Introduction
Advancements in clinical care and increased public
awareness have been made; however, heart failure (HF) remains a
leading cause of morbidity and mortality globally (1). HF is considered as the terminal
pathological manifestation of various cardiovascular diseases,
including coronary artery disease, hypertension and valvular
disease. HF frequently leads to cardiac overload or myocardial
injury, which ultimately results in an insufficient blood supply to
meet the metabolic demands of the body. HF is pathologically
characterized by cardiomyocyte loss, leading to cardiac apoptosis,
hypertrophy, fibrosis and dysfunctional ventricular remodeling,
which impairs the contraction and/or relaxation of the ventricles,
and reduces cardiac ejection fraction (2,3).
Numerous mechanisms have been hypothesized to contribute to the
development of HF, including impaired calcium homeostasis,
increased reactive oxygen species (ROS) concentration, accelerated
cardiomyocyte apoptosis and autophagy, and renin-angiotensin-system
and sympathetic nerve activation (4–8);
however, understanding of the precise mechanisms underlying the
pathogenesis of HF remains limited.
The transforming growth factor β (TGF-β)
superfamily, including TGF-β1-3, bone morphogenetic proteins
(BMPs), growth differentiation factors (GDFs), activins and
nodal-associated proteins (9), has
been proposed to serve important roles in the progression of HF
(10,11). Of these factors, TGF-β1 is the most
widely studied, and serves various roles in HF that may affect cell
growth, apoptosis and differentiation, including promotion of
collagen and matrix protein production, maintenance of fibroblast
viability and inhibition of collagen degradation (12,13).
GDF15, a member of the TGF-β superfamily, is a potential diagnostic
marker for HF; increased circulating levels of GDF15 are associated
with the severity and progression of HF (14). A recent study reported that the
expression levels of BMP9, another member of the TGF-β superfamily,
were increased in patients with HF; furthermore, increasing BMP9
activity by treatment with recombinant (r)BMP9 or inhibiting the
BMP9 receptor attenuated cardiac fibrosis, and improved cardiac
function in HF (15).
GDF11, also known as BMP11, is a member of the TGF-β
superfamily and contributes to the regulation of cell growth and
differentiation in embryonic and adult tissues (16). Furthermore, it serves important
roles in diverse biological processes and diseases (17–19).
The biological role of GDF11, particularly in the cardiovascular
system, is heavily debated. Loffredo et al (20) reported that circulating levels of
GDF11 reduced with aging, and restoration of GDF11 expression in
older mice using heterochronic parabiosis reversed age-associated
cardiac hypertrophy via cardiomyocyte regeneration, and
downregulation of brain natriuretic peptide (BNP) and atrial
natriuretic peptide. Data from large human cohorts demonstrated
that increased levels of GDF11 expression are associated with a
reduced risk of cardiovascular events and mortality in patients
with stable ischemic heart diseases (21). Conversely, Smith et al
(22) reported that restoring
levels of GDF11 in old mice did not improve aging-associated
pathological cardiac hypertrophy; however, high levels of GDF11
induced neonatal rat ventricular myocyte hypertrophy. Schafer et
al (23) used a
specifically-developed liquid chromatography-tandem mass
spectrometry assay to quantify GDF11 expression, and revealed that
GDF11 levels did not decrease with aging in healthy men, and that
individuals with increased GDF11 levels were more likely to be
frail with diabetes or prior cardiac conditions than those with
reduced GDF11 levels. Thus, the role of GDF11 in cardiovascular
diseases remains controversial. Further investigation is required
to validate GDF11 as a therapeutic target in the treatment of
cardiovascular diseases, particularly HF. The present study aimed
to determine the effects of GDF11 on isoproterenol (ISO)-induced HF
and investigate the underlying molecular mechanisms.
Materials and methods
Animals and treatment
Twelve male Sprague-Dawley rats, aged 8-weeks-old
and weighing 220–260 g, were supplied by Beijing Vital River
Laboratory Animal Technology Co., Ltd. (Beijing, China). The rats
were housed at constant temperature (22±2°C) and humidity (60%)
under a 12-h light/dark cycle, and provided with ad libitum
access to water and food for 1 week prior to initiation of the
experiment. All animal care and experimental procedures were
performed according to the Guide for the Care and Use of Laboratory
Animals of the US National Institutes of Health (National
Institutes of Health publication no. 85-23) (24). The present study was approved by
the Ethics Committee of Third Hospital of Shijiazhuang
[Shijiazhuang, China; approval no. 054(2018)].
Following an acclimatization period of one week, the
rats were randomly assigned to control and ISO groups (n=6). Rats
in the ISO and control groups were intraperitoneally administered
ISO (5 mg/kg) or equal volume of saline daily for one week to
induce HF as previously described (25).
Hemodynamic measurement
After 4 weeks following successful establishment of
the model of HF, the rats were intraperitoneally anesthetized using
urethane (1.2 g/kg). To evaluate left ventricular function, a
heparin-filled catheter connected to a pressure transducer (Chengdu
Taimeng Technology Co., Ltd., Chengdu, China) was inserted into the
left ventricle from the right carotid artery to measure hemodynamic
parameters, including left ventricular systolic pressure (LVSP),
left ventricular end-diastolic pressure (LVEDP), heart rate (HR),
maximum contraction velocity (+dp/dtmax) and maximum
relaxation velocity (-dp/dtmax). Following recording,
rats were sacrificed, and their hearts were immediately dissected
and frozen at −80°C for subsequent experimentation. Additionally,
blood was collected from the right common carotid artery and
centrifuged at 1,370 × g at 4°C for 10 min to obtain plasma, which
was collected and stored at −80°C for subsequent analysis.
Biochemical analysis
Plasma lactate dehydrogenase (LDH), creatine kinase
(CK) and CK-muscle/brain (CK-MB) levels were determined using the
corresponding assay kits (cat. nos. A020-2, A032, H197,
respectively; Nanjing Jiancheng Bioengineering Institute, Nanjing,
China) according to the manufacturer's protocols.
Measurement of plasma BNP and
GDF11
The plasma levels of BNP (cat. no. JYM00013; Wuhan
Colorful Gene Biological Technology, Wuhan, China) and GDF11 (cat.
no. E-EL-R0463c; Elabscience Biotechnology Co., Ltd, Wuhan, China)
were determined using the corresponding assay kits according to the
manufacturer's protocols.
Cell culture and treatment
H9C2 rat cardiac myoblasts, a cardiomyoblast cell
line derived from the heart tissue of embryonic rats (Chinese
Academy of Sciences Cell Bank, Shanghai, China), were cultured in
Dulbecco's Modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (HyClone; GE Healthcare, Logan, UT, USA) and 1%
penicillin-streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.) at 37°C in 5% CO2. H9C2 cells were categorized
into three groups based on treatment: i) Control group (cells were
cultured in DMEM for 24 h and then treated with saline at 37°C for
24 h); ii) ISO group (cells were cultured in DMEM for 24 h and then
treated with 10 µM ISO at 37°C for 24 h); and iii) ISO + rGDF11
group [cells were pre-incubated with different concentrations (0.5,
5 or 50 nM) of rGDF11 protein (cat. no. 1958-GD; R&D Systems,
Inc., Minneapolis, MN, USA) (22)
at 37°C for 24 h and then treated with 10 µM ISO at 37°C for 24
h].
