Introduction
Anti-phospholipid syndrome (APS), also known as
anti-phospholipid antibody syndrome, is a non-organ-specific
autoimmune and hypercoagulable disease. APS is characterized by the
presence of anti-phospholipid antibodies (aPL), venous and/or
arterial thrombosis, thrombocytopenia and recurrent fetal loss
(1,2). The aPL have been reported to have an
important role in the etiology of thrombosis (3,4), the
formation of a blood clot inside a blood vessel, leading to the
obstruction of the blood flow through the circulatory system,
which, in turn, can lead to a number of complications, including
heart attack, stroke and miscarriage. In addition to the anionic
phospholipids, phospholipid-binding proteins may be target
antigens. β2-glycoprotein I (β2GPI), also known as apolipoprotein
H, is a 38–50 kDa multifunctional plasma apolipoprotein that clears
lipopolysaccharide (LPS) and dead cell remnants via interactions
with phospholipids. In fact, the aPLs including anti-β2GPI
antibodies are known to be involved in the pathogenesis of
thrombosis (5,6). Anti-β2GPI antibodies are a
heterogeneous group of antibodies and their common recognition of a
single β2GPI domain I epitope around amino acids G40-R43 is
associated with the observed clinical manifestation of APS
(7).
Mononuclear cells (or simply monocytes) and vascular
endothelial cells are the primary effector cells involved in
thrombus formation. In these cells, the coagulation cascade is
activated by high expression of tissue factor (8). Mononuclear cells can be activated to
have a pro-inflammatory role via the secretion of various
inflammatory cytokines [including tumor necrosis factor-α (TNF-α),
interleukin (IL)-1β and IL-6 (9)],
inhibition of physiological anti-coagulant system, activation of
platelets and endothelial cells, and further promotion of
thrombosis (10,11). Endothelial cells can be activated
in vivo by enhancing the expression of adhesion molecules,
and enhancing the adhesion of leukocytes and/or platelets (9,12,13).
While the cellular and molecular mechanisms by which
anti-phospholipid antibodies lead to thromboembolic events are
still not entirely clear, Toll-like receptor-4 (TLR-4) has been
suggested to have an important role in thrombosis. For instance,
previous studies have reported that a β2GPI/anti-β2GPI complex is
able of inducing the expression of inflammatory cytokines (TNF-α,
IL-1β and IL-6) in monocytes via the activation of TLR-4 (14). In vitro studies have also
demonstrated that anti-β2GPI can activate endothelial cells via
TLR-4, as determined by measuring the increased expression levels
of adhesion molecules, including intercellular cell adhesion
molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and
E-selectin (15). These previous
studies suggest the important role of TLR-4 in
aPL/anti-β2GPI-mediated pathogenesis of thrombosis.
TLR-4 belongs to the TLR family type I transmembrane
receptors. TLR-4 is widely expressed in various cell types,
including macrophages, endothelial cells, lymphocytes and dendritic
cells, and it actively participates in immune defense responses
(16–18). Following activation by
pathogen-associated molecular pattern (PAMPs), a signal
transduction cascade is initiated. TLR-4 is an important receptor
involved in mediating the responses to lipopolysaccharides (LPS), a
polysaccharide composed of O-antigen that is present in the outer
membrane of Gram-negative bacteria and elicits strong immune
responses in animals. Upon binding to TLR-4, the
β2GPI/anti-β2GPI-immunoglobulin G (IgG) complex activates a
signaling cascade, which is characterized by phosphorylation of p38
mitogen-activated protein kinase (MAPK) and nuclear factor-κB
(NF-κB), the key factors involved in inflammatory responses.
Activation of TLR-4 mainly induces the expression of inflammatory
factors and chemokines (14,19,20).
Myeloid differentiation primary response gene 88 (Myd88) and
TIR-domain-containing adapter-inducing interferon-β (TRIF) are the
adapters that respond to the activation of TLRs. Myd88 and TRIF
induce intracellular signal transduction following TLR-4 activation
(21). Thus, the role of TLR-4 in
the aPL/anti-β2GPI-mediated pathogenesis of thrombosis deserves to
be further investigated.
Currently, there is limited understanding of whether
TLR-4 has the same roles in vivo. In the current study, it
was aimed to further elucidate the roles of TLR-4 in
aPL/anti-β2GPI-mediated pathogenesis of thrombosis by determining
whether TLR4 is involved in anti-β2GPI–IgG-mediated activation of
endothelial cells and macrophages in vivo. The effects of
anti-β2GPI on the expression levels of inflammatory factors (TNF-α,
IL-1β and IL-6) and adhesion molecules (ICAM-1, VCAM-1 and
E-selectin) were examined and compared in TLR-4 intact C3H/HeN mice
and TLR-4 defective C3H/HeJ mice.
Materials and methods
Animals
96 male C3H/HeN mice (TLR4-intact) were purchased
from Vital River Laboratory Animal Technology (Beijing, China) and
24 male C3H/HeJ mice (TLR-4-defective) were obtained from the Model
Animal Research Center of Nanjing University (Nanjing, China).
C3H/HeJ mice carry a mutant, nonfunctional TLR-4 and thus, are
hyporesponsive to the lethal effects of LPS. The body weights of
mice were 20–25 g and the mice were used at 8–12 weeks of age. The
animals were bred in the Laboratory Animal Research Center of
Jiangsu University (Zhenjiang, China) at standardized specific
pathogen free conditions (12-h light/dark cycle, with 22±2°C
temperature and 50±10% humidity). All the experiments involving
animals were approved by the Laboratory Animal Administration
Committee of Jiangsu University and conducted in accordance with
the Guide for the Care and Use of Laboratory Animals (2011)
published by the US National Institutes of Health (Bethesda, MD,
USA) (22).
Treatment of animals with IgGs
The polyclonal anti-β2GPI antibodies were purified
from sera of New Zealand rabbits immunized with human β2GPI peptide
sequence (35GYVSRGGMRKFICPLTG51) according to our previous methods
(23). The purified anti-β2GPI IgG
could recognize human and mouse β2GPI as demonstrated by western
blotting and ELISA (23). Our
previous study demonstrated that this anti-β2GPI IgG could induce
an APS mouse model. The isotype control antibodies (NR-IgG) from
sera of normal rabbits were purified by Protein G Sepharose columns
(GE Healthcare, Chicago, IL, USA). All the IgG samples and reagents
were subjected to Detoxi-Gel™ (Pierce; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) to remove endotoxin contamination (<0.03
EU/ml) using the Limulus amebocyte lysate assay (Associates of Cape
Cod, Inc., Falmouth, MA, USA).
C3H/HeN and C3H/HeJ mice (n=8 per treatment group)
were twice injected intraperitoneally with anti-β2GPI (100 µg) or
NR-IgG (100 µg) at 0 and 48 h. Surgical procedures to obtain
peritoneal macrophages or aortas were performed at 72 h after the
first injection. In addition, other groups of C3H/HeN and C3H/HeJ
mice (n=8 in each group) were challenged with LPS (1 µg/g body
weight; E. coli serotype O111:B4; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) 2 h before surgical procedures as the positive
control.
Preparation of mouse peritoneal
macrophages
Both C3H/HeN mice and C3H/HeJ mice were sacrificed
via cervical dislocation at 72 h after the first injection.
Peritoneal macrophages of the mice were obtained by flushing the
peritoneal cavity of the mice with 10 ml PBS solution for 5 min.
The peritoneal cells were centrifuged at 400 × g, 25°C, for 5 min.
The cells were collected and washed twice with PBS, and suspended
in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 1% penicillin/streptomycin and 10% (v/v) fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.). The cells
(2×106 cells/well) were seeded into 6-well culture
plates and incubated at 37°C in a humid atmosphere of 5%
CO2. Following incubation for 4 h, the non-adherent
cells were removed and fresh RPMI-1640 was added, then the
remaining cells were used as mouse peritoneal macrophages for the
subsequent experiments.
Immunofluorescence staining for
inflammatory cytokines in macrophages
The cultured peritoneal macrophages were fixed in 4%
paraformaldehyde for 20 min and then washed with PBS three times.
Following permeabilization with ice-cold 0.3% Triton X-100 for 10
min, the cells were blocked in 5% (m/v) bovine serum albumin (BSA,
cat. no. A1993; Sigma-Aldrich; Merck KGaA) for 1 h at room
temperature and then incubated with primary rabbit anti-mouse
antibodies as follows: TNF-α (diluted 1:200 with PBS/5% BSA; cat.
no. BS6000; Bioworld Technology, Inc., St. Louis Park, MN, USA),
IL-1β (diluted 1:50 with PBS/5% BSA; cat. no. 31202; Cell Signaling
Technology, Inc., Danvers, MA, USA), IL-6 (diluted 1:200 with
PBS/5% BSA; cat. no. 12912; Cell Signaling Technology, Inc.)
overnight at 4°C. Subsequently, cells were washed with PBS three
times, followed by incubation with corresponding secondary
phycoerythrin-conjugated goat anti-rabbit IgG (1:200 diluted with
PBS/5% BSA; cat. no. sc-3739; Santa Cruz Biotechnology, Inc.
Dallas, TX, USA) for 1 h at room temperature. For nuclear staining,
cells were covered with 10 µg/ml DAPI for 2 min at room
temperature. The stained cells were visualized under a fluorescence
microscope at ×400 magnification with green excitation light.
