Introduction
Nance-Horan syndrome (NHS; Online Mendelian
Inheritance in Man no. 302350), also known as cataract-dental
syndrome, is a rare X-linked inherited disease first described in
1974 (1–3). It is characterized by congenital
cataracts, dental and craniofacial abnormalities, and mental
retardation in ~30% of affected males (4–6). All
affected males have bilateral severe congenital cataracts, usually
requiring cataract surgery at an early age, and the majority also
have congenital microcornea, nystagmus, strabismus or
microphthalmia (7,8). Dental anomalies are a typical feature
that may distinguish NHS from other types of congenital cataracts.
Dental anomalies observed in patients with NHS include
screwdriver-shaped incisors, supernumerary maxillary incisors
(mesiodens) and a space between two teeth (diastema) (2). Dysmorphic facial features are usually
mild and are often overlooked by ophthalmologists, but include a
prominent nose and nasal bridge, a long narrow face, and large
anteverted pinnae (2,9). Heterozygous carrier females often
exhibit similar but milder, symptoms compared with affected males,
including posterior Y-sutural lens opacities, little or no loss of
vision, and occasional dental and facial abnormalities (10,11).
In the present study, a clinical and genetic
analysis of a Chinese family with congenital cataracts and no
obvious dental or craniofacial abnormalities was performed. A novel
frameshift mutation in the NHS actin remodeling regulator
(NHS) gene (c.302dupA; p.Ala102fs) was identified using
whole exome sequencing (WES), and validated by direct sequencing.
This result broadens the known phenotypic and genotypic spectrum of
NHS-associated diseases.
Materials and methods
Subjects
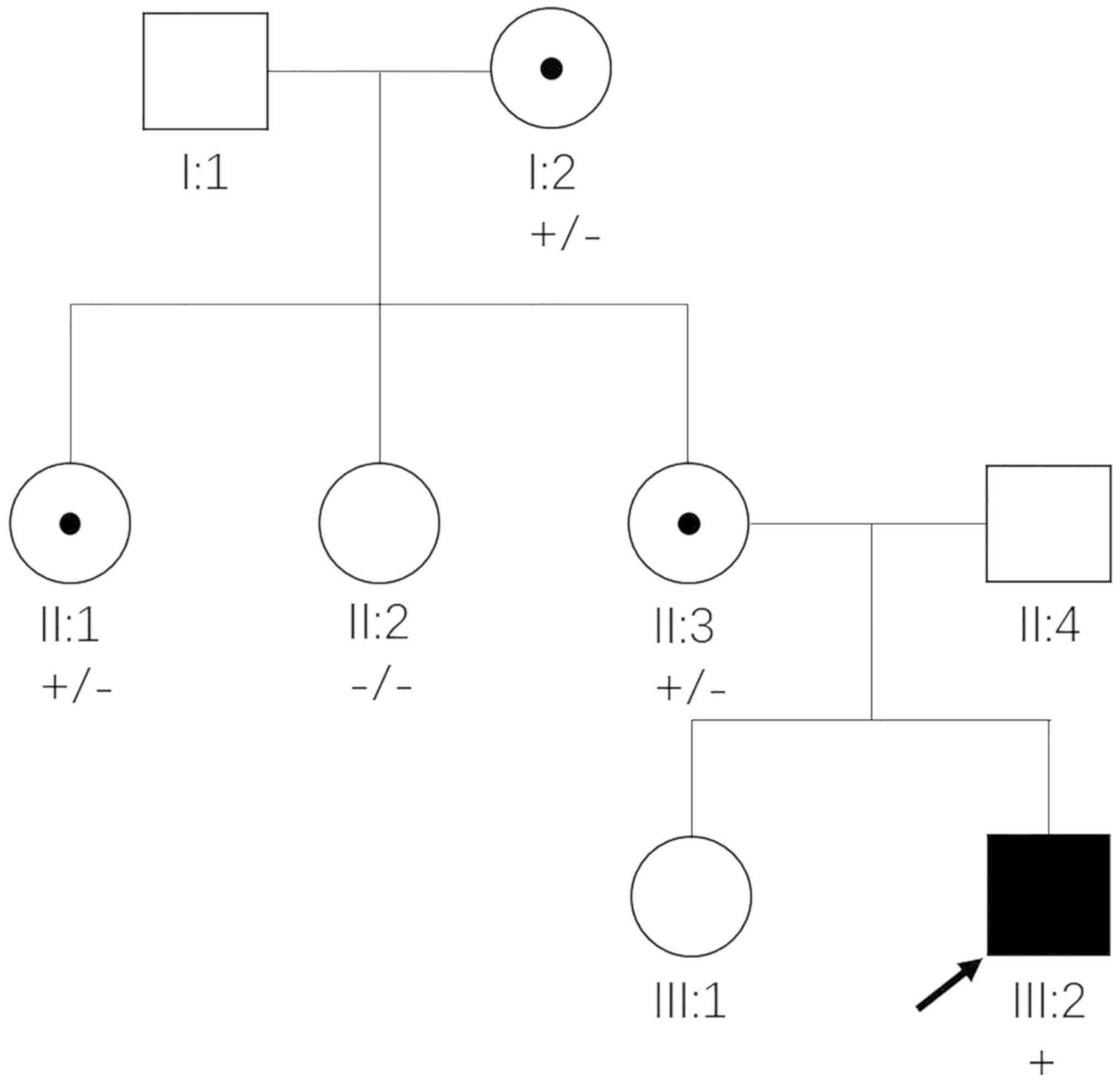
A three-generation Chinese family was enrolled in
the present study (Fig. 1).
Ophthalmic examinations and disease diagnosis were performed in
Liuzhou Maternal and Child Health Care Hospital (Liuzhou, China).
The study adhered to the guidelines of the Declaration of Helsinki
and the Association for Research in Vision and Ophthalmology
statement on human subjects. All procedures were approved by the
Ethics Committee of the National Research Institute for Family