GDF11 silencing using small
interfering RNA (siRNA)
H9C2 cells were inoculated in 6-well culture plates
and transfections were performed using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Specific siRNA against GDF11
(5′-GCCAGUGCGAGUACAUGUUTTdTdT-3′) and scrambled siRNA
(5′-UUCUCCGAACGUGUCACGUdTdT-3′; negative control) were obtained
from Ambion (cat. no. AM16708; Thermo Fisher Scientific, Inc.). For
each reaction, 5 µl of siRNA, 5 µl of Lipofectamine 2000 and 95 µl
of Opti-MEM™ reduced-serum medium (Invitrogen; Thermo Fisher
Scientific, Inc.) were mixed at room temperature followed by
incubation at 37°C for 20 min. Opti-MEM (800 µl) was subsequently
added drop-wise to each well. H9C2 cells were then added to the
resultant mixture. Following transfection for 6 h, the cell culture
medium was replaced and cells were further incubated at 37°C for 24
h prior to treatment with ISO.
CCK-8 assays
The proliferation of H9C2 cells cultured at a
density of 1×104 cells/well in 96-well plates was
measured using the Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) according to the
manufacturer's protocols. The absorbance of the cells at 450 nm was
determined using a microplate reader and normalized to that of the
control group.
LDH activity
The extent of cellular injury was monitored by LDH
release. H9C2 cells were cultured at a density of 1×106
cells/well in 6-well plates. Following treatment, the culture
medium was analyzed to determine LDH activity using a LDH assay kit
(Nanjing Jiancheng Bioengineering Institute) with a microplate
reader.
Caspase-3 activity
Caspase-3 activity was determined to investigate the
apoptosis of cells. Following treatment, H9C2 cells cultured at a
density of 1×106 cells/well in 6-well plates were
homogenized using 50 mM of potassium phosphate buffer. Following
centrifugation at 10,000 × g at 4°C for 10 min, the supernatant was
analyzed using the caspase-3 activity kit (Beyotime Institute of
Biotechnology, Shanghai, China) according to the manufacturer's
protocols. Briefly, 50 µl supernatant was mixed with 10 µl
caspase-3 substrate Ac-DEVD-pNA and 40 µl buffer solution, and then
incubated at 37°C for 2 h, and absorbance at 450 nm was measured,
reflecting cleavage of the colorimetric substrate. Finally, values
were normalized to those of the control group.
Hoechst 33258 staining
Apoptotic cell death was determined by Hoechst 33258
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) staining. In brief,
H9C2 cells were cultured at a density of 1×106
cells/well in 6-well plates. Following treatment, the cells were
fixed with 1 ml of 4% paraformaldehyde at 37°C for 30 min. Then,
the cells were incubated in 1 ml PBS containing 10 µM Hoechst 33258
at 37°C for 30 min and observed using fluorescence microscopy
(Olympus Corporation, Tokyo, Japan) from five random fields (×200).
Normal nuclei stained blue and apoptotic nuclei were identified as
condensed or fragmented nuclei stained bright blue. The rate of
apoptosis was calculated as follows: Apoptotic rate=No. of
apoptotic cells/Total no. of cells ×100.
Detection of intracellular ROS
Intracellular ROS generation was estimated using
dihydroethidium (DHE; Sigma-Aldrich; Merck KGaA) fluorescent
staining. Briefly, following treatment as aforementioned, H9C2
cells cultured at a density of 1×104 cells/well in
96-well plates were washed with PBS and incubated with DHE (10 µM)
at 37°C for 30 min in the dark. Then, DHE was removed by washing
with PBS, and the fluorescence intensity was measured using a
fluorescence plate reader (Tecan Infinite M200, Mannedorf,
Switzerland) at excitation/emission wavelength of 488/610 nm,
respectively. The values were normalized to those of the control
group.
Determination of intracellular
malondialdehyde (MDA)
Following treatment as aforementioned, H9C2 cells
cultured at a density of 1×106 cells/well in 6-well
plates were homogenized using 50 mM potassium phosphate buffer.
Following centrifugation at 10,000 × g at 4°C for 10 min, the MDA
concentration of the supernatant was analyzed using an MDA assay
kit (Nanjing Jiancheng Bioengineering Institute) according to the
manufacturer's protocols and the optical density (OD) was measured
at 532 nm using a microplate reader. The obtained values were
standardized to total protein content, which was determined via a
bicinchoninic acid (BCA) protein assay kit.
Western blot analysis
Protein extracted from heart tissues or H9C2 cells
was quantified using a BCA protein assay kit. Proteins (50 µg) were
separated via 10% SDS-PAGE, transferred to polyvinylidene
difluoride membranes, and blocked using 5% non-fat milk at room
temperature for 1 h. Subsequently, the membranes were incubated
overnight at 4°C with the following primary antibodies: Anti-B-cell
lymphoma 2 (Bcl-2; 1:1,000; cat. no. 3498; Cell Signaling
Technology, Inc., Danvers, MA, USA); anti-Bcl-2-associated X
protein (Bax; 1:1,000; cat. no. 5023; Cell Signaling Technology,
Inc.); anti-nicotinamide adenine dinucleotide phosphate oxidase 2
(Nox2; 1:1,000; cat. no. 19013-1; ProteinTech Group, Inc., Chicago,
IL, USA); anti-Nox4 (1:1,000; cat. no. 14347-1; ProteinTech Group,
Inc.); anti-GDF11 (1:1,000; cat. no. AF1958; R&D Systems, Inc.)
and anti-GAPDH as an internal control (1:2,000; cat. no. 10494-1;
ProteinTech Group, Inc.). Following three washes in TBS-Tween 20
(0.5 ml/l), the membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (1:1,000; cat. no.
A0208; Beyotime Institute of Biotechnology) at room temperature for
1 h. Target bands were detected using the SuperSignal West Pico
Chemiluminescent Substrate (Pierce; Thermo Fisher Scientific,
Inc.), and band intensity was quantified using ImageJ 1.48i
software (National Institutes of Health, Bethesda, MD, USA). The
experiment was repeated three times.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from heart tissues or H9C2
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and 1 µg RNA was subjected to reverse
transcription using first-strand cDNA synthesis kit (Promega
Corporation, Madison, WI, USA) according to the manufacturer's
protocols. The temperature protocol was as follows: 42°C for 30 min
and 85°C for 5 min. Real-time PCR analysis was performed with the
ABI 7500 FAST system, using an SYBR® Green RT-PCR kit
(Toyobo Life Science, Osaka, Japan) according to the manufacturer's
protocols. The thermocycling conditions consisted of an initial,
single cycle of 2 min at 94°C, followed by 40 cycles of 15 sec at
94°C, 20 sec at 60°C and 30 sec at 70°C. As an internal control,
β-actin primers were used for RNA template normalization. All PCRs
were performed in triplicate. The primers used to determine gene
expression were as follows: Rat GDF11, forward
5′-TGGGGAGCAGGCAAGGGGTAG-3′, reverse, 5′-TGCCCGTGGTAAGTGCTCAGAA-3′;
and rat β-actin, forward 5′-TACCACATCCAAGGAAGGCAGCA-3′ and reverse,
5′-TGGAATTACCGCGGCTGCTGGCA-3′. Fold changes in GDF11 expression
normalized to β-actin expression were calculated using the
2−ΔΔCq method (26).
Statistical analysis
Data were presented as the mean ± standard error of
the mean. Statistical analysis was performed using SPSS version
17.0 (SPSS, Inc., Chicago, IL, USA). Comparisons between two rat
groups were conducted using Student's t-tests. Comparisons between
>2 groups were performed using one-way analyses of variance
followed by a Tukey's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
ISO induces HF in rats
The results of hemodynamic analysis revealed that
ISO treatment induced a significant increase in LVEDP, and a
decrease in LVSP and ± dp/dtmax in the ISO group
compared with the control group (Fig.
1A-D). A significant difference in HR was not observed;
however, the mean value was markedly lower in the ISO group than in
the control group (Fig. 1E). The
results indicated that ISO treatment induced cardiac dysfunction in
rats.
