 mM butyrate for 48 h. Activity of
mTOR/S6K1 signaling in cells was detected by western blot analysis.
The results demonstrated that SIRT1 silencing increased the
acetylation of S6K1 and decreased the phosphorylation of mTOR at
Ser2448 and S6K1 at Thr389. In addition, butyrate treatment further
reduced the phosphorylations of mTOR at Ser2448 and S6K1 at Thr389
in SIRT1-silenced HCT116 cells (Fig.
3C). Additionally, cell proliferation assay demonstrated that
SIRT1 silencing further reduced cell proliferation of HCT116 cells
treated with butyrate (Fig. 3D).
The cellular apoptosis assay revealed that SIRT1 silencing promoted
the inducing effect of butyrate on apoptosis in HCT116 cells
(Fig. 3E). In addition, it was
identified that butyrate could still inhibit proliferation and
induce apoptosis. These data suggested that SIRT1 may be involved
in the regulation of butyrate in mTOR/S6K1 signaling, and thus
affect cell proliferation and apoptosis, but it may not be the only
target of butyrate; it is possible that the acetylation of S6K1 is
associated with the inhibition of butyrate on mTOR/S6K1
signaling.
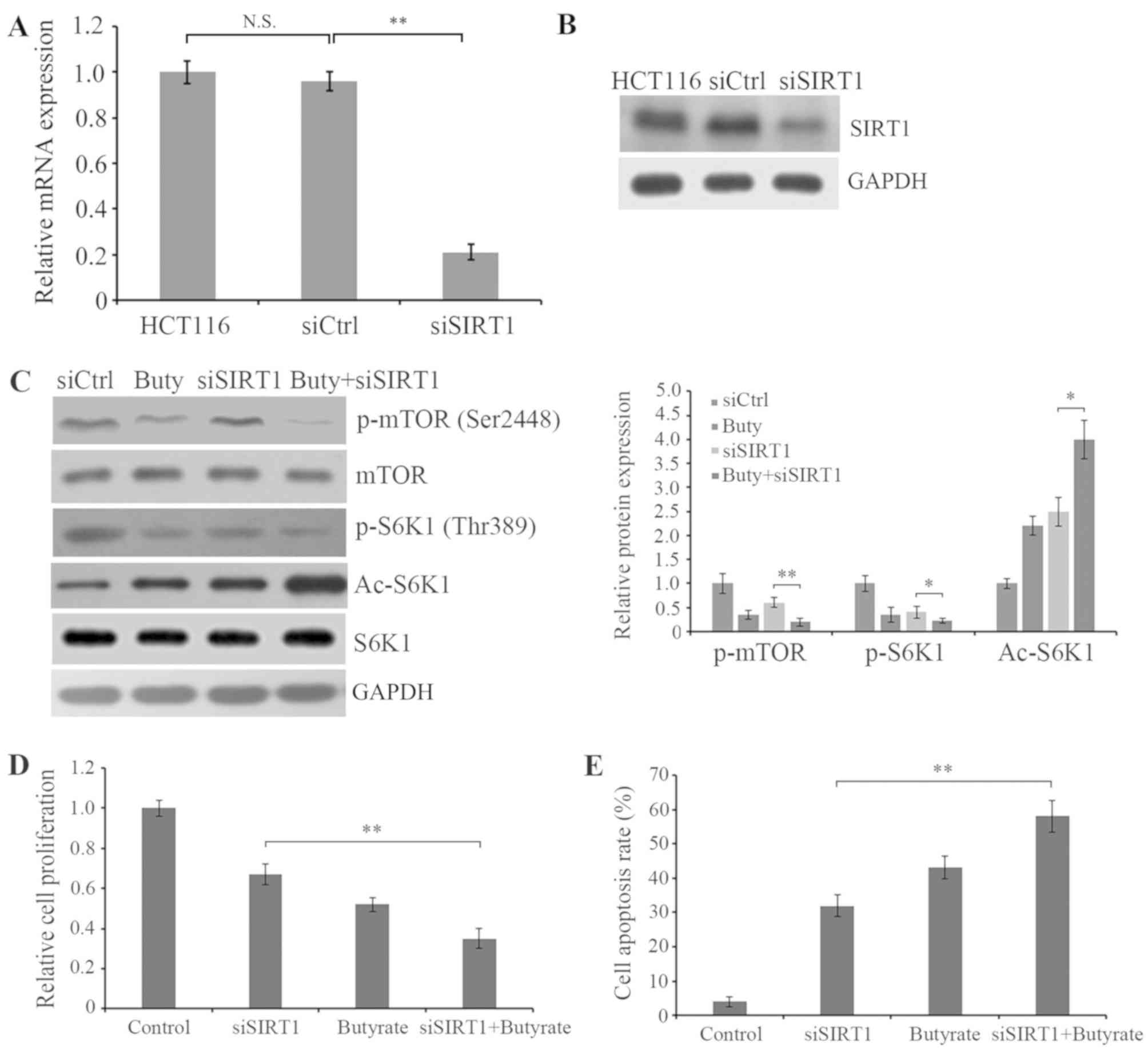 | Figure 3.Butyrate treatment enhances the
inhibition of cell proliferation and the activity of mTOR/S6K1
induced by SIRT1 silencing. Cells were transfected with siCtrl or
siSIRT1 for 48 h and the (A) mRNA and (B) protein expressions of
SIRT1 cells were analyzed by reverse transcription-quantitative
polymerase chain reaction and western blot analyses, respectively.
GAPDH was detected as the internal standard. N.S., no significance;
**P<0.01 vs. siCtrl. (C) SIRT1-silenced and non-silenced HCT116
cells were treated with or without 1 mM butyrate for 48 h. The
protein expression of p-mTOR, mTOR, p-S6K1, Ac-S6K1 and S6K1 in
cells was assessed by western blot analysis using the corresponding