Different groups of images were taken in the same software
settings.
Harvesting of aortas
The mice were sacrificed via cervical dislocation,
and the midline of the chest was incised to expose the heart and
lungs. The abdominal aorta was cut to release the blood. The aorta
was dissected from the aortic arch. The fat tissues and connecting
tissue were dissociated from aorta under the microscope, and washed
with PBS three times.
Immunohistochemistry analysis of
aortas
The aortas were fixed with 10% formalin at 4°C for
48 h, embedded in paraffin and sectioned transversely. For
immunohistochemistry analysis, paraffin sections of 4 µm were
deparaffinized, rehydrated, incubated with 0.3%
H2O2 in PBS for 20 min at 25°C, and then
blocked with 10% goat serum for 30 min at room temperature.
Subsequently, tissue sections were washed with PBS and incubated
with rabbit anti-mouse polyclonal antibody: ICAM-1 (diluted 1:150
with PBS; cat. no. BS7138; Bioworld Technology, Inc.), VCAM-1
(diluted 1:150 with PBS; cat. no. BS6005; Bioworld Technology,
Inc.) or E-selectin (diluted 1:200 with PBS; cat. no. ab18981;
Abcam, Cambridge, MA, USA) overnight at 4°C. Following washing with
PBS three times, antibody reactivity was detected using
peroxidase-conjugated goat anti-rabbit IgG (diluted 1:2,000 with
PBS; cat. no. TA130017; OriGene Technologies, Inc., Beijing,
China). The sections were developed with 50 and 100%
diaminobenzidine solution (Sigma-Aldrich; Merck KGaA) diluted in
ethanol, for 15 min each at 25°C. The tissue sections were
visualized under a light microscope at ×40 magnification and
analyzed using ImageJ software (ver.1.51J8; National Institutes of
Health, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was performed according to established
protocols (24). In brief, aortas
and peritoneal macrophages from C3H/HeN mice and C3H/HeJ mice
(treated as described above) were homogenized in TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.). For inhibitory
assays, the peritoneal macrophages from C3H/HeN mice were treated
with 5 µM TAK-242 (TLR-4 inhibitor; Invitrogen; Thermo Fisher
scientific, Inc.) or 20 µM pyrrolidine dithiocarbamate (PDTC; NF-κB
inhibitor; Sigma-Aldrich; Merck KGaA) or 10 µM SB203508 (p38 MAPK
inhibitor; Sigma-Aldrich; Merck KGaA) for 2 h, then stimulated with
NR-IgG (100 µg/ml), anti-β2GPI–IgG (100 µg/ml) or LPS (500 ng/ml)
for 6 h. Total RNA was isolated using TRIzol® and cDNA
synthesis was performed using the Vazyme Reverse Transcription
System (Vazyme, Piscataway, NJ, USA): HiScript II qRT SuperMix II
was added into the RNA and reverse transcribed using the following
temperature protocol: 25°C for 10 min, 42°C for 30 min and 85°C for
5 min. The primer sequences used for RT-qPCR are listed in Table I. mRNA levels of mouse TNF-α,
IL-1β, IL-6, ICAM-1, VCAM-1 and E-selectin were measured by RT-qPCR
using cDNA obtained from the RT reactions as the templates, with
SYBR-Green I dye (Vazyme, Piscataway). RT-qPCR was conducted using
the following parameters: Denaturation at 94°C for 5 min, followed
by 40 cycles at 75°C for 30 sec each, and a final cycle at 72°C for
10 min. Amplification of cDNA for GAPDH was used as an internal
control. The relative mRNA expressions of target genes compared to
GAPDH were calculated by 2−ΔΔCq method (25).
 | Table I.Sequences of primers used for reverse
transcription-quantitative polymerase chain reaction analyses. |
Table I.
Sequences of primers used for reverse
transcription-quantitative polymerase chain reaction analyses.
| Gene | Primer
sequences | Product length
(bp) | Annealing
temperature (°C) |
|---|
| TNF-α |
F:ATTATGGCTCAGGGTCCAAC | 197 | 60 |
|
|
R:GACAGAGGCAACCTGACCAC |
|
|
| IL-1β |
F:GCTGCTTCCAAACCTTTGACC | 110 | 56 |
|
|
R:AGCCACAATGAGTGATACTGCC |
|
|
| IL-6 |
F:GACTTCCATCCAGTTGCCTT | 150 | 59 |
|
|
R:ATGTGTAATTAAGCCTCCGACT |
|
|
| ICAM-1 |
F:CTCACTTGCAGCACTACGG | 138 | 59 |
|
|
R:TTCATTCTCAAAACTGACAGGC |
|
|
| VCAM-1 |
F:GCCACCCTCACCTTAATTGCT | 188 | 61 |
|
|
R:GCACACGTCAGAACAACCGAA |
|
|
| E-selectin |
F:ATAACGAGACGCCATCATGC | 191 | 58 |
|
|
R:TGTCCACTGCCCTTGTGC |
|
|
| GAPDH |
F:GGCATTGCTCTCAATGACAA | 200 | 58 |
|
|
R:TGTGAGGGAGATGCTCAGTG |
|
|
Western blot analysis
The mice were treated with different stimulants as
described above. Protein samples were isolated from peritoneal
macrophages and homogenate of aortas using a proteome extraction
kit (radioimmunoprecipitation assay; Thermo Fisher Scientific
Inc.), and the protein concentration of samples were measured using
Pierce™ bicinchoninic acid protein assay kit (Thermo Fisher
Scientific Inc.). The proteins and respective primary antibodies
used in this assay were as follows: β-actin (cat. no. 4970), TNF-α
(cat. no. 11948), IL-1β (cat. no. 31202), IL-6 (cat. no. 12912; all
from Cell Signaling Technology, Inc.), ICAM-1 (cat. no. ab179707;
Abcam), VCAM-1 (cat. no. 39036; Cell Signaling Technology, Inc.),
E-selectin (cat. no. ab18981; Abcam), p38-MAPK (cat. no. 8690,
p-p38-MAPK (cat. no. 4511), NF-κB (cat. no. 8242) and NF-κB
phosphorylation (cat. no. 3033) (all from Cell Signaling
Technology, Inc.). Equal amounts of protein (5 µg/well) from the
samples under different experimental conditions were
electrophoresed by SDS-PAGE on 12% gels. The gels were transferred
to a polyvinylidene difluoride membrane (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The membranes were blocked in PBS
containing 5% (m/v) non-fat milk powder for 1 h at room
temperature, washed with Tris aminomethane-buffered saline
(TBS)/0.1% Triton X-100 (TBS/T) three times, and probed with
corresponding rabbit anti-mouse antibodies described above (1:1,000
diluted with TBS/T) at 4°C overnight. Following three washes with
TBS/T, the membranes were exposed to horseradish
peroxidase-conjugated goat anti-rabbit antibodies (1:4,000 diluted
with TBS/T; cat. no. sc-2004; Santa Cruz Biotechnology, Inc.) at
room temperature for 1 h. Finally, the immunoblots were exposed and
visualized with an Amersham Typhoon 9400 Fluor-S MultiImager (GE
Healthcare Life Sciences, Little Chalfont, UK) using enhanced
chemiluminescence western blotting detection reagents (GE
Healthcare Life Sciences). Densitometry analysis was performed
using ImageJ v1.8.0 software (National Institutes of Health).