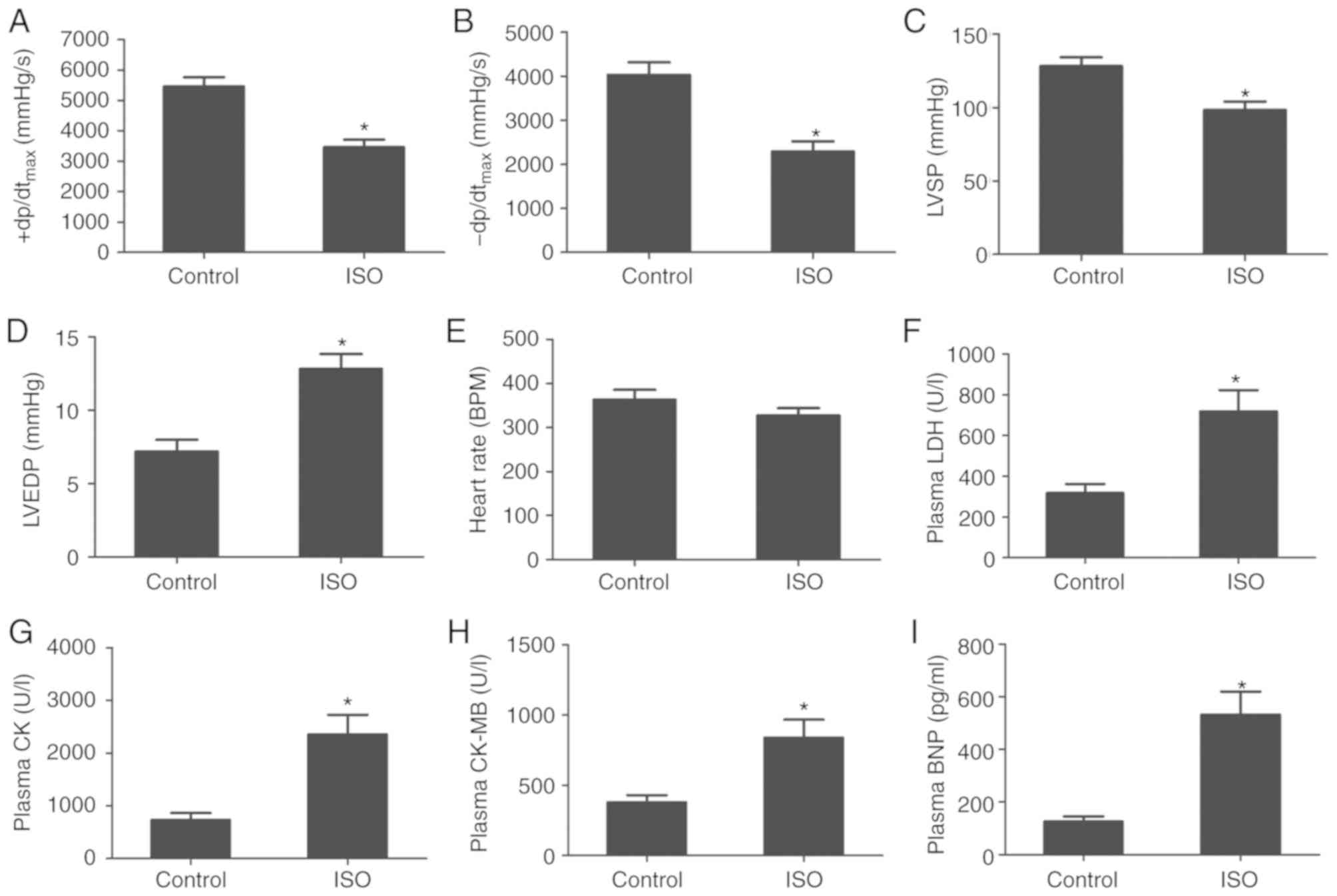 | Figure 1.ISO induces heart failure in rats.
(A) +dp/dtmax, (B) -dp/dtmax, (C) LSVP, (D)
LVEDP and (E) heart rate of rats following control or ISO
treatment. (F) LDH, (G) CK, (H) CK-MB and (I) BNP expression levels
in plasma following control or ISO treatment. Data are presented as
the mean ± standard error of the mean. *P<0.01 vs. control
group. BNP, brain natriuretic peptide; CK-MB, creatine kinase
muscle/brain; ISO, isoproterenol; LDH, lactate dehydrogenase; LSVP,
left ventricular systolic pressure; LVEDP, left ventricular
end-diastolic pressure; +dp/dtmax, maximum contraction
velocity; -dp/dtmax, maximum relaxation velocity. |
ISO-induced HF was associated with increased plasma
levels of myocardial injury markers, including LDH, CK and CK-MB.
Compared with the control group, the plasma levels of LDH, CK and
CK-MB increased significantly following ISO treatment (Fig. 1F-H). Additionally, plasma BNP
levels, an important biomarker of HF, were significantly elevated
following ISO treatment compared with the control group (Fig. 1I). These findings suggested that
ISO treatment induced HF in rats.
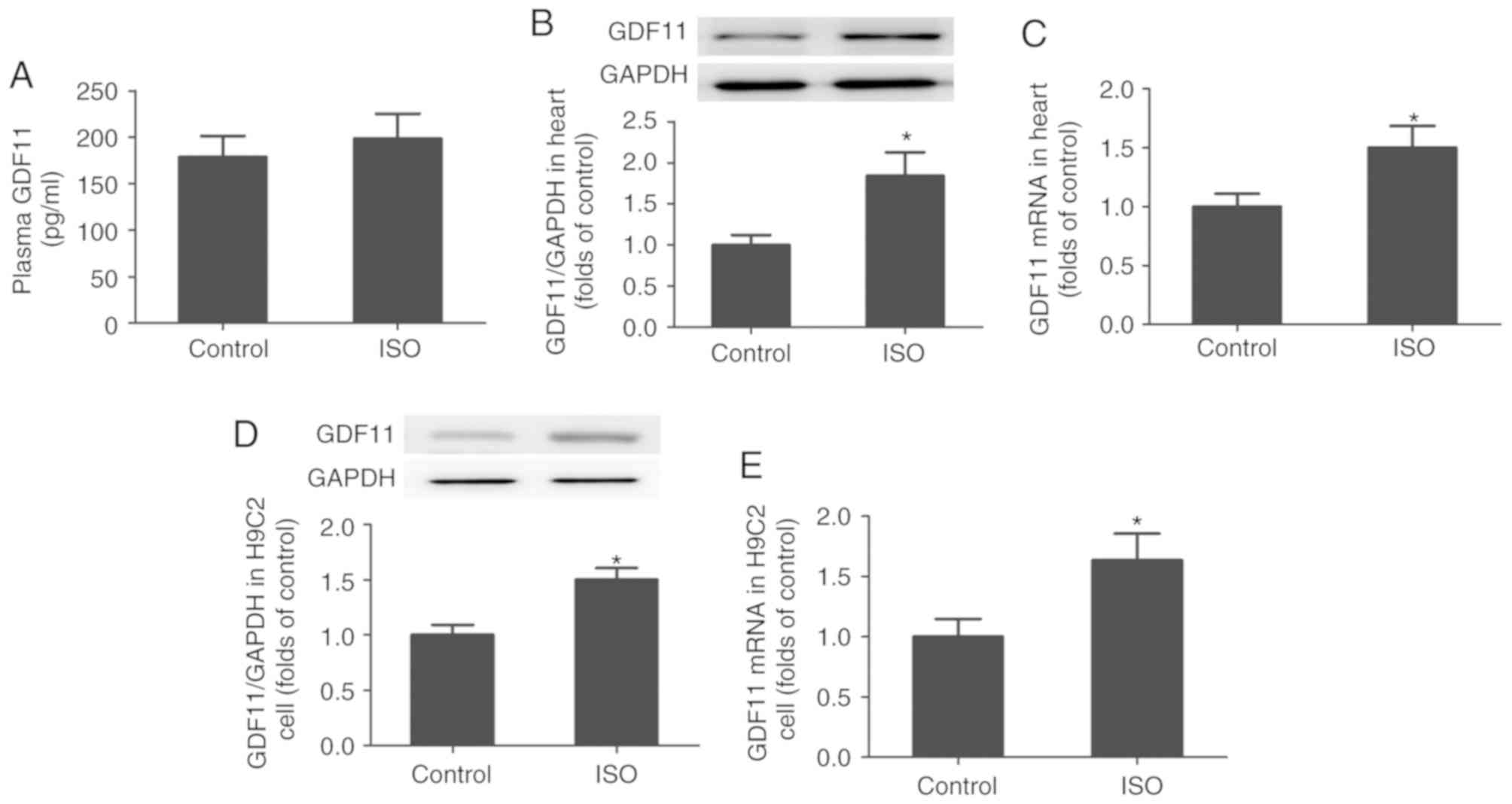
ISO increases the generation of GDF11
in vivo and in vitro
As presented in Fig.