antibodies. GAPDH was detected as an internal standard. The blots
were quantified by densitometry, and data were presented as the
ratios of p-mTOR/mTOR, p-S6K1/S6K1 and Ac-S6K1/S6K1. The results
were expressed as a ratio relative to the value obtained in control
cells. (D) SIRT1-silenced and non-silenced HCT116 cells were
incubated with fresh medium with or without 1 mM butyrate for 48 h.
Cell proliferation was detected by an MTT assay. (E) Cellular
apoptosis was evaluated by detecting the fold induction of DNA
fragmentation using ELISA assay. *P<0.05 and **P<0.01, as
indicated. SIRT, silent mating type information regulation 2
homolog; mTOR, mammalian target of rapamycin; S6K1, S6 kinase β-1;
siCtrl, control small interfering RNA; siSIRT1, SIRT1 small
interfering RNA; p-, phosphorylated; Ac-, acetylated; Buty,
butyrate. |
Butyrate treatment attenuates the
induction of SIRT1 overexpression in the proliferation of HCT116
cells
To verify the role of SIRT1 in the modulation of
butyrate on mTOR/S6K1 signaling and cell proliferation, SIRT1 was
overexpressed in HCT116 cells and the effect of butyrate on
mTOR/S6K1 signaling in SIRT1-overexpressed cells was detected.
Western blotting demonstrated that SIRT1 overexpression increased
the phosphorylations of mTOR at Ser2448 and S6K1 at Thr389, while
butyrate treatment attenuated the promotive effect (Fig. 4A). In addition, the cell
proliferation assay indicated that butyrate treatment also
significantly suppressed the enhancement of proliferation induced
by SIRT1 overexpression and the upregulation of SIRT1 attenuated
the effect of butyrate on cell proliferation (Fig. 4B). Therefore, these data
demonstrated that butyrate treatment inhibited cell proliferation
by deactivating mTOR/S6K1 signaling, which was possibly mediated by
SIRT1 reduction.