Statistical analysis
Normally distributed variables were expressed as the
mean ± standard deviation. One-way analyses of variance (ANOVA)
with Newman-Keuls post-hoc test was used to compare three or more
groups, and two-factor treatment results were analyzed by two-way
ANOVA using SPSS statistical software package (version 20.0; IBM
Corp., Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
TLR-4 mediates the expression of
inflammatory cytokines in peritoneal macrophages induced by
anti-β2GPI–IgG
A previous study demonstrated the role of TLR-4 in
the anti-β2GPI/β2GPI-induced expression of inflammatory cytokines
in THP-1 cells and monocytes (14). The current study focused on
examining the effect of TLR-4 in the activation of peritoneal
macrophages in mice stimulated with anti-β2GPI–IgG. As demonstrated
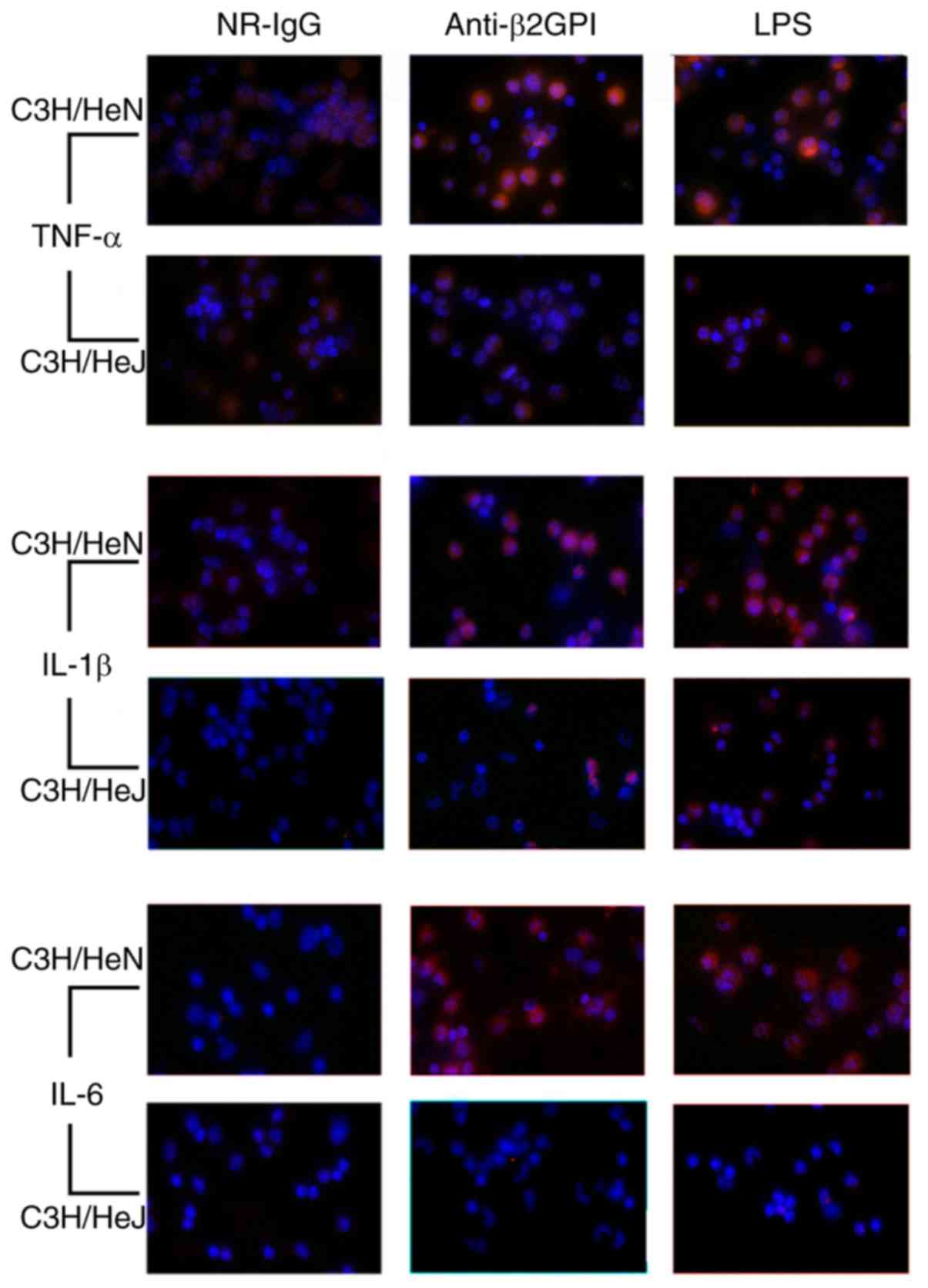
in Fig. 1, immunostaining revealed
that anti-β2GPI–IgG significantly increased expression levels of
TNF-α, IL-1β, and IL-6 in peritoneal macrophages isolated from
C3H/HeN mice as compared with those of the NR-IgG group, as no
significant fluorescence was detected in the isotype control.
Notably, the increased expression levels of these cytokines were
even higher than those of the LPS-stimulated positive control
groups. However, in peritoneal macrophages isolated from C3H/HeJ
mice (TLR-4-defective) stimulated with anti-β2GPI–IgG, the
intensity of red fluorescence was far weaker than those of
peritoneal macrophages derived from C3H/HeN received the same
stimuli.
TLR-4 mediates the expression of
adhesion molecules in vascular endothelial cells induced by
anti-β2GPI–IgG
It has been reported that aPLs are able to enhance
the adhesion of white blood cells to endothelial cells (26,27);
thus, in the current study it was determined whether the expression
levels of ICAM-1, E-selectin and VCAM-1 in mouse arteries were
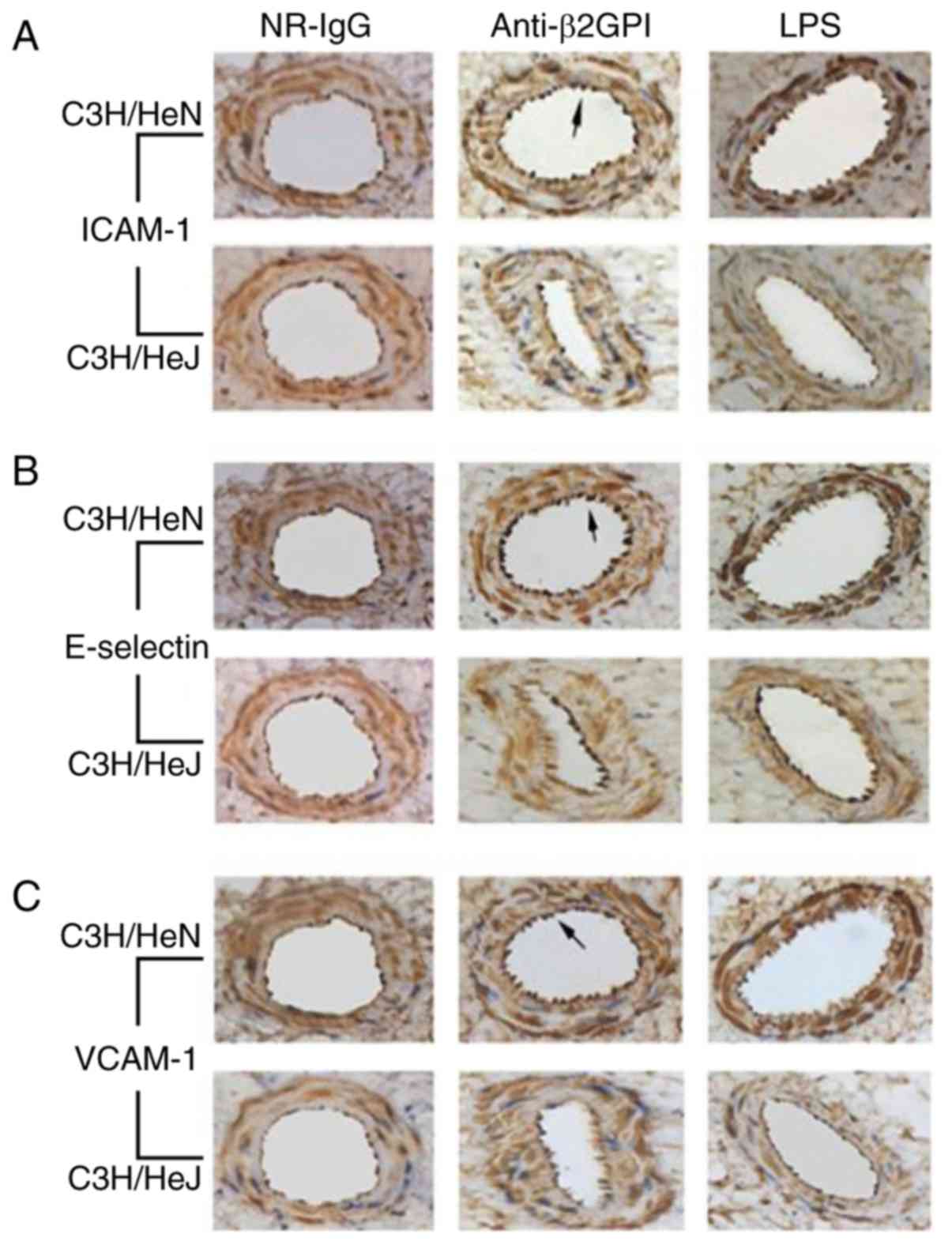
affected by anti-β2GPI–IgG injection. As presented in Fig. 2, brown granular staining on the
aorta intima was produced using antibodies against ICAM-1,
E-selectin and VCAM-1 from C3H/HeN mice treated with
anti-β2GPI–IgG. In addition, endothelial cells of C3H/HeN mice
stimulated with anti-β2GPI–IgG displayed more intense of brown
color compared to those of the NR-IgG group, as no significant
brown granular particles were detected in the isotype control. By
contrast, endothelial cells derived from C3H/HeJ (TLR-4-defective)
mice injected with anti-β2GPI–IgG exhibited lower expression of
ICAM-1, E-selectin, and VCAM-1, as indicated by the lower intensity
of brown granular particles on their intimal, compared with those
derived from anti-β2GPI–IgG injected C3H/HeN mice.
TLR-4 mediates the mRNA expression
levels of inflammatory cytokines in peritoneal macrophages, and
adhesion molecules in vascular endothelial cells derived from mice
induced by anti-β2GPI–IgG
To investigate the roles of TLR-4 in the
anti-β2GPI–IgG-induced expression of inflammatory cytokines and
adhesion molecules, the mRNA levels of inflammatory cytokines
(TNF-α, IL-1β and IL-6) were measured in peritoneal macrophages,
and adhesion molecules (ICAM-1, E-selectin, and VCAM-1) were
measured in artery endothelial cells with RT-qPCR. As presented in
Fig. 3, NR-IgG-treated mice
exhibited the basal expression levels of TNF-α, IL-1β, and IL-6 and
ICAM-1, VCAM-1 and E-selectin, with very low levels. Treatment of
C3H/HeN mice with anti-β2GPI–IgG or with LPS significantly enhanced
the mRNA expression levels of all these effector molecules;
however, in C3H/HeJ (TLR-4-defective) mice treated with
anti-β2GPI–IgG or LPS, the mRNA expression levels of these effector
molecules were significantly lower than those from C3H/HeN mice
treated with the same stimuli.