2A-C, the levels of GDF11 protein and mRNA expression were
significantly increased in the heart tissues of ISO-treated rats
compared with in the control group; however, the mean plasma GDF11
levels were markedly unchanged. Additionally, to demonstrate the
effects of ISO on GDF11 production in cardiomyocytes, H9C2 cells
were treated with ISO for 24 h. As presented in Fig. 2D and E, the levels of GDF11 protein
and mRNA expression in H9C2 cells were significantly increased
following ISO treatment compared with the control.
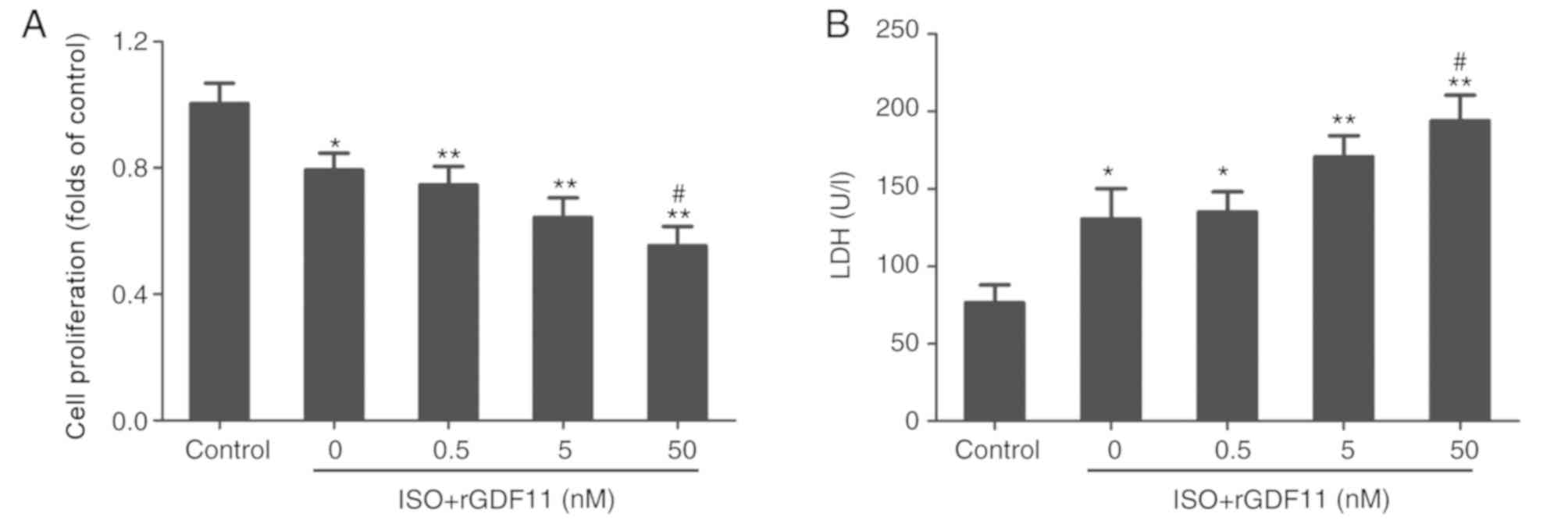
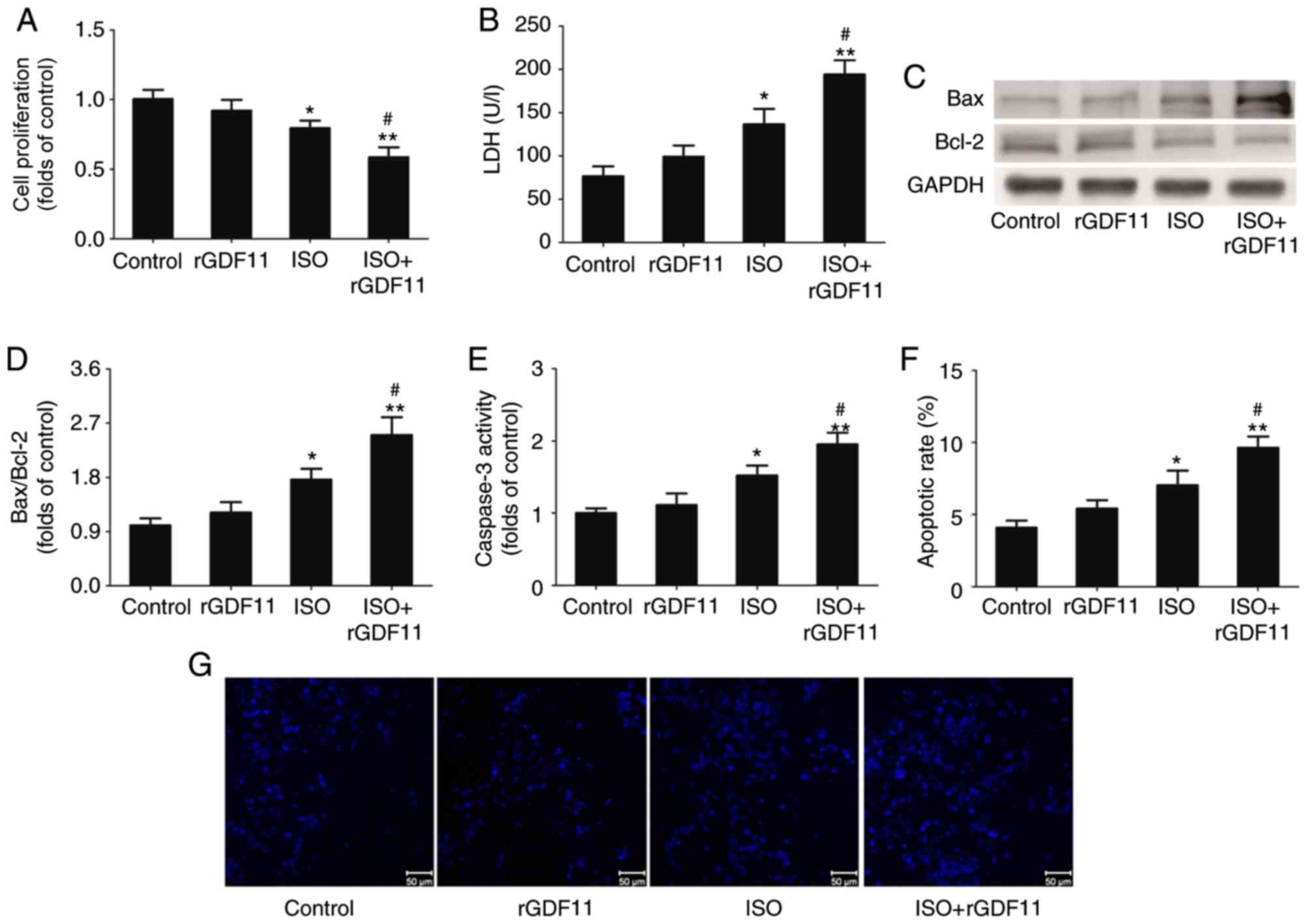
GDF11 aggravates ISO-induced damage in
H9C2 cells
To investigate the roles of GDF11 in ISO-treated
H9C2 cells, rGDF11 was used in the following experiments. rGDF11
significantly reduced the proliferation (Fig. 3A) and increased LDH levels
(Fig. 3B) in ISO-treated H9C2
cells in a dose-dependent manner compared with ISO treatment only;
50 nM rGDF11 was selected for use in subsequent experiments. It was
revealed that, compared with ISO treatment, 50 nM rGDF11
significantly reduced the proliferation (Fig. 4A), and increased the LDH release
(Fig. 4B) and apoptosis of H9C2
cells, represented by the increase in Bax/Bcl-2 protein expression,
caspase-3 activity and apoptotic rate in Hoechst 33258 staining
(Fig. 4C-G).