Planning (Beijing, China) and informed consent was obtained from
the patients.
Whole exome sequencing and variant
screening
The genomic DNA of 5 individuals of the family was
extracted using the HiPure Blood DNA Mini kit (Qiagen GmbH, Hilden,
Germany). WES of the proband (III:2) was performed using the
SureSelect Human All Exon V5 kit (Agilent Technologies, Inc., Santa
Clara, CA, USA) to capture all exons and enrich the DNA fragments.
Sequencing was then performed using the HiSeq4000 sequencer
(Illumina, Inc., San Diego, CA, USA), with Burrows-Wheeler
Alignment software (bio-bwa.sourceforge.net/bwa.shtml; version bwa-0.7.12)
to align the sequences against the human reference genome. Variants
were labelled using the Genome Analysis Toolkit (www.broadinstitute.org/gatk/; version
GATKv3.4.0), including single nucleotide variants and indels, and
annotated using SnpEff (snpeff.sourceforge.net/SnpEff.html; version
SnpEff_v4.1g).
Annotated variants were filtered by a series of
processes: Firstly, exonic non-synonymous variants of
cataracts-causing genes (12) were
extracted, including missense, nonsense, frameshift and splice site
mutations. Secondly, high frequency mutations with minor allele
frequencies >1% in 1000Genomes (www.ncbi.nlm.nih.gov/variation/tools/1000genomes/),
ESP6500 (evs.gs.washington.edu/EVS/) or ExAC (exac.broadinstitute.org/) databases were
excluded. Then, genes with inheritance patterns that did not match
the family were removed. Finally, genes responsible for syndromic
phenotypes where cataracts are one of the features, but not the
primary symptom, were excluded.
Candidate mutation validation and
co-segregation analysis
The candidate mutation of the proband was validated
by Sanger sequencing (performed by Beijing Liuhe Huada Gene
Technology Co., Ltd., Beijing, China) with primers designed using
Primer Premier 5 software (version 5.00; PREMIER Biosoft
International, Palo Alto, CA, USA). Forward and reverse primers
used to identify the NHS mutation were:
5′-GGACTGGCTGGACTGATTTGCT-3′ and 5′-CGCTCCCTTTCTGCTCCTTCT-3′,
respectively. Sanger sequencing was then used to perform
co-segregation analysis of mutation and disease phenotypes.