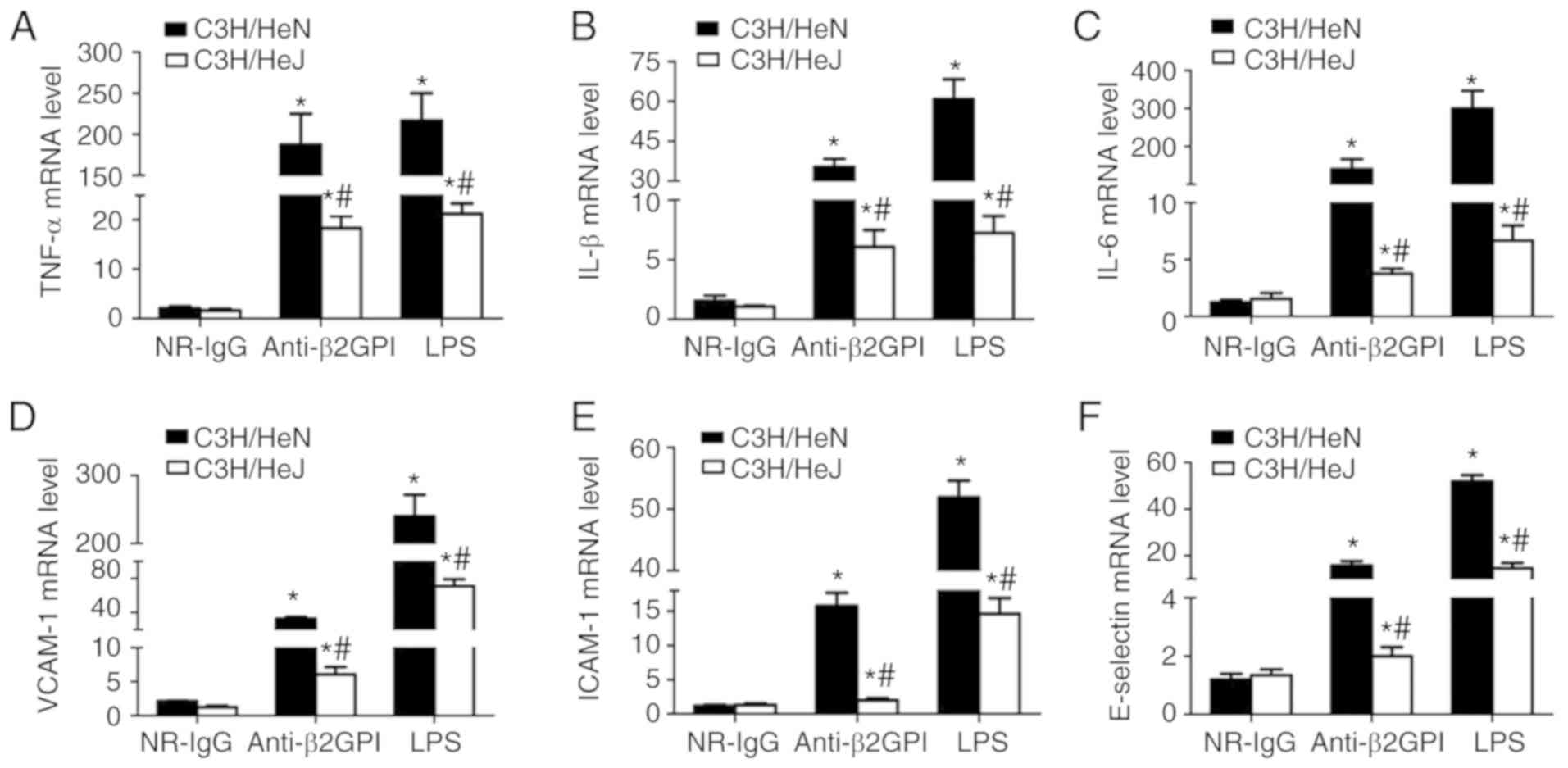 | Figure 3.TLR-4 mediates the mRNA expression
levels of inflammatory cytokines in peritoneal macrophages and
adhesion molecules in vascular endothelial cells from mice induced
by anti-β2GPI–IgG. C3H/HeN mice (TLR-4 intact; n=8 in each group)
and C3H/HeJ mice (TLR-4 defective; n=8 in each group) were treated
by intraperitoneal injection of NR-IgG (100 µg antibody per
injection), anti-β2GPI–IgG (100 µg antibody per injection) at time
0 and 48 h. The peritoneal macrophages and aortas were collected at
72 h after the first injection, and the positive control group was
challenged with LPS (1 µg/g body weigh) 2 h before the experiments.
Total RNA was extracted from peritoneal macrophages
(2×106) and mRNA expression levels of (A) TNF-α, (B)
IL-1β and (C) IL-6 were detected by RT-qPCR. The total RNA was
extracted from vascular tissue (aortas) and the mRNA levels of (D)
VCAM-1, (E) ICAM-1 and (F) E-selectin were detected by RT-qPCR. The
data are representative of three experiments. *P<0.05 vs.
control NR-IgG; #P<0.05 vs. corresponding C3H/HeN
stimulation group. TLR-4, Toll-like receptor-4; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; NR-IgG,
isotype control antibody; Anti-β2GPI, anti-β2-glycoprotein I; LPS,
lipopolysaccharide; TNF-α, tumor necrosis factor-α; IL,
interleukin; ICAM-1, intercellular cell adhesion molecule-1;
VCAM-1, vascular cell adhesion molecule-1. |
TLR-4 mediates the protein expression
levels of inflammatory cytokines in peritoneal macrophages, and
adhesion molecules in vascular endothelial cells from mice induced
by anti-β2GPI–IgG
To further demonstrate the roles of TLR-4 in the
anti-β2GPI–IgG-induced expression of inflammatory cytokines and
adhesion molecules, the protein expression levels of inflammatory
cytokines (TNF-α, IL-1β and IL-6) were examined in peritoneal
macrophages, and adhesion molecules (ICAM-1, E-selectin and VCAM-1)
were examined in artery endothelial cells via western blot analysis
with corresponding antibodies. As demonstrated in Fig. 4, the protein levels of inflammatory
cytokines (TNF-α, IL-1β and IL-6) and adhesion molecules (ICAM-1,
E-selectin and VCAM-1) are basically consistent with their mRNA
levels. NR-IgG-treated mice exhibited low basal levels of all
above-mentioned molecules. Treatment of C3H/HeN mice with
anti-β2GPI–IgG or with LPS significantly enhanced the protein
expression level of these effector molecules; however, in C3H/HeJ
mice treated with anti-β2GPI–IgG or LPS, the protein expression
levels of these effector molecules were significantly lower than
those from C3H/HeN mice treated with the same stimuli.
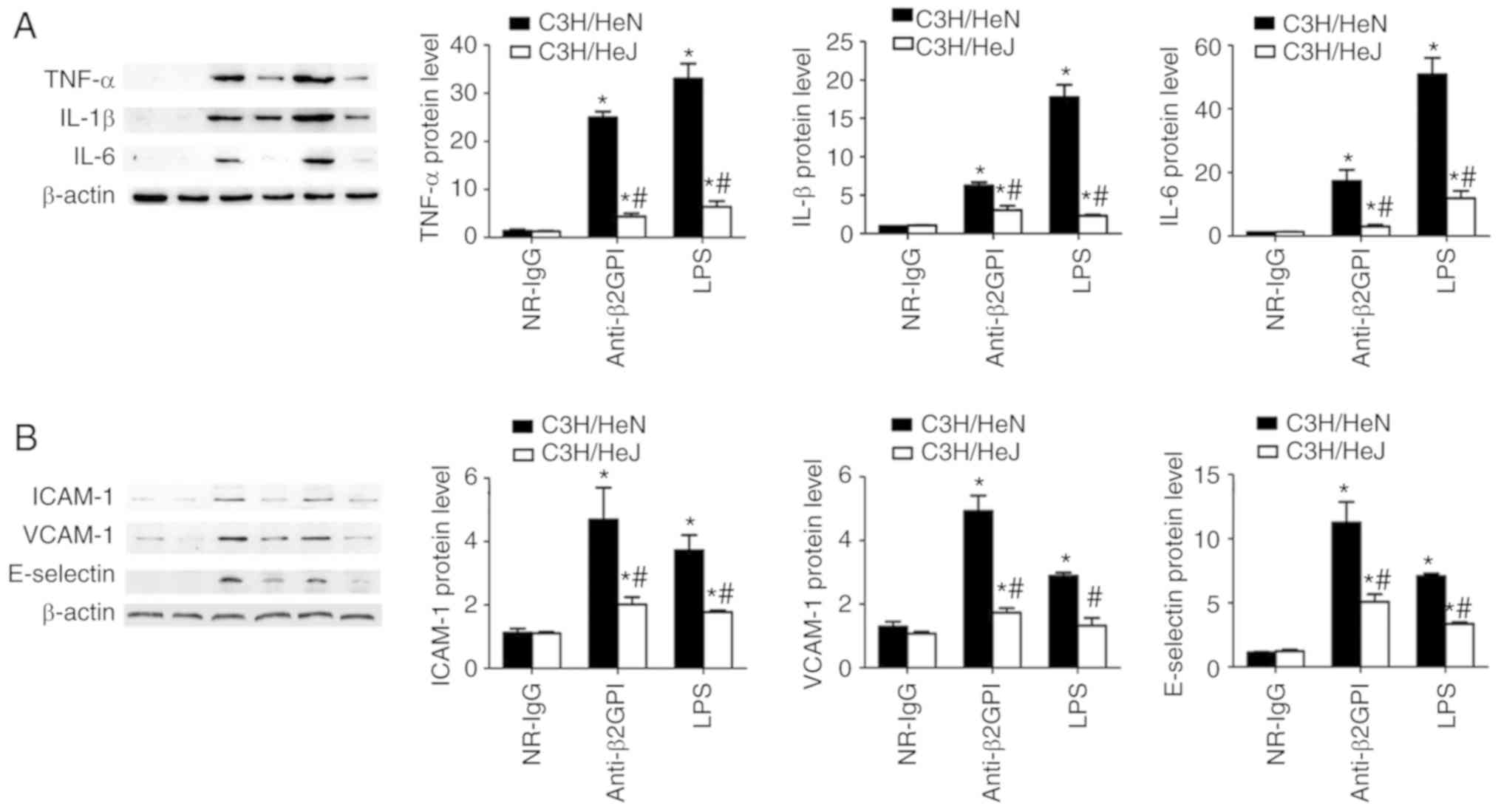 | Figure 4.TLR-4 mediated the protein expression
of inflammatory cytokines in peritoneal macrophages and adhesion
molecules in vascular endothelial cells from mice induced by
anti-β2GPI–IgG. C3H/HeN mice (n=8 in each group) and C3H/HeJ mice
(n=8 in each group) were treated by intraperitoneal injection of
NR-IgG (100 µg antibody per injection), anti-β2GPI–IgG (100 µg
antibody per injection) at time 0 and 48 h. The peritoneal
macrophages and aortas were collected at 72 h after the first
injection, and positive control group were challenged with LPS (1
µg/g body weigh) 2 h before the experiments. Protein samples were
isolated from peritoneal macrophages and homogenate of aortas using
a proteome extraction kit. The levels of (A) TNF-α, IL-1β, and IL-6
in peritoneal macrophages and (B) ICAM-1, VCAM-1, and E-selectin in
vascular tissue were measured by western blotting using their
corresponding antibodies. The data shown are the pooled data
representative of three separated experiments. *P<0.05 vs.