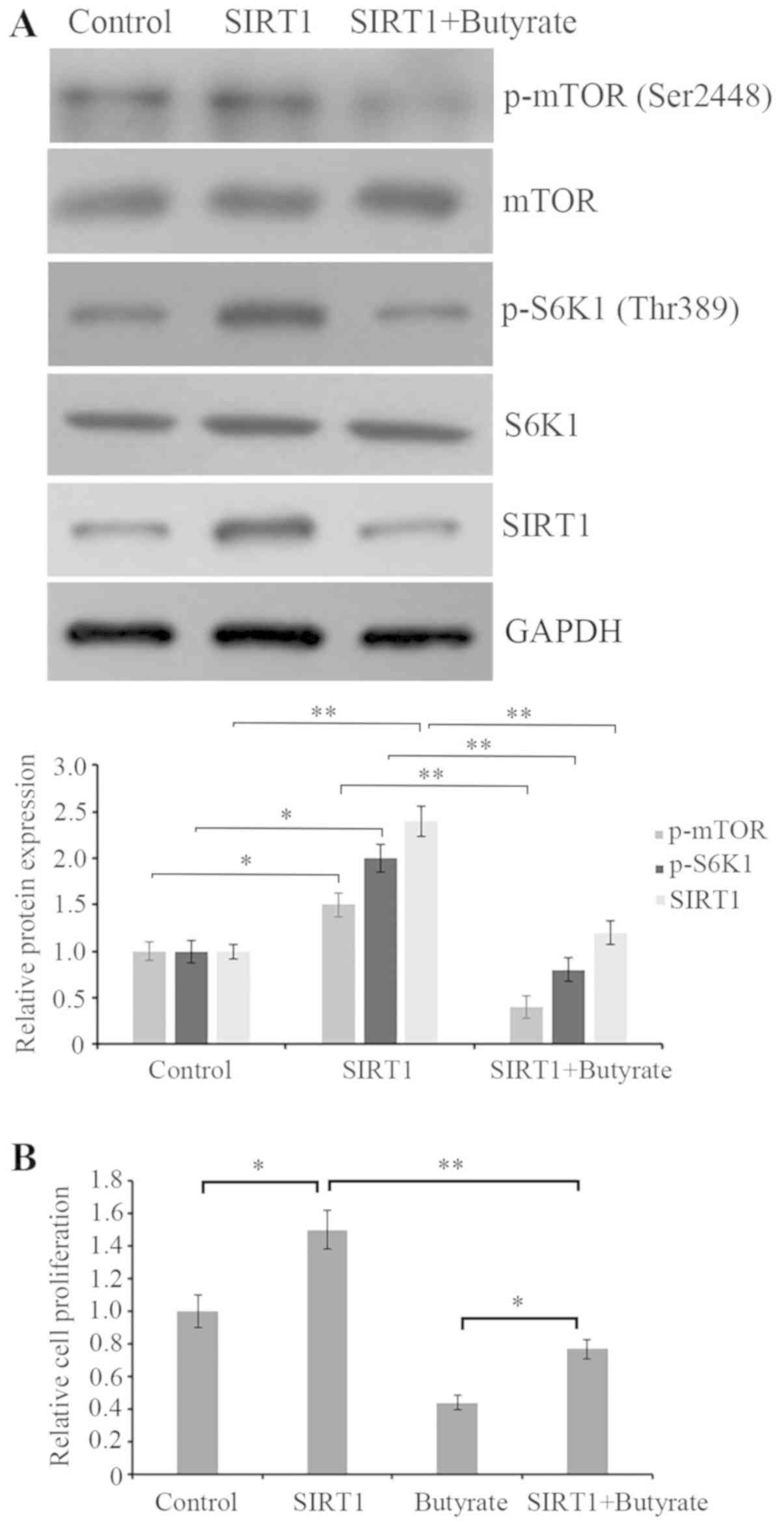 | Figure 4.Butyrate treatment attenuates SIRT1
overexpression mediated activation of mTOR/S6K1 and proliferation
elevation. (A) Cells were transfected with or without
SIRT1-expressing plasmid for 48 h, and then the protein expression
of p-mTOR, mTOR, p-S6K1, S6K1 and SIRT1 in cells was assessed by
western blot analysis using the corresponding antibodies. GAPDH was
detected as an internal standard. The western blots were quantified
by densitometry and data were presented as the ratio of
p-mTOR/mTOR, p-S6K1/S6K1 and SIRT1/GAPDH. The results were
expressed as a ratio relative to the value obtained in control
cells. (B) Cell proliferation was detected by an MTT assay.