Results
Clinical manifestations
The proband (III:2), a 2-year-old boy, had diagnosed
cataracts in both eyes following birth. Ophthalmic examinations
revealed congenital cataracts with microcornea (horizontal corneal
diameter =8 mm) and nystagmus. No other facial or dental
abnormalities were observed, with the exception of slightly large
anteverted pinnae. He underwent lentectomy without artificial lens
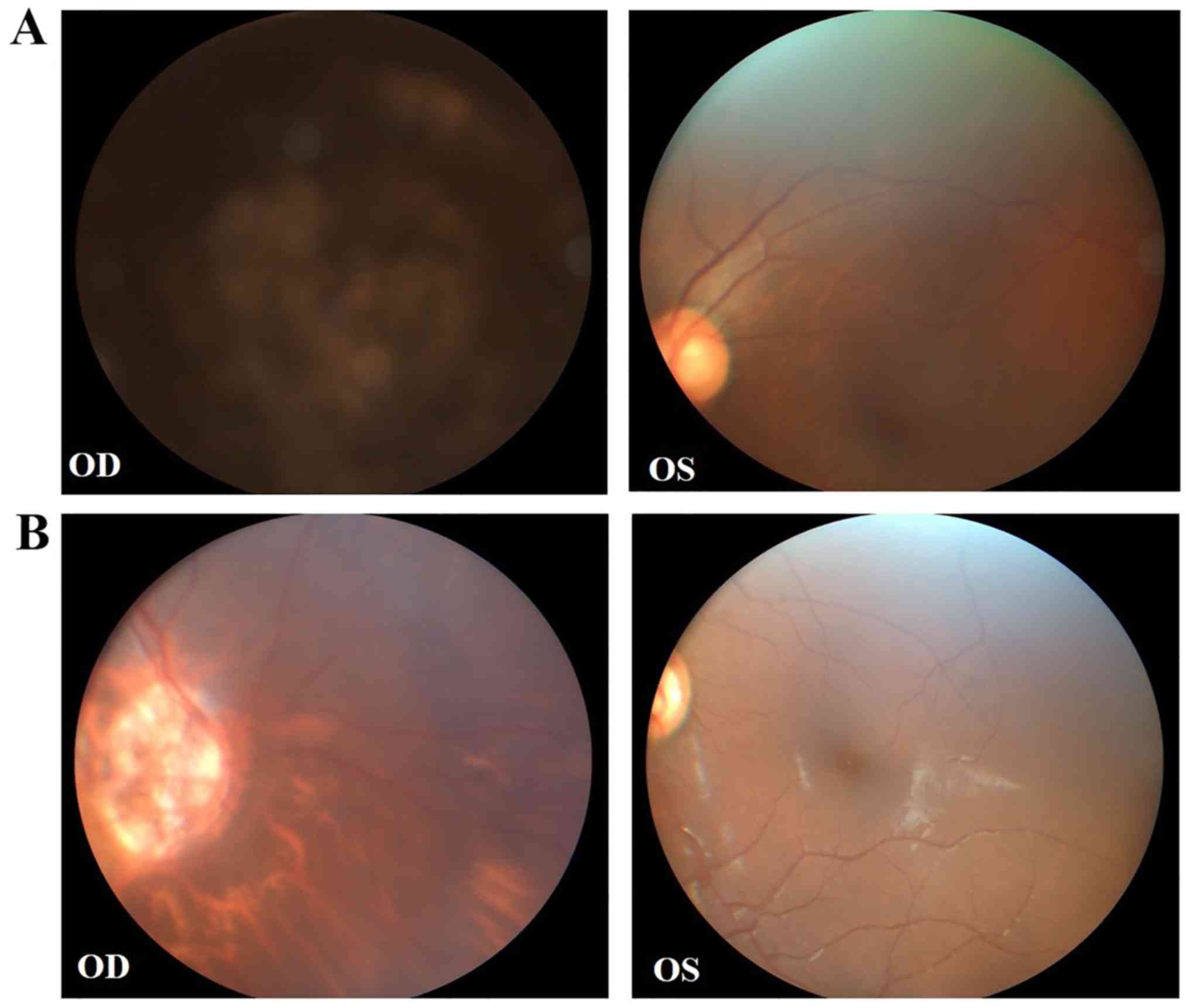
implantation due to his young age. His mother (II:3) exhibited a
congenital cataract with microcornea and strabismus of the right
eye, and received cataract surgery. Fundus examination demonstrated
unclear optic atrophy and tigroid fundus of the right eye, and the
left eye was normal (Fig. 2A). The
maternal aunt of the proband (II:1) also exhibited a congenital
cataract of the right eye, and compound myopic astigmatism of the
left eye with no obvious cataract. Fundus examination revealed
similar results to those of II:3 (Fig.