control NR-IgG; #P<0.05 vs. corresponding C3H/HeN
stimulation group. TLR-4, Toll-like receptor-4; TNF-α, tumor
necrosis factor-α; IL, interleukin; NR-IgG, isotype control
antibody; Anti-β2GPI, anti-β2-glycoprotein I; LPS,
lipopolysaccharide; ICAM-1, intercellular cell adhesion molecule-1;
VCAM-1, vascular cell adhesion molecule-1. |
Role of TLR-4 in
anti-β2GPI–IgG-stimulated phosphorylation of p38 MAPK and NF-κB
p65
p38 MAPK and the p65 subunit of NF-κB are key
molecules involved in triggering the expression of pro-inflammatory
cytokines and pro-coagulant factors during the inflammatory
response (28,29). Our previous experiments (19,20)
demonstrated that p38 MAPK and NF-κB p65 were involved in inducing
the expression of pro-inflammatory cytokines and pro-coagulant
factors in human monocytes (THP-1 cells) stimulated by
β2GPI/anti-β2GPI–IgG complex. Based on these preliminary
experimental results, the effects of anti-β2GPI–IgG treatment on
the phosphorylation status of p38 MAPK and NF-κB p65 in peritoneal
macrophages and artery endothelial cells derived from C3H/HeN mice
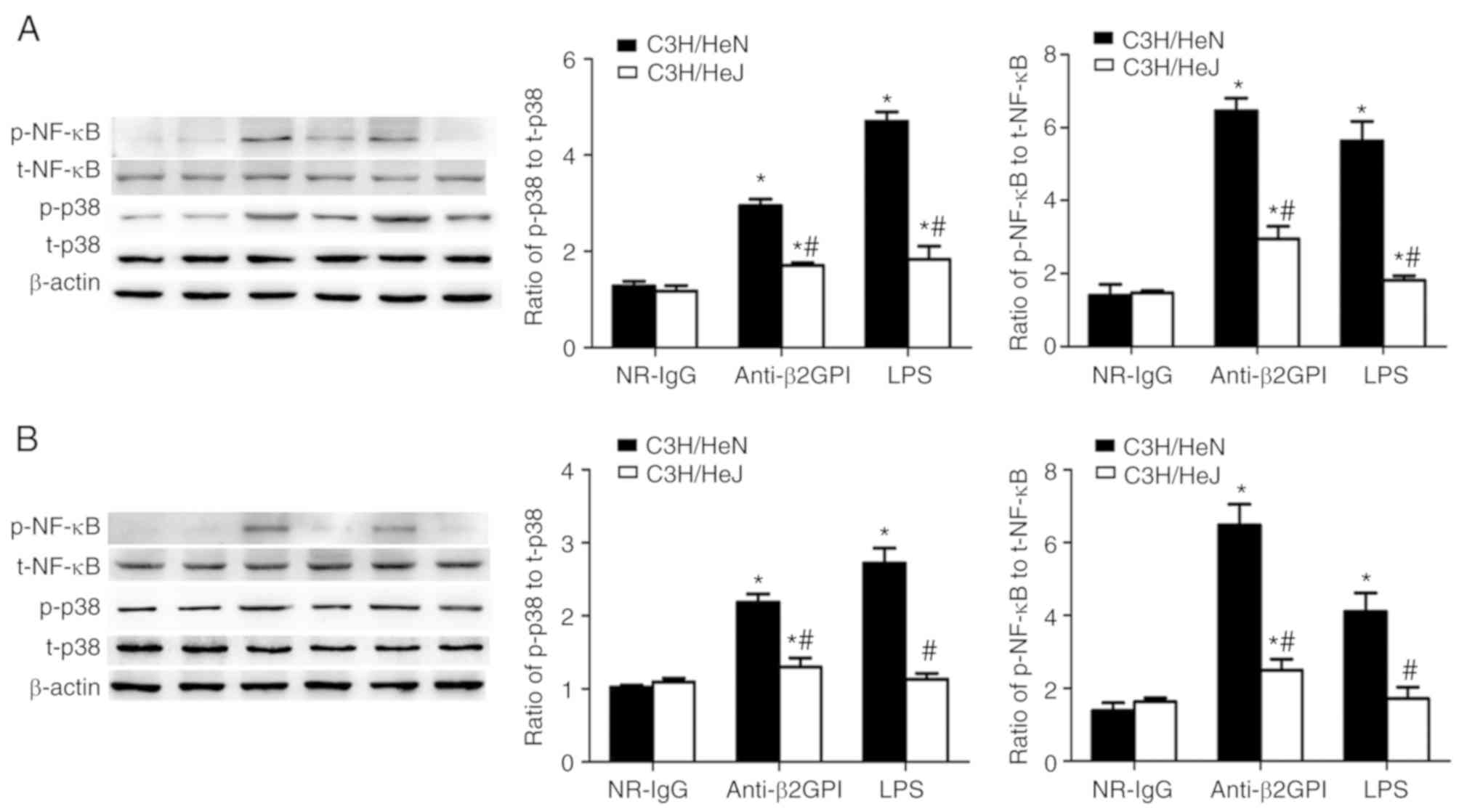
were examined. As presented in Fig.
5, treatment with anti-β2GPI–IgG or LPS (as the positive
control) increased the phosphorylation of p38 MAPK and NF-κB p65 in
peritoneal macrophages and artery endothelial cells from C3H/HeN
mice, as compared with the NR-IgG treatment (as isotype control).
Additionally, the phosphorylation of these two signaling molecules
in peritoneal macrophages and artery endothelial cells derived from
C3H/HeJ (TLR-4 defective) mice was significantly lower than in
C3H/HeN mice, indicating the involvement of p38 MAPK and NF-κB p65
in the TLR-4-mediated anti-β2GPI–IgG-induced activation of mouse
peritoneal macrophages and endothelial cells.
To further confirm the roles of a TLR-4/p38
MAPK/NF-κB signal transduction pathway in anti-β2GPI–IgG-induced
activation of mouse peritoneal macrophages, the inhibitors of
TLR-4, p38 MAPK and NF-κB were used in subsequent assays. As
demonstrated in Fig. 6, TLR-4
inhibitor (TAK-242), NF-κB inhibitor (PDTC) and p38 MAPK inhibitor
(SB203508) significantly decreased the expression of TNF-α, IL-6
and IL-1β mRNA in the peritoneal macrophages from anti-β2GPI–IgG
and LPS-injected C3H/HeN mice (P<0.05 vs. untreated); however,
none of these inhibitors could fully block the effects of
anti-β2GPI–IgG or LPS on peritoneal macrophages, suggesting that
TLR-4/p38 MAPK/NF-κB signal transduction pathway plays a part of
roles in the process.