*P<0.05 and **P<0.01, as indicated. SIRT, silent mating type
information regulation 2 homolog; mTOR, mammalian target of
rapamycin; S6K1, S6 kinase β-1; p-, phosphorylated. |
Discussion
Butyrate, a HDAC inhibitor, has been considered as a
promising antitumor agent for a variety of different types of human
cancers (11,23,24).
Chemotherapy commonly kills tumor cells by activating apoptosis. It
has been reported that butyrate is able to reduce cell
proliferation and stimulate cell apoptosis in multiple types of
cancers (25–27). However, the exact role of butyrate
in CRC and the underlying mechanism of butyrate-induced apoptosis
remains unclear. The present study investigated the effect of
butyrate on cell proliferation and apoptosis, and explored the
possible molecular mechanism in human CRC HCT116 cells. It was
identified that butyrate significantly inhibited cell proliferation
and induced apoptosis in HCT116 cells. mTOR/S6K1 signaling was
revealed to be regulated by butyrate treatment and SIRT1 expression
was demonstrated to be involved in the regulation.
In addition, it was also revealed that butyrate
treatment induced apoptosis in CRC HCT116 cells and significantly
increased the expression of the pro-apoptosis protein Bax and
decreased the expression the anti-apoptosis protein Bcl-2. In the
Bcl-2 family, the ratio of pro-apoptotic to anti-apoptotic proteins
is a key factor in determining the occurrence and level of
apoptosis. The proportion of pro-apoptotic proteins in cells also
determines the cellular response to death signals and cell destiny
(28,29). SIRT1 is commonly highly expressed
in cancer cells and serves an important role in promoting cell
proliferation and blocking apoptosis (13,30).
It has been demonstrated that downregulation of SIRT1 expression
mediated by chemotherapeutic drugs can inhibit tumor growth and
induce cell death (24,31). SIRT1 downregulation by butyrate
reduced Bcl-2 expression and elevated caspase-3 expression in
hepatic cancer cells (17). In the
present study, SIRT1 expression was decreased by butyrate
treatment. Therefore, it is possible that butyrate treatment
regulated apoptosis-associated proteins by decreasing SIRT1
expression.
It has also been reported that SIRT1, as a
deacetylase, can inhibit the acetylation of S6K1 and thereby
promote the phosphorylation of Thr389 (a critical phosphorylation
site for S6K1 activity) (20).
S6K1, one of the substrates of mTOR, is responsible for modulating
multiple cell biological functions, including protein synthesis,
cell survival and growth (19). In
the present study, it was demonstrated that SIRT1 silencing
markedly increased the acetylation of S6K1, decreased its
phosphorylation at Thr389 and slightly decreased the
phosphorylation of mTOR at Ser2448 (the activity site for mTOR).
However, SIRT1 overexpression induced the opposite effect to SIRT1
silencing. Butyrate treatment further enhanced the promotive effect
of SIRT1 silencing on S6K1 acetylation and the inhibitory effect on
S6K1 activity, whereas butyrate treatment reversed the promotion of
SIRT1 overexpression on mTOR/S6K1 signaling and cell proliferation.
However, it was identified that butyrate treatment further
inhibited the proliferation and induced cell apoptosis in
SIRT1-silenced HCT116 cells, suggesting that SIRT1 may not be the
only target of butyrate to regulate cell proliferation and
apoptosis via the mTOR/S6K1 signaling pathway, and that there may
be other signaling pathways involved in this regulation. It has
also been reported that the Lys484, Lys485 and Lys493 sites of S6K1
are acetylated (20). Whether
these Lys sites of S6K1 are involved in the modulation of SIRT1 on
S6K1 activity and the mechanism underlying the regulation of mTOR
activity have yet to be elucidated.