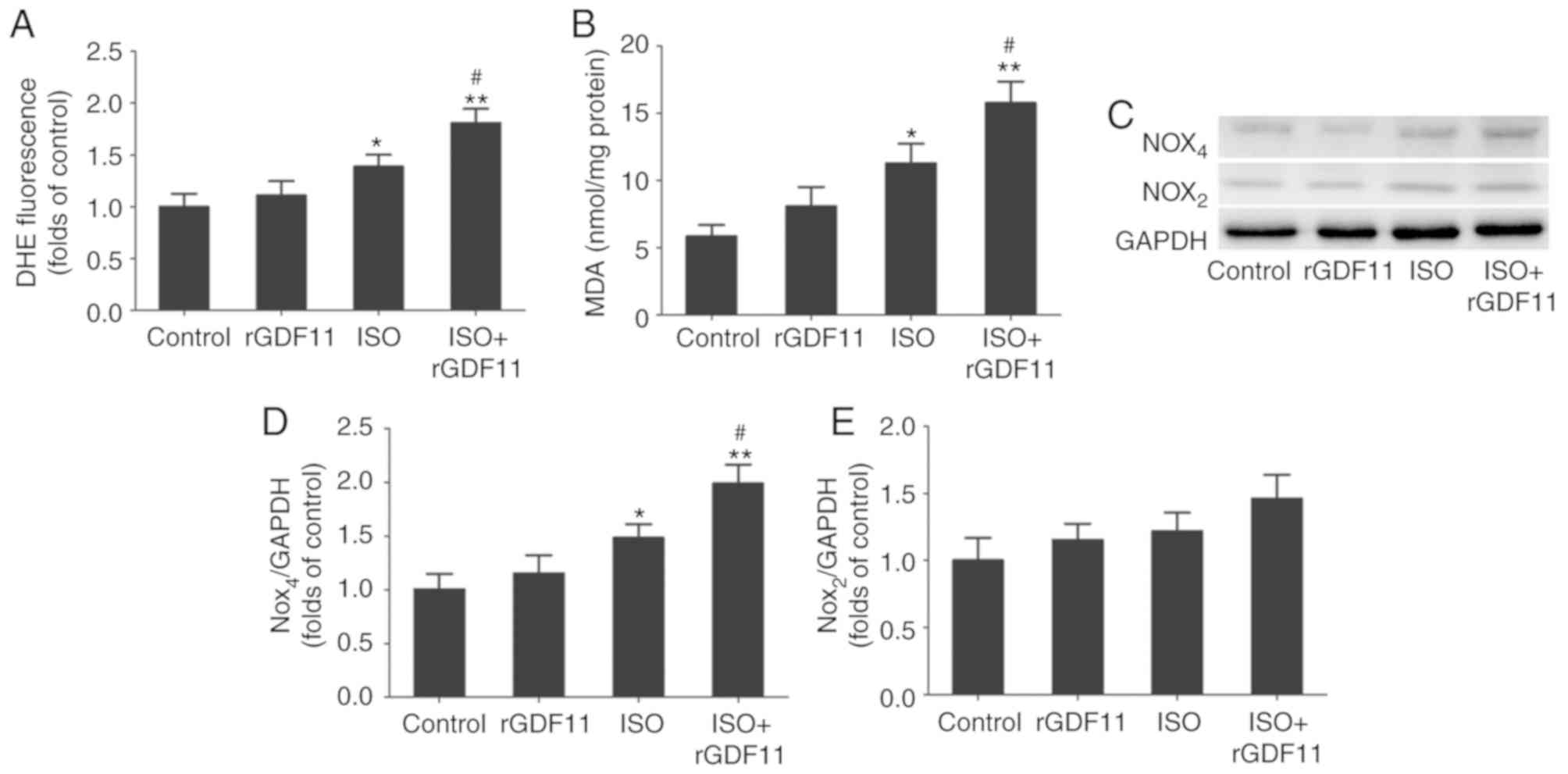
GDF11 increases ISO-induced oxidative
stress by upregulating Nox4 in H9C2 cells
ISO treatment often induces oxidative stress injury
that is reflected by increased ROS and MDA concentrations (27). As presented in Fig. 5A and B, the concentrations of ROS
and MDA were significantly increased in the ISO group compared with
in the control group. rGDF11 in addition to ISO treatment further
increased ROS and MDA concentrations compared with ISO treatment
only.
To further investigate the potential molecular
mechanisms underlying ISO-induced HF, the expression levels of the
Nox subunits, Nox2 and Nox4, in H9C2 cells were determined via
western blot analysis (Fig. 5C).
The results revealed that Nox4 protein levels were significantly
increased in the ISO group compared with in the control group and
were further upregulated following rGDF11 treatment; however, no
significant alterations in Nox2 expression were observed (Fig. 5D and E).