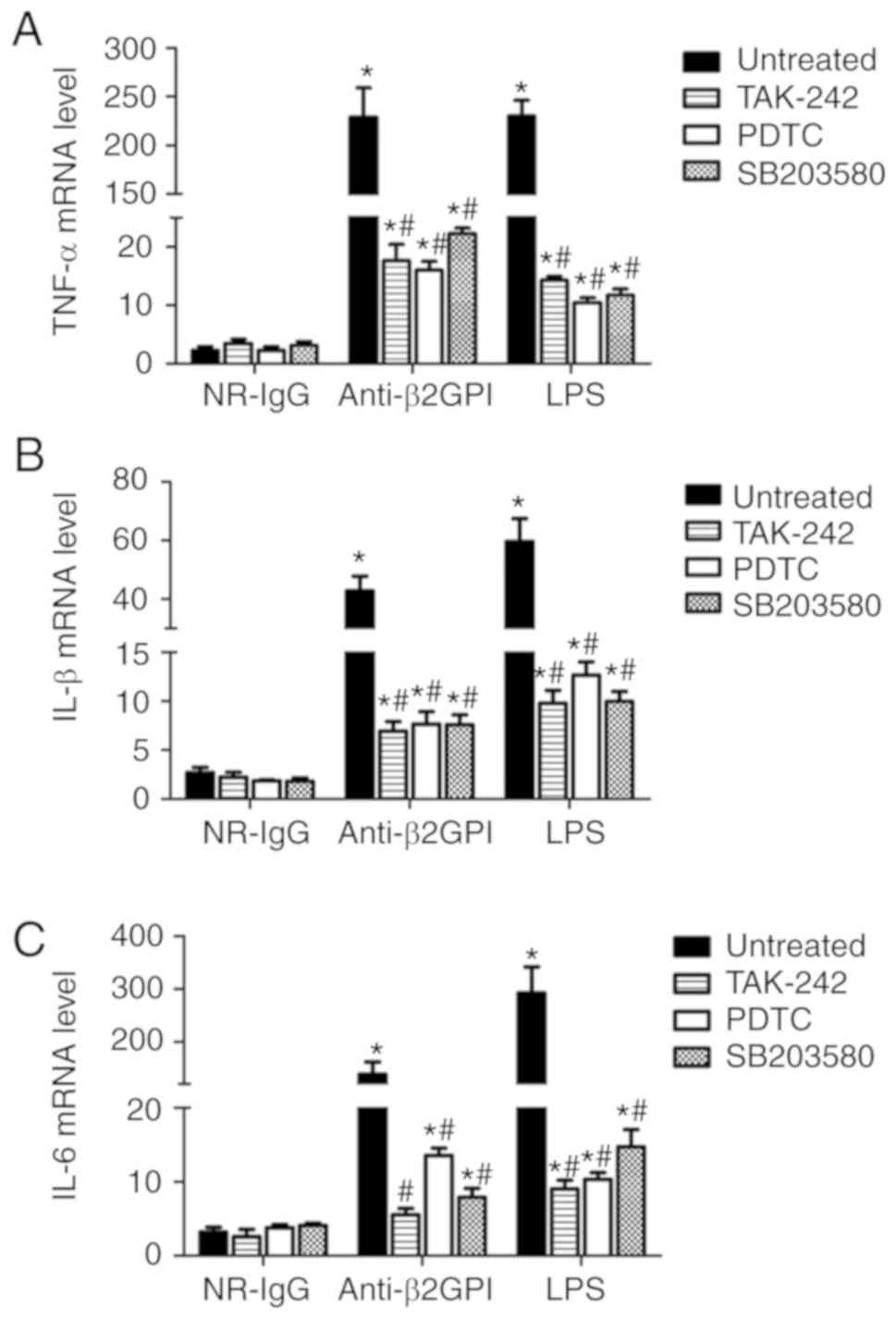 | Figure 6.Inhibitors of TLR-4, p38 MAPK and
NF-κB reduce the expression of TNF-α, IL-1β, and IL-6 mRNAs in
peritoneal macrophages from C3H/HeN mice stimulated with
anti-β2GPI–IgG. The peritoneal macrophages (2×106) from
C3H/HeN mice (TLR-4 intact; n=8 in each group) were incubated with
5 µM TAK-242 (TLR-4 inhibitor) or 20 µM PDTC (NF-κB inhibitor) or
10 µM SB203508 (p38 MAPK inhibitor) for 2 h, then treated with
NR-IgG (100 µg/ml), anti-β2GPI–IgG (100 µg/ml) or LPS (500 ng/ml)
for 6 h. The total RNA were extracted from the macrophages and mRNA
expression levels of (A) TNF-α, (B) IL-1β and (C) IL-6 were
detected by reverse transcription-quantitative polymerase chain
reaction. The data are representative of three separated
experiments with the similar results. *P<0.05 vs. control
NR-IgG; #P<0.05 vs. untreated. TLR-4, Toll-like
receptor-4; TNF-α, tumor necrosis factor-α; PDTC, pyrrolidine
dithiocarbamate; NR-IgG, isotype control antibody; Anti-β2GPI,
anti-β2-glycoprotein I; LPS, lipopolysaccharide; IL,
interleukin. |
Discussion
The findings of the present study demonstrated that
TLR-4 was involved in anti-β2GPI-induced activation of mouse
peritoneal macrophages and vascular endothelial cells in
vivo. These results indicate that anti-β2GPI antibodies
stimulate the expression of pro-inflammatory molecules in
macrophages and adhesion molecules in endothelial cells in mice.
Furthermore, it was demonstrated that TLR-4 is involved in
anti-β2GPI–IgG-stimulated phosphorylation of p38 MAPK and p65 NF-κB
that mediates the induced expression of inflammatory cytokines in
peritoneal macrophages, and adhesion molecules in vascular
endothelial cells. This TLR-4/p38 MAPK/NF-κB signal transduction
pathway has an important role in the anti-β2GPI–IgG-induced
expression of pro-inflammatory molecules in macrophages.
The role of TLR-4 in anti-β2GPI-induced activation
of mouse peritoneal macrophages and vascular endothelial cells
in vivo is supported by the following findings. Firstly, the
expression levels of inflammatory cytokines (TNF-α, IL-1β, and
IL-6) were significantly upregulated at the mRNA and protein levels
by anti-β2GPI–IgG in peritoneal macrophages derived from
TLR-4-intact C3H/HeN mice, but were not induced in peritoneal
macrophages derived from TLR-4-defective C3H/HeJ mice, as
demonstrated by immunostaining, RT-qPCR analysis and immunoblot
analysis. Additionally, the expression levels of adhesion molecules
(ICAM-1, E-selectin and VCAM-1) were significantly upregulated by
anti-β2GPI–IgG in vascular endothelial cells derived from
TLR-4-intact C3H/HeN mice, but were not induced in vascular
endothelial cells derived from TLR-4-defective C3H/HeJ mice, as
indicated by immunostaining, RT-qPCR analysis and immunoblotting.
These results clearly indicate that TLR-4 has a key role in
mediating of the expression levels of inflammatory cytokines in
mouse peritoneal macrophages, and adhesion molecules in vascular
endothelial cells induced by anti-β2GPI–IgG. The results are
consistent with the report by He et al (24), which demonstrated that TLR-4 was
involved in the interaction of aPLs with endothelial cells in
vivo and showed that anti-β2GPI thrombogenic activity was
strongly reduced in mice expressing defective TLR4 (27). These results have also enriched the
findings of our previous study (23), in which TLR-4 was involved in
IgG-APS or anti-β2GPI-induced activation of peritoneal macrophages
and vascular endothelium, as well as thrombosis induced by
FeCl3.
The aPL/anti-β2GPI antibodies are known to
contribute to thrombosis pathogenesis, pregnancy morbidity and
accelerated atherosclerosis in patients with APS and systemic lupus
erythematosus (30). By binding to
Annexin A2 (ANX2), anti-β2GPI triggers activation of peritoneal
macrophages and vascular endothelial cells, and regulates the
production of pro-inflammatory molecules and pro-coagulant factors
(31–36); however, as ANX2 is not a
transmembrane protein, it is unable to recruit downstream signaling
molecules. Instead, TLR-4 was considered as an ‘adaptor’ in cells
expressing anti-β2GPI-induced effector molecules (35). Consistent with the proposed role of
TLR-4 as an ‘adaptor’, the results of the current study further
suggest the close co-operation between TLR-4 and anti-β2GPI
antibody in mediating the pathogenesis of APS in vivo by
inducing the expression of inflammatory cytokines and adhesion
molecules.
In C3H/HeN mice, peritoneal macrophages and
endothelial cells derived from anti-β2GPI treatment upregulated the
expression of inflammatory cytokines (TNF-α, IL-1β and IL-6) and
adhesion molecules (ICAM-1, VCAM-1 and E-selectin); however, this
phenomenon was not been present in C3H/HeN mice (TLR-4 defective)
treated with NR-IgG. Our previous study demonstrated that targeting
β2GPI using F(ab)2 fragments, but not the Fc segment, of anti-β2GPI
was affected by TLR-4 (31,34).
A significant protective effect was observed in TLR-4 defective
C3H/HeJ mice, i.e. the increased expression levels of inflammatory
cytokines and adhesion molecules did not occur in C3H/HeJ mice
treated with anti-β2GPI. These results clearly indicate that TLR-4
is involved in mediating the effects of anti-β2GPI in the
pathogenesis of thrombosis in APS. Inflammatory cytokines (TNF-α,
IL-1β and IL-6) inhibit the physiological anti-coagulant system,
transforming endothelial cells from the anti-coagulant state into
the pro-coagulant state (37). The
increased expression of adhesion molecules (e.g. ICAM-1, VCAM-1 and
E-selectin) can induce the adhesion of monocytes and platelets to
endothelial cells, thus promoting the inflammation and coagulation
responses. Increased adhesion of leukocytes to the endothelium of
mouse cremaster muscle, an indication of activation of endothelial
cells in vivo, and enhanced thrombosis in vivo was
found to be closely associated with upregulated expression of
ICAM-1, VCAM-1 and P-selectin on endothelial cells stimulated by
aPL (13,27,38).