In conclusion, the results of the present study
demonstrated that butyrate treatment inhibited cell proliferation
and induced apoptosis by inhibiting the activation of the mTOR/S6K1
signaling pathway, partly via SIRT1 inhibition. Therefore, the
present study provides a promising target for butyrate to suppress
the proliferation and induce apoptosis in CRC HCT116 cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Health
Department of Heilongjiang Province (grant no. 2016-022),
Innovative Science Research Project of Harbin Medical University
(grant nos. 2016LCZX51 and 2016LCZX40), Scientific Foundation of
the First Affiliated Hospital of Harbin Medical University (grant
nos. 2016B008 and 2017B001) and Heilongjiang Postdoctoral Science
Foundation (grant no. LBH-Z15159).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
XL was the major contributor to the conception and
design of the research, and critically revised the manuscript for
important intellectual content. MC and ZZ performed the experiments
and acquired the data. SH performed statistical analysis, and
analyzed and interpreted the data. XL and MC obtained the funding.
MC and ZZ drafted the manuscript and gave final approval of the
version to be published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bandyopadhyay A, Wang L, Agyin J, Tang Y,
Lin S, Yeh IT, De K and Sun LZ: Doxorubicin in combination with a
small TGFβ inhibitor: A potential novel therapy for metastatic
breast cancer in mouse models. PLoS One. 5:e103652010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–17.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ben-Eliyahu S: The promotion of tumor
metastasis by surgery and stress: Immunological basis and
implications for psychoneuroimmunology. Brain Behav Immun.
17:S27–S36. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goldfarb Y and Ben-Eliyahu S: Surgery as a
risk factor for breast cancer recurrence and metastasis: Mediating
mechanisms and clinical prophylactic approaches. Breast Dis.
26:99–114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Biswas S, Guix M, Rinehart C, Dugger TC,
Chytil A, Moses HL, Freeman ML and Arteaga CL: Inhibition of
TGF-beta with neutralizing antibodies prevents radiation-induced
acceleration of metastatic cancer progression. J Clin Invest.
117:1305–1313. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaliski A, Maggiorella L, Cengel KA, Mathe
D, Rouffiac V, Opolon P, Lassau N, Bourhis J and Deutsch E:
Angiogenesis and tumor growth inhibition by a matrix
metalloproteinase inhibitor targeting radiation-induced invasion.
Mol Cancer Ther. 4:1717–1728. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhai GG, Malhotra R, Delaney M, Latham D,
Nestler U, Zhang M, Mukherjee N, Song Q, Robe P and Chakravarti A:
Radiation enhances the invasive potential of primary glioblastoma
cells via activation of the Rho signaling pathway. J Neurooncol.
76:227–237. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
den Besten G, van Eunen K, Groen AK,
Venema K, Reijngoud DJ and Bakker BM: The role of short-chain fatty
acids in the interplay between diet, gut microbiota, and host
energy metabolism. J Lipid Res. 54:2325–2340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Louis M, Rosato RR, Brault L, Osbild S,
Battaglia E, Yang XH, Grant S and Bagrel D: The histone deacetylase
inhibitor sodium butyrate induces breast cancer cell apoptosis
through diverse cytotoxic actions including glutathione depletion
and oxidative stress. Int J Oncol. 25:1701–1711. 2004.PubMed/NCBI
|
|
10
|
Louis P, Hold GL and Flint HJ: The gut
microbiota, bacterial metabolites and colorectal cancer. Nat Rev
Microbiol. 12:661–672. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lane AA and Chabner BA: Histone
deacetylase inhibitors in cancer therapy. J Clin Oncol.