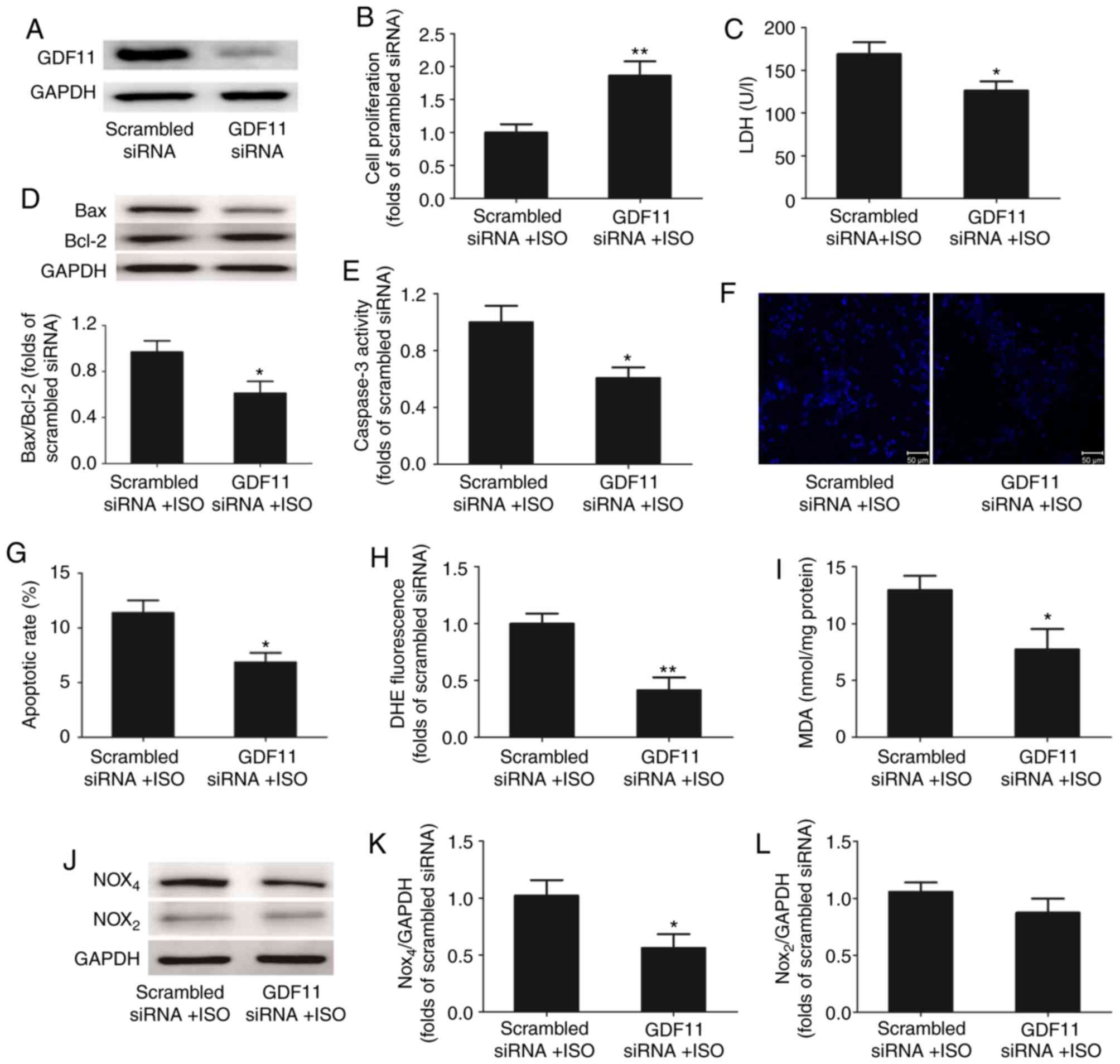
GDF11 knockdown alleviates ISO-induced
apoptosis by inhibiting oxidative stress injury
To further investigate the effects of GDF11 on
ISO-treated H9C2 cells, GDF11 was downregulated by siRNA-mediated
knockdown (Fig. 6A). Compared with
the scrambled siRNA group, silencing of GDF11 significantly
increased the proliferation (Fig.
6B), and significantly reduced the LDH release (Fig. 6C) and apoptosis of ISO-treated
cells, represented by the decrease in Bax/Bcl-2 protein expression,
caspase-3 activity and apoptotic rate in Hoechst 33258 staining
(Fig. 6D-G).
GDF11 knockdown also significantly decreased the
concentrations of ROS (Fig. 6H)
and MDA (Fig. 6I) in ISO-treated
H9C2 cells compared with the control. Additionally, western blot
analysis demonstrated that the levels of Nox4 protein expression
were downregulated in the GDF11 knockdown group compared with in
the scrambled siRNA group; however, the levels of Nox2 expression
were not significantly altered (Fig.
6J-L).
Discussion
The present study revealed that the production of
GDF11 was increased in ISO-induced rats with HF and ISO-treated
H9C2 cells. Furthermore, rGDF11 treatment of H9C2 cells promoted
ISO-induced oxidative stress injury by upregulating the levels of
Nox4 expression, whereas GDF11 knockdown downregulated the
expression of Nox4. These findings suggested that GDF11 may be a
potential target in the treatment of HF.
HF is considered to be a progressive and
irreversible disease characterized by failure of cardiac pumping,
which is induced by cardiac injury and pathological cardiac
remodeling, ultimately leading to ischemia, apoptosis and cell
necrosis; thus cardiac pump failure is aggravated. HF is known as
the terminal pathological manifestation of a variety of organic
heart diseases, such as myocardial infarction (MI). MI involves a
restriction in the flow of blood and oxygen to the heart, which
induces sudden cardiomyocyte loss. This leads to cardiac
fibroblasts and activated myofibroblasts promoting post-MI cardiac
remodeling, which contributes to the development of ventricular
dysfunction and HF. ISO-induced MI in rats is a widely accepted
non-invasive and reliable model for investigating the molecular
mechanisms underlying HF (2,3,28).
ISO results in non-uniform, predominantly subendocardial myocardial
necrosis with subsequent hypertrophy and remodeling, leading to the
presentation of HF similar to that observed in patients with MI
following the infarction episode (29). In the present study, rats were
intraperitoneally injected with 5 mg/kg ISO once daily for 7 days
to establish the model of HF. Hemodynamic parameters, including
LVSP, ± dp/dtmax, LVEDP and HR were measured 4 weeks
later to evaluate left ventricular function. The results revealed
that ISO treatment induced a significant increase in LVEDP, and a
decrease in LVSP and ± dp/dtmax, with no significant alterations in
HR. The plasma levels of myocardial injury markers, including LDH,
CK and CK-MB, were increased following ISO treatment. Furthermore,
the plasma levels of BNP, an important biomarker of HF, were
significantly increased following ISO treatment. These results
indicated that ISO treatment successfully induced HF, consistent
with previous studies (30,31).
Members of the TGF-β superfamily, which are produced
and secreted from cardiac myocytes, serve numerous roles in the
development and progression of HF by modulating various phenomena,
including the differentiation, proliferation and apoptosis of cells
(32). For example, it was
reported that GDF15, a member of the TGF-β superfamily, exhibited a
protective role in an animal model of HF due to its antiapoptotic
and antihypertrophic properties (33). Conversely, numerous human studies
have reported that the levels of GDF15 expression are positively
associated with the severity of HF; thus, GDF15 expression may be
regarded as a potential biomarker for the diagnosis, prognosis
and/or risk stratification of patients with HF (34,35).
In the present study, an ISO-induced rat model of HF was used to
determine alterations in the expression of GDF11, a member of the
TGF-β superfamily. GDF11 is known as a rejuvenation factor that
reverses aging and aging-associated dysfunction in muscles, nerves
and the cardiovascular system (20,36,37).
Conversely, certain studies determining the age-associated decline
of levels of the circulating GDF11 reported that the expression of
GDF11 increased with age, inhibiting muscle regeneration rather
than promoting rejuvenation (38,39);
however, the effects of GDF11 on ISO-induced HF require further
investigation. Thus, it was revealed that levels of GDF11 protein
and mRNA expression were significantly increased in rat heart
tissues following ISO treatment. Of note, the plasma expression
levels of GDF11 were markedly increased in the ISO group compared
with in the control group; however, this difference was not
significant. This result may be due to the high expression GDF11 in
a number of organs, including the pancreas, intestine, kidney,
skeletal muscle and heart (40,41);
therefore, increased myocardial expression of GDF11 induced by ISO
may be insufficient to affect plasma GDF11 levels.
To determine the effects of ISO on GDF11 production
in cardiomyocytes, H9C2 cells were treated with ISO for 24 h.
Compared with control treatment, the ISO group demonstrated
significantly increased levels of GDF11 protein and mRNA
expression. To investigate the effects of GDF11 on ISO-treated H9C2
cells, rGDF11 was administered to ISO-treated H9C2 cells, inducing
a further decrease in proliferation, and increase in the LDH
release and the apoptosis of cells. These findings are consistent
with those of previous studies reporting that GDF11 did not reduce
neonatal rat ventricular myocyte hypertrophy, but instead induced
hypertrophy (22), and cardiac and
skeletal muscle dysfunction in vivo (42). Conversely, the findings from the
present study opposed those of a previous report, which revealed
that GDF11 modulated Ca2+ signaling and the mothers
against decapentaplegic homolog family member 2/3 (Smad2/3) pathway
to prevent cardiomyocyte hypertrophy (43). The results of the present study
indicated that GDF11 aggravated ISO-induced cell damage in HF;
however, the molecular mechanisms underlying the role of GDF11 in
HF, such as the signaling pathway involved in the pathogenesis of
HF, remain unclear.