Furthermore, ICAM-1, VCAM-1 and P-selectin were shown to be
involved in mediating the activation of endothelial cells and
enhanced thrombosis by aPL in vivo, as demonstrated by
reduced adhesion of leukocytes in ICAM-1-defective mice and
completely abrogated leukocyte adhesion in ICAM-1/P-selectin double
defective mice treated with IgG-APS, compared with wild-type mice
treated with IgG-APS (38). These
observations indicate that increased expression levels of ICAM-1,
VCAM-1 and E-selectin are involved in the thrombotic complications
mediated by aPL antibodies.
LPS is an important TLR-4 ligand. In the current
study, the expression levels of inflammatory cytokines and adhesion
molecules in TLR-4-intact C3H/HeN mice stimulated with anti-β2GPI
were higher than those stimulated by LPS, suggesting that in
addition to TLR-4, there may be other receptors mediating the
effects of anti-β2GPI in vivo. In addition, the expression
levels of inflammatory cytokines and adhesion molecules were not
fully abolished in TLR-4 defective C3H/HeJ mice. Furthermore, the
TLR-4 inhibitor TAK-242 blocked the effects of anti-β2GPI or LPS on
the expression of inflammatory cytokines in C3H/HeN mouse
peritoneal macrophages, but did not fully abrogate the effects.
These results suggested that other receptors may participate in the
antibody binding and associated activation pathways. In fact, it
was also reported that TLR-8, another member of TLR family, induced
upregulation of TNF-α following aPL stimulation (39). Furthermore, TLR-2, which has a
fundamental role in pathogen recognition and activation of innate
immunity, was also reported to have a role in the pathogenesis of
APS (40). Our previous study
investigated the relationship between TLR-2 and the
β2GPI/anti-β2GPI complex, and revealed that TLR-2 blockade could
reduce TNF-α expression induced by β2GPI/anti-β2GPI complex in
mouse peritoneal macrophages (41). Additionally, it has also been
previously demonstrated that apolipoprotein E receptor (apoER) and
platelet factor 4 (PF4) are able to induce the activation of
endothelial cells and platelets (42); thus, more studies are required to
define the precise roles of TLR-4, TLR-8, TLR-2, apoER, PF4 and
other factors in mediating the pathogenesis of APS caused by
anti-β2GPI.
In the present study, further experiments were
performed to identify molecules downstream of TLR-4. It has been
previous reported that the interaction between TLR-4 and anti-β2GPI
aPLs led to the activation of Myd88-dependent and TRIF-dependent
signaling pathways, which, in turn, induced phosphorylation of
NF-κB p65 and p38 MAPK, and finally the upregulation of TNF-α,
IL-1β, IFN-γ and IL-6 protein expression (43). Consistent with this finding, in the
current study, the phosphorylated levels of p38 MAPK and NF-κB p65
in C3H/HeN cells stimulated by anti-β2GPI were significantly
increased compared with those of C3H/HeJ mice, suggesting that p38
MAPK and NF-κB p65 are involved in TLR-4-mediated β2GPI-induced
activation in peritoneal macrophages and vascular endothelial
cells. Subsequently, it was demonstrated that NF-κB inhibitor,
PDTC, and p38 MAPK inhibitor, SB203508, significantly decreased the
expression of TNF-α, IL-1β and IL-6 mRNA in peritoneal macrophages
of C3H/HeN mice. p38 MAPK and NF-κB p65 are the key factors
involved in mediating inflammatory responses in several types of
cells induced by different stimuli. For instance, transcriptional
activation of NF-κB and TNF-α production in response to Borrelia
burgdorferi antigens was found to be controlled by RelA
phosphorylation, which was mediated by mitogen- and
stress-activated protein kinase 1 (44). Activation of the p38 MAPK/NF-κB
pathway has been reported to contribute to doxorubicin-induced
inflammation and cytotoxicity in H9c2 cardiac cells (45). TLR-4 is also involved in mediating
anti-β2GPI/β2GPI-induced expression of tissue factor (TF) in THP-1
cells (34). Circulating levels of
TF and pro-inflammatory cytokines, including IL-6, IL-6 receptor,
TNF-α and interferon-γ, were reported to be higher in patients with
primary APS or leprosy associated with aPLs. It is proposed that
the imbalance of cytokines and upregulation of the TF pathway may
be potential mechanisms of thrombosis in APS.
Anti-coagulant therapy has been the mainstay of
management in APS; however, the therapeutic effects of the current
treatments are not satisfactory (46). Therefore, effective alternative or
additional strategies for treatment of APS are urgently needed.
Blockade of TLR-4 signaling pathway with genetic and/or
pharmacological approaches in combination with anti-inflammatory
drugs and anti-coagulant agents may be a new strategy for
management in APS. In term of pharmacological approaches, it was
reported that resatorvid (or TAK-242), a small-molecule inhibitor,
bound selectively to TLR-4 that interferes with interactions
between TLR-4 and its adaptor molecules, can serve as an inhibitor
for TLR-4 (47). TLR-4 activation
is reported to be a potent inducer of signaling pathways in the
nervous system, causing chronic pain, opioid tolerance and
dependence. Small-molecule modulators of TLR-4, including MD2-I and
tricyclic antidepressants, can be developed as therapeutic agents
to target TLR-4-mediated neuroinflammation (48). Several other approaches, including
inhibition of intracellular pathways, anti-cytokine therapies and
hydroxychloroquine, have been also suggested (49).
TLR-4 is involved in the activation of peritoneal
macrophages and endothelial cells stimulated by anti-β2GPI in
vivo. TLR-4-mediated pro-inflammatory and pro-coagulant events
via p38 MAPK/NF-κB pathway are involved in the aPL-mediated
pathogenic effects in APS. Blockage of TLR-4/p38 MAPK/NF-κB signal
transduction pathways may be a promising potential alternative or
additional treatment strategy for APS.
Acknowledgements
The authors would like to acknowledge all members of
Hong Zhou team for collaboration to the project.
Funding
This research was supported by National Natural
Science Foundation of China (grant no. 81370614), a grant awarded
to HZ.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MW and HZ conceived and designed the experiments.
MW, XK, YX and CH performed the experiments: MW, XK, HZ, TW and CH
analyzed the data. MW, XK, YX, HZ, TW and CH contributed
reagents/materials/analysis tools. MW, XK and HZ wrote the paper.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All the experiments involving animals were approved
by the Laboratory Animal Administration Committee of Jiangsu
University (Zhenjiang, China; UJS-LAER-2014072301).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chaturvedi S and McCrae KR: Diagnosis and
management of the antiphospholipid syndrome. Blood Rev. 31:406–417.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McNeil HP, Chesterman CN and Krilis SA:
Immunology and clinical importance of antiphospholipid antibodies.
Adv Immunol. 49:193–280. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Willis R, Harris EN and Pierangeli SS:
Pathogenesis of the antiphospholipid syndrome. Semin Thromb Hemost.
38:305–321. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanne D, Katzav A, Beilin O, Grigoriadis
NC, Blank M, Pick CG, Landenberg Pv, Shoenfeld Y and Chapman J:
Interaction of inflammation, thrombosis, aspirin and enoxaparin in
CNS experimental antiphospholipid syndrome. Neurobiol Dis.
30:56–64. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McNeil HP, Simpson RJ, Chesterman CN and
Krilis SA: Anti-phospholipid antibodies are directed against a
complex antigen that includes a lipid-binding inhibitor of
coagulation: Beta 2-glycoprotein I (apolipoprotein H). Proc Natl
Acad Sci USA. 87:4120–4124. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Galli M, Comfurius P, Maassen C, Hemker
HC, de Baets MH, van Breda-Vriesman PJ, Barbui T, Zwaal RF and
Bevers EM: Anticardiolipin antibodies (ACA) directed not to
cardiolipin but to a plasma protein cofactor. Lancet.
335:1544–1547. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Laat B, Derksen RH, Urbanus RT and de
Groot PG: IgG antibodies that recognize epitope Gly40-Arg43 in
domain I of beta 2-glycoprotein I cause LAC, and their presence
correlates strongly with thrombosis. Blood. 105:1540–1545. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Forastiero RR, Martinuzzo ME and de
Larrañaga GF: Circulating levels of tissue factor and
proinflammatory cytokines in patients with primary antiphospholipid
syndrome or leprosy related antiphospholipid antibodies. Lupus.
14:129–136. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Clemens N, Frauenknecht K, Katzav A,
Sommer C and von Landenberg P: In vitro effects of antiphospholipid
syndrome-IgG fractions and human monoclonal antiphospholipid IgG
antibody on human umbilical vein endothelial cells and monocytes.
Ann NY Acad Sci. 1173:805–813. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Levi M and van der Poll T: Two-way
interactions between inflammation and coagulation. Trends
Cardiovasc Med. 15:254–259. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
ten Cate JW, van der Poll T, Levi M, ten
Cate H and van Deventer SJ: Cytokines: Triggers of clinical
thrombotic disease. Thromb Haemost. 78:415–419. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meroni PL, Raschi E, Testoni C, Tincani A,
Balestrieri G, Molteni R, Khamashta MA, Tremoli E and Camera M:
Statins prevent endothelial cell activation induced by
antiphospholipid (anti-beta2-glycoprotein I) antibodies: Effect on
the proadhesive and proinflammatory phenotype. Arthritis Rheum.