27:5459–5468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berni Canani R, Di Costanzo M and Leone L:
The epigenetic effects of butyrate: Potential therapeutic
implications for clinical practice. Clin Epigenetics. 4:42012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen HC, Jeng YM, Yuan RH, Hsu HC and Chen
YL: SIRT1 promotes tumorigenesis and resistance to chemotherapy in
hepatocellular carcinoma and its expression predicts poor
prognosis. Ann Surg Oncol. 19:2011–2019. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang T, Rong N, Chen J, Zou C, Jing H,
Zhu X and Zhang W: SIRT1 expression is associated with the
chemotherapy response and prognosis of patients with advanced
NSCLC. PLoS One. 8:e791622013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Osama A, Sabry D, Hassany SM, Abdelmoneim
SS and Sabry A: SIRT-1expression is associated with expression of
NANOG in patients with colorectal adenocarcinoma. Cancer Biomark.
17:155–163. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cao B, He X, Wang W and Shi M: SIRT1
Influences the sensitivity of A549 non-small cell lung cancer cell
line to cisplatin via modulating the Noxa expression. Zhongguo Fei
Ai Za Zhi. 19:57–63. 2016.(In Chinese). PubMed/NCBI
|
|
17
|
Pant K, Yadav AK, Gupta P, Islam R, Saraya
A and Venugopal SK: Butyrate induces ROS-mediated apoptosis by
modulating miR-22/SIRT-1 pathway in hepatic cancer cells. Redox
Biol. 12:340–349. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hong S, Zhao B, Lombard DB, Fingar DC and
Inoki K: Cross-talk between sirtuin and mammalian target of
rapamycin complex 1 (mTORC1) signaling in the regulation of S6
kinase 1 (S6K1) phosphorylation. J Biol Chem. 289:13132–13141.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oberdoerffer P, Michan S, McVay M,
Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner
A, Loerch P, et al: SIRT1 redistribution on chromatin promotes
genomic stability but alters gene expression during aging. Cell.
135:907–918. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma X, Ezzeldin HH and Diasio RB: Histone
deacetylase inhibitors: Current status and overview of recent
clinical trials. Drugs. 69:1911–1934. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tailor D, Hahm ER, Kale RK, Singh SV and
Singh RP: Sodium butyrate induces DRP1-mediated mitochondrial
fusion and apoptosis in human colorectal cancer cells.
Mitochondrion. 16:55–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Medina V, Edmonds B, Young GP, James R,
Appleton S and Zalewski PD: Induction of caspase-3 protease
activity and apoptosis by butyrate and trichostatin A (inhibitors
of histone deacetylase): Dependence on protein synthesis and
synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer
Res. 57:3697–3707. 1997.PubMed/NCBI
|
|
27
|
Tang Y, Chen Y, Jiang H and Nie D:
Short-chain fatty acids induced autophagy serves as an adaptive
strategy for retarding mitochondria-mediated apoptotic cell death.
Cell Death Differ. 18:602–618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiao D, Vogel V and Singh SV: Benzyl
isothiocyanate-induced apoptosis in human breast cancer cells is
initiated by reactive oxygen species and regulated by Bax and Bak.
Mol Cancer Ther. 5:2931–2945. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiao D, Lew KL, Kim YA, Zeng Y, Hahm ER,
Dhir R and Singh SV: Diallyl trisulfide suppresses growth of PC-3
human prostate cancer xenograft in vivo in association with Bax and
Bak induction. Clin Cancer Res. 12:6836–6843. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saunders LR and Verdin E: Sirtuins:
Critical regulators at the crossroads between cancer and aging.
Oncogene. 26:5489–5504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Portmann S, Fahrner R, Lechleiter A, Keogh
A, Overney S, Laemmle A, Mikami K, Montani M, Tschan MP, Candinas D
and Stroka D: Antitumor effect of SIRT1 inhibition in human HCC
tumor models in vitro and in vivo. Mol Cancer Ther. 12:499–508.
2013. View Article : Google Scholar : PubMed/NCBI
|