ISO treatment generates highly cytotoxic ROS,
resulting in the peroxidation of membrane phospholipids, inducing
marked impairment of the myocardial membrane associated with
infarct-like necrosis of the heart muscle (44). Excessive ROS generation also
activates various intracellular signaling pathways regulating
myocyte survival, apoptosis and cardiac remodeling (45). Additionally, accumulating evidence
suggests that Nox, the major source of ROS in the cardiovascular
system, is an important downstream effector of the TGF-β signaling
pathway, whereas Nox-dependent redox signaling regulates TGF-β/Smad
signaling in a feedforward manner (46). Qin et al (47) reported that GDF11 overexpression
and ROS overproduction was observed in patients with metastatic
oral cancer; however, treatment with the antioxidant,
N-acetylcysteine, suppressed the GDF11-induced
epithelial-mesenchymal transition and migration of tumor cells. Of
note, the present study demonstrated that ISO treatment induced
oxidative stress injury, reflected by increased ROS and MDA
concentrations, which was exacerbated by rGDF11 treatment in
ISO-treated H9C2 cells.
The levels of Nox2 and Nox4 expression in H9C2
cells, the major isoforms in cardiomyocytes, were determined via
western blot analysis. The results revealed that Nox4 protein
levels following ISO treatment were increased compared with in the
control group; rGDF11 treatment further upregulated Nox4 expression
in the ISO group. No significant alterations in the levels of Nox2
expression were observed. Conversely, siRNA-mediated knockdown of
GDF11 downregulated Nox4 expression, suppressing oxidative stress
injury and alleviating the ISO-induced apoptosis of H9C2 cells.
These findings suggested that GDF11 increased ISO-induced oxidative
stress injury by upregulating Nox4 in H9C2 cells, in agreement with
the findings of Zhang et al (48), in which GDF11 treatment increased
the levels of Nox4 protein expression and ROS production in human
umbilical vein endothelial cells. Additionally, TGF-β increased the
levels of Nox4 expression without affecting Nox1, Nox2 or Nox5
expression in cardiac fibroblasts (49,50).
In conclusion, the present study demonstrated that
GDF11 production is increased in ISO-induced rats with HF and
ISO-treated H9C2 cells. Furthermore, rGDF11 treatment increased
ISO-induced oxidative stress injury by upregulating Nox4 in H9C2
cells, whereas GDF11 knockdown exhibited opposing effects. These
findings suggested that GDF11 may be a potential target in the
treatment of HF.
Acknowledgements
Not applicable.
Funding
The present study was supported by the project of
Science and Technology Research and Development Guidance Plan of
Shijiazhuang City (grant no. 171461913).
Availability of data and materials
The datasets used or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
X-J Z conceived the study, performed research and
wrote the manuscript; HT and Z-F S performed research and analyzed
data; NL, YJ and ZH conducted the statistical analysis and
participated in the critical discussion. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal care and experimental procedures were
performed according to the Guide for the Care and Use of Laboratory
Animals of the US National Institutes of Health (24). The study was approved by the Ethics
Committee of Third Hospital of Shijiazhuang [Shijiazhuang, China;
no. 054(2018)].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ambrosy AP, Fonarow GC, Butler J, Chioncel
O, Greene SJ, Vaduganathan M, Nodari S, Lam CSP, Sato N, Shah AN
and Gheorghiade M: The global health and economic burden of
hospitalizations for heart failure: Lessons learned from
hospitalized heart failure registries. J Am Coll Cardiol.
63:1123–1133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tanai E and Frantz S: Pathophysiology of
heart failure. Compr Physiol. 6:187–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Luedde M, Spehlmann MW and Frey N:
Progress in heart failure treatment in Germany. Clin Res Cardiol.
107:105–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Peana D and Domeier TL: Cardiomyocyte
Ca2+ homeostasis as a therapeutic target in heart
failure with reduced and preserved ejection fraction. Curr Opin
Pharmacol. 33:17–26. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uchihashi M, Hoshino A, Okawa Y, Ariyoshi
M, Kaimoto S, Tateishi S, Ono K, Yamanaka R, Hato D, Fushimura Y,
et al: Cardiac-specific bdh1 overexpression ameliorates oxidative
stress and cardiac remodeling in pressure overload-induced heart
failure. Circ Heart Fail. 10:e0044172017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang S, Lin X, Li G, Shen X, Niu D, Lu G,
Fu X, Chen Y, Cui M and Bai Y: Knockout of Eva1a leads to rapid
development of heart failure by impairing autophagy. Cell Death
Dis. 8:e25862017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma NM, Nandi SS, Zheng H, Mishra PK
and Patel KP: A novel role for miR-133a in centrally mediated
activation of the renin-angiotensin system in congestive heart
failure. Am J Physiol Heart Circ Physiol. 312:H968–H979. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Patel KP, Xu B, Liu X, Sharma NM and Zheng
H: Renal denervation improves exaggerated sympathoexcitation in
rats with heart failure: A role for neuronal nitric oxide synthase
in the paraventricular nucleus. Hypertension. 68:175–184. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Budi EH, Duan D and Derynck R:
Transforming growth factor-β receptors and smads: Regulatory
complexity and functional versatility. Trends Cell Biol.
27:658–672. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Heger J, Schulz R and Euler G: Molecular
switches under TGFβ signalling during progression from cardiac
hypertrophy to heart failure. Br J Pharmacol. 173:3–14. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goletti S and Gruson D: Personalized risk
assessment of heart failure patients: More perspectives from
transforming growth factor super-family members. Clin Chim Acta.
443:94–99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo Y, Gupte M, Umbarkar P, Singh AP, Sui
JY, Force T and Lal H: Entanglement of GSK-3β, β-catenin and TGF-β1
signaling network to regulate myocardial fibrosis. J Mol Cell
Cardiol. 110:109–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan Z, Shen D, Liao J, Zhang Y, Chen Y,
Shi G and Gao F: Hypoxia suppresses TGF-β1-induced cardiac myocyte
myofibroblast transformation by inhibiting Smad2/3 and rhoa
signaling pathways. Cell Physiol Biochem. 45:250–257. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anand IS, Kempf T, Rector TS, Tapken H,
Allhoff T, Jantzen F, Kuskowski M, Cohn JN, Drexler H and Wollert
KC: Serial measurement of growth-differentiation factor-15 in heart
failure: Relation to disease severity and prognosis in the
valsartan heart failure trial. Circulation. 122:1387–1395. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morine KJ, Qiao X, York S, Natov PS,
Paruchuri V, Zhang Y, Aronovitz MJ, Karas RH and Kapur NK: Bone
morphogenetic protein 9 reduces cardiac fibrosis and improves
cardiac function in heart failure. Circulation. 138:513–526. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McPherron AC, Lawler AM and Lee SJ:
Regulation of anterior/posterior patterning of the axial skeleton
by growth/differentiation factor 11. Nat Genet. 22:260–264. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H, Li Y, Xiang L, Zhang J, Zhu B, Xiang
L, Dong J, Liu M and Xiang G: GDF11 attenuates development of type
2 diabetes via improvement of islet β-cell function and survival.
Diabetes. 66:1914–1927. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu X, Chen X, Zheng XD, Zhang J, Zhao X,
Liu Y, Zhang H, Zhang L, Yu H, Zhang M, et al: Growth
differentiation factor 11 promotes abnormal proliferation and
angiogenesis of pulmonary artery endothelial cells. Hypertension.
71:729–741. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rochette L, Zeller M, Cottin Y and Vergely
C: Growth and differentiation factor 11 (GDF11): Functions in the
regulation of erythropoiesis and cardiac regeneration. Pharmacol
Ther. 156:26–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Loffredo FS, Steinhauser ML, Jay SM,
Gannon J, Pancoast JR, Yalamanchi P, Sinha M, Dall'Osso C, Khong D,
Shadrach JL, et al: Growth differentiation factor 11 is a
circulating factor that reverses age-related cardiac hypertrophy.