44:2870–2878. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pierangeli SS, Colden-Stanfield M, Liu X,
Barker JH, Anderson GL and Harris EN: Antiphospholipid antibodies
from antiphospholipid syndrome patients activate endothelial cells
in vitro and in vivo. Circulation. 99:1997–2002. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou H, Sheng L, Wang H, Xie H, Mu Y, Wang
T and Yan J: Anti-β2GPI/β2GPI stimulates activation of THP-1 cells
through TLR4/MD-2/MyD88 and NF-κB signaling pathways. Thromb Res.
132:742–749. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vega-Ostertag ME, Ferrara DE,
Romay-Penabad Z, Liu X, Taylor WR, Colden-Stanfield M and
Pierangeli SS: Role of p38 mitogen-activated protein kinase in
antiphospholipid antibody-mediated thrombosis and endothelial cell
activation. J Thromb Haemost. 5:1828–1834. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Medzhitov R, Preston-Hurlburt P and
Janeway CA Jr: A human homologue of the Drosophila Toll protein
signals activation of adaptive immunity. Nature. 388:394–397. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Medzhitov R and Janeway CA Jr: Innate
immunity: The virtues of a nonclonal system of recognition. Cell.
91:295–298. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Werling D and Jungi TW: TOLL-like
receptors linking innate and adaptive immune response. Vet Immunol
Immunopathol. 91:1–12. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xia L, Zhou H, Hu L, Xie H, Wang T, Xu Y,
Liu J, Zhang X and Yan J: Both NF-κB and c-Jun/AP-1 involved in
anti-β2GPI/β2GPI-induced tissue factor expression in monocytes.
Thromb Haemost. 109:643–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou H, Chen D, Xie H, Xia L, Wang T, Yuan
W and Yan J: Activation of MAPKs in the anti-β2GPI/β2GPI-induced
tissue factor expression through TLR4/IRAKs pathway in THP-1 cells.
Thromb Res. 130:e229–e235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie HX, Zhou H, Wang HB, Chen DD, Wang T,
Zhang XM, Xia LF and Mu Y: The activation of TRIF-dependent
signaling pathway in THP-1 cells induced by β2
GPI/anti-β2 GPI antibodies complex. Xi Bao Yu Fen Zi
Mian Yi Xue Za Zhi. 27:1280–1283, 1287. 2011.(In Chinese).
PubMed/NCBI
|
|
22
|
Ethics and Animal Use, . Guide for the
Care and Use of Laboratory Animals. 8th. National Academies Press
(US); Washington (DC): 2011, PubMed/NCBI
|
|
23
|
Xie H, Kong X, Zhou H, Xie Y, Sheng L,
Wang T, Xia L and Yan J: TLR4 is involved in the pathogenic effects
observed in a murine model of antiphospholipid syndrome. Clin
Immunol. 160:198–210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He C, Zhang G, Zhou H, Cheng S and Farwa
A: Effects of Toll-like receptor 4 on β2-glycoprotein I-induced
splenic T cell subsets differentiation. Immunol Lett. 198:17–25.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brandt KJ, Kruithof EK and de Moerloose P:
Receptors involved in cell activation by antiphospholipid
antibodies. Thromb Res. 132:408–413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pierangeli SS, Vega-Ostertag ME, Raschi E,
Liu X, Romay-Penabad Z, De Micheli V, Galli M, Moia M, Tincani A,
Borghi MO, et al: Toll-like receptor and antiphospholipid mediated
thrombosis: In vivo studies. Ann Rheum Dis. 66:1327–1333. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao MY, Chen L, Yang L, Yu X, Kou JP and
Yu BY: Berberine inhibits LPS-induced TF procoagulant activity and
expression through NF-κB/p65, Akt and MAPK pathway in THP-1 cells.
Pharmacol Rep. 66:480–484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li X, Zheng Z, Li X and Ma X:
Unfractionated heparin inhibits lipopolysaccharide-induced
inflammatory response through blocking p38 MAPK and NF-κB
activation on endothelial cell. Cytokine. 60:114–121. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Willis R and Pierangeli SS:
Anti-β2-glycoprotein I antibodies. Ann NY Acad Sci. 1285:44–58.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou H, Wolberg AS and Roubey RA:
Characterization of monocyte tissue factor activity induced by IgG
antiphospholipid antibodies and inhibition by dilazep. Blood.
104:2353–2358. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou H, Wang H, Li N, Yu Y, Huang H, Yan Y
and Wang T: Annexin A2 mediates anti-beta 2 GPI/beta 2 GPI-induced
tissue factor expression on monocytes. Int J Mol Med. 24:557–562.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou H, Ling S, Yu Y, Wang T and Hu H:
Involvement of Annexin A2 in anti-beta2GPI/beta2GPI-induced tissue
factor expression on monocytes. Cell Res. 17:737–739. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou H, Yan Y, Xu G, Zhou B, Wen H, Guo D,
Zhou F and Wang H: Toll-like receptor (TLR)-4 mediates
anti-β2GPI/β2GPI-induced tissue factor expression in THP-1 cells.
Clin Exp Immunol. 163:189–198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xie H, Zhou H, Wang H, Chen D, Xia L, Wang
T and Yan J: Anti-β(2)GPI/β(2)GPI induced TF and TNF-α expression
in monocytes involving both TLR4/MyD88 and TLR4/TRIF signaling
pathways. Mol Immunol. 53:246–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hurst J, Lorenz M, Prinz N and von
Landenberg P: The roll of Toll-like receptors in the
antiphospholipid syndrome. Curr Rheumatol Rep. 12:58–63. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zelaya H, Rothmeier AS and Ruf W: Tissue
factor at the crossroad of coagulation and cell signaling. J Thromb
Haemost. 16:1941–1952. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pierangeli SS, Espinola RG, Liu X and
Harris EN: Thrombogenic effects of antiphospholipid antibodies are
mediated by intercellular cell adhesion molecule-1, vascular cell
adhesion molecule-1, and P-selectin. Circ Res. 88:245–250. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ghosh TK, Mickelson DJ, Solberg JC, Lipson
KE, Inglefield JR and Alkan SS: TLR-TLR cross talk in human PBMC
resulting in synergistic and antagonistic regulation of type-1 and
2 interferons, IL-12 and TNF-alpha. Int Immunopharmacol.
7:1111–1121. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Benhamou Y, Bellien J, Armengol G,
Brakenhielm E, Adriouch S, Iacob M, Remy-Jouet I, Le Cam-Duchez V,
Monteil C, Renet S, et al: Role of Toll-like receptors 2 and 4 in
mediating endothelial dysfunction and arterial remodeling in
primary arterial antiphospholipid syndrome. Arthritis Rheumatol.
66:3210–3220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu Y, Zhou H, Xia L, Kong X, Xie Y, Xie H,
He C and Cheng S: TLR2 blockade reduces TNF-α expression induced by
β2GP1/anti-β2GP1 complex in mouse peritoneal macrophages. Xi Bao Yu
Fen Zi Mian Yi Xue Za Zhi. 32:446–450, 456. 2016.(In Chinese).
PubMed/NCBI
|
|
42
|
Sikara MP, Routsias JG, Samiotaki M,
Panayotou G, Moutsopoulos HM and Vlachoyiannopoulos PG: {beta}2
Glycoprotein I ({beta}2GPI) binds platelet factor 4 (PF4):
Implications for the pathogenesis of antiphospholipid syndrome.
Blood. 115:713–723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kawai T and Akira S: Signaling to
NF-kappaB by Toll-like receptors. Trends Mol Med. 13:460–469. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Olson CM, Hedrick MN, Izadi H, Bates TC,
Olivera ER and Anguita J: p38 mitogen-activated protein kinase
controls NF-kappaB transcriptional activation and tumor necrosis
factor alpha production through RelA phosphorylation mediated by
mitogen- and stress-activated protein kinase 1 in response to
Borrelia burgdorferi antigens. Infect Immun. 75:270–277. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guo RM, Xu WM, Lin JC, Mo LQ, Hua XX, Chen
PX, Wu K, Zheng DD and Feng JQ: Activation of the p38 MAPK/NF-κB
pathway contributes to doxorubicin-induced inflammation and
cytotoxicity in H9c2 cardiac cells. Mol Med Rep. 8:603–608. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ruiz-Irastorza G and Khamashta MA: Lupus
and pregnancy: Integrating clues from the bench and bedside. Eur J
Clin Invest. 41:672–678. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Matsunaga N, Tsuchimori N, Matsumoto T and
Ii M: TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like
receptor (TLR) 4 signaling, binds selectively to TLR4 and
interferes with interactions between TLR4 and its adaptor
molecules. Mol Pharmacol. 79:34–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li J, Csakai A, Jin J, Zhang F and Yin H:
Therapeutic developments targeting Toll-like receptor-4-mediated
neuroinflammation. ChemMedChem. 11:154–165. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Comarmond C and Cacoub P: Antiphospholipid
syndrome: From pathogenesis to novel immunomodulatory therapies.
Autoimmun Rev. 12:752–757. 2013. View Article : Google Scholar : PubMed/NCBI
|