Cell. 153:828–839. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Olson KA, Beatty AL, Heidecker B, Regan
MC, Brody EN, Foreman T, Kato S, Mehler RE, Singer BS, Hveem K, et
al: Association of growth differentiation factor 11/8, putative
anti-ageing factor, with cardiovascular outcomes and overall
mortality in humans: Analysis of the heart and soul and HUNT3
cohorts. Eur Heart J. 36:3426–3434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smith SC, Zhang X, Zhang X, Gross P,
Starosta T, Mohsin S, Franti M, Gupta P, Hayes D, Myzithras M, et
al: GDF11 does not rescue aging-related pathological hypertrophy.
Circ Res. 117:926–932. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schafer MJ, Atkinson E, Vanderboom PM,
Kotajarvi B, White TA, Moore MM, Bruce CJ, Greason KL, Suri RM,
Khosla S, et al: Quantification of GDF11 and myostatin in human
aging and cardiovascular disease. Cell Metab. 23:1207–1215. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bayne K: Revised Guide for the Care and
Use of Laboratory Animals available. American Physiological
Society. Physiologist. 39:199–208, 11. 1996.PubMed/NCBI
|
|
25
|
Simko F, Bednarova KR, Krajcirovicova K,
Hrenak J, Celec P, Kamodyova N, Gajdosechova L, Zorad S and
Adamcova M: Melatonin reduces cardiac remodeling and improves
survival in rats with isoproterenol-induced heart failure. J Pineal
Res. 57:177–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Patil AS, Singh AD, Mahajan UB, Patil CR,
Ojha S and Goyal SN: Protective effect of omeprazole and
lansoprazole on β-receptor stimulated myocardial infarction in
Wistar rats. Mol Cell Biochem. 2019. View Article : Google Scholar
|
|
28
|
Wong ZW, Thanikachalam PV and Ramamurthy
S: Molecular understanding of the protective role of natural
products on isoproterenol-induced myocardial infarction: A review.
Biomed Pharmacother. 94:1145–1166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Teerlink JR, Pfeffer JM and Pfeffer MA:
Progressive ventricular remodeling in response to diffuse
isoproterenol-induced myocardial necrosis in rats. Circ Res.
75:105–113. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang JJ, Rau C, Avetisyan R, Ren S, Romay
MC, Stolin G, Gong KW, Wang Y and Lusis AJ: Genetic dissection of
cardiac remodeling in an isoproterenol-induced heart failure mouse
model. PLoS Genet. 12:e10060382016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mohamed SS, Ahmed LA, Attia WA and Khattab
MM: Nicorandil enhances the efficacy of mesenchymal stem cell
therapy in isoproterenol-induced heart failure in rats. Biochem
Pharmacol. 98:403–411. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Morikawa M, Derynck R and Miyazono K:
TGF-β and the TGF-β Family: Context-dependent roles in cell and
tissue physiology. Cold Spring Harb Perspect Biol. 8:a0218732016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu J, Kimball TR, Lorenz JN, Brown DA,
Bauskin AR, Klevitsky R, Hewett TE, Breit SN and Molkentin JD:
GDF15/MIC-1 functions as a protective and antihypertrophic factor
released from the myocardium in association with SMAD protein
activation. Circ Res. 98:342–350. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meijers WC, van der Velde AR, Muller
Kobold AC, Dijck-Brouwer J, Wu AH, Jaffe A and de Boer RA:
Variability of biomarkers in patients with chronic heart failure
and healthy controls. Eur J Heart Fail. 19:357–365. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wollert KC, Kempf T and Wallentin L:
Growth differentiation factor 15 as a biomarker in cardiovascular
disease. Clin Chem. 63:140–151. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Katsimpardi L, Litterman NK, Schein PA,
Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ
and Rubin LL: Vascular and neurogenic rejuvenation of the aging
mouse brain by young systemic factors. Science. 344:630–634. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sinha M, Jang YC, Oh J, Khong D, Wu EY,
Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, et al:
Restoring systemic GDF11 levels reverses age-related dysfunction in
mouse skeletal muscle. Science. 344:649–652. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Egerman MA, Cadena SM, Gilbert JA, Meyer
A, Nelson HN, Swalley SE, Mallozzi C, Jacobi C, Jennings LL, Clay
I, et al: GDF11 increases with age and inhibits skeletal muscle
regeneration. Cell Metab. 22:164–174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rodgers BD and Eldridge JA: Reduced
circulating GDF11 is unlikely responsible for age-dependent changes
in mouse heart, muscle, and brain. Endocrinology. 156:3885–3888.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Walker RG, Poggioli T, Katsimpardi L,
Buchanan SM, Oh J, Wattrus S, Heidecker B, Fong YW, Rubin LL, Ganz
P, et al: Biochemistry and biology of GDF11 and myostatin:
Similarities, differences, and questions for future investigation.
Circ Res. 118:1125–1142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jamaiyar A, Wan W, Janota DM, Enrick MK,
Chilian WM and Yin L: The versatility and paradox of GDF 11.
Pharmacol Ther. 175:28–34. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zimmers TA, Jiang Y, Wang M, Liang TW,
Rupert JE, Au ED, Marino FE, Couch ME and Koniaris LG: Exogenous
GDF11 induces cardiac and skeletal muscle dysfunction and wasting.
Basic Res Cardiol. 112:482017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Duran J, Troncoso MF, Lagos D, Ramos S,
Marin G and Estrada M: GDF11 modulates Ca2+-dependent
smad2/3 signaling to prevent cardiomyocyte hypertrophy. Int J Mol
Sci. 19:E15082018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mukherjee D, Roy SG, Bandyopadhyay A,
Chattopadhyay A, Basu A, Mitra E, Ghosh AK, Reiter RJ and
Bandyopadhyay D: Melatonin protects against isoproterenol-induced
myocardial injury in the rat: Antioxidative mechanisms. J Pineal
Res. 48:251–262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fan D, Yang Z, Liu FY, Jin YG, Zhang N, Ni
J, Yuan Y, Liao HH, Wu QQ, Xu M, et al: Sesamin protects against
cardiac remodeling via Sirt3/ROS pathway. Cell Physiol Biochem.
44:2212–2227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Samarakoon R, Overstreet JM and Higgins
PJ: TGF-β signaling in tissue fibrosis: Redox controls, target
genes and therapeutic opportunities. Cell Signal. 25:264–268. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qin X, Kuang H, Chen L, Wei S, Yu D and
Liang F: Coexpression of growth differentiation factor 11 and
reactive oxygen species in metastatic oral cancer and its role in
inducing the epithelial to mesenchymal transition. Oral Surg Oral
Med Oral Pathol Oral Radiol. 123:697–706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang YH, Cheng F, Du XT, Gao JL, Xiao XL,
Li N, Li SL and Dong DL: GDF11/BMP11 activates both smad1/5/8 and
smad2/3 signals but shows no significant effect on proliferation
and migration of human umbilical vein endothelial cells.
Oncotarget. 7:468322016.PubMed/NCBI
|
|
49
|
Cucoranu I, Clempus R, Dikalova A, Phelan
PJ, Ariyan S, Dikalov S and Sorescu D: NAD(P)H oxidase 4 mediates
transforming growth factor-beta1-induced differentiation of cardiac
fibroblasts into myofibroblasts. Circ Res. 97:900–907. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chan EC, Peshavariya HM, Liu GS, Jiang F,
Lim SY and Dusting GJ: Nox4 modulates collagen production
stimulated by transforming growth factor β1 in vivo and in vitro.
Biochem Biophys Res Commun. 430:918–925. 2013. View Article : Google Scholar : PubMed/NCBI
|