2B). Detailed clinical features are summarized in Table I.
 | Table I.Clinical symptoms of carriers and
affected members in the Chinese Nance-Horan Syndrome family. |
Table I.
Clinical symptoms of carriers and
affected members in the Chinese Nance-Horan Syndrome family.
| Characteristics | Disease
phenotype | II:1 (carrier) | II:3 (carrier) | III:2 (affected) |
|---|
| Age, years/sex | − | 41/F | 36/F | 2/M |
| Ocular features | Bilateral congenital
cataracts | + | + (right) | + |
|
| Microcornea | − | + | + |
|
| Nystagmus | − | − | + |
|
| Strabismus | − | + (right) | − |
|
| Microphthalmia | − | − | − |
| Non-ocular
features | Dental anomalies | + (slight) | + (slight) | − |
|
| Long narrow face | − | − | − |
|
| Prominent nose | − | − | − |
|
| Large anteverted
pinnae | − | − | + (slight) |
|
| Mental
retardation | − | − | − |
Identification of the pathogenic
mutation
WES identified 79,533 single nucleotide
polymorphisms and 9,307 indels in the proband. A total of 7
mutations in different genes were identified following the first
two filtering processes, including 5 missense mutations, one splice
acceptor mutation and one frameshift mutation (Table II). Variants of genes inherited in
an autosomal recessive pattern were excluded, as the family tree
suggested an autosomal dominant or X-linked inheritance pattern. A
total of 3 genes, NHS, aldehyde dehydrogenase 18 family
member A1 (ALDH18A1) and myosin heavy chain 9 (MYH9),
were identified as candidates. However, ALDH18A1 and
MYH9 are syndromic cataract genes. ALDH18A1 is one of
the pathogenic genes of autosomal dominant or autosomal recessive
inherited cutis laxa (13), which
is primarily characterized by redundant, sagging, inelastic and
wrinkled skin with variable systemic involvement (14). MYH9 is the pathogenic gene
of Fechtner syndrome, which is characterized by hereditary
nephritis, hearing loss, cataracts, macrothrombocytopenia and
leucocyte inclusions, present in varying combinations (15). In cutis laxa and Fechtner syndrome,
cataracts are one of the clinical manifestations, but are not the
main phenotype. In addition, members of this family with cataracts
did not present other clinical symptoms of cutis laxa and Fechtner
syndrome. Therefore, ALDH18A1 and MYH9 were excluded.
NHS is the pathogenic gene of NHS, which is characterized by
congenital cataracts, and dental and craniofacial abnormalities,
and mental retardation in ~30% of affected males (5,11).
However, there are also certain NHS families with atypical clinical
manifestations, that only exhibit congenital cataracts, and
occasionally other concurrent ocular abnormalities (16). Tug et al (8) suggested that 100% of patients with
NHS have bilateral congenital cataracts, 90% have large anteverted
pinnae and only 65% have dental abnormalities. Therefore,
NHS was identified as the only candidate gene.
| Table II.Candidate mutations identified
following the first two filtering processes. |
Table II.
Candidate mutations identified
following the first two filtering processes.
| Gene | Locus | Inheritance | Mutation | Group of genes in a
cataract review (12) |
|---|
| PEX5 | 12p13.3 | AR | NM_001300789.1;
c.362G>A; p.Arg121His | Syndromic cataract
genes |
| RECQL4 | 8q24.3 | AR | NM_004260.3;
c.2296+1delC | Syndromic cataract
genes |
| NHS | Xp22.13 | XL | NM_001291867.1;
c.302dupA; p.Ala102fs | Syndromic cataract
genes Syndromic genes only with cataract |
| LRP2 | 2q24q31 | AR | NM_004525.2;
c.1199A>G; p.Asn400Ser | Syndromic cataract
genes |
|
ALDH18A1 | 10q24.3 | AD/AR | NM_002860.3;
c.868G>A; p.Gly290Arg | Syndromic cataract
genes |
| MYH9 | 22q13.1 | AD | NM_002473.4;
c.3676C>T; p.Arg1226Trp | Syndromic cataract
genes |
| GALK1 | 17q24 | AR | NM_000154.1;
c.735A>T; p.Glu245Asp | Only cataract
genes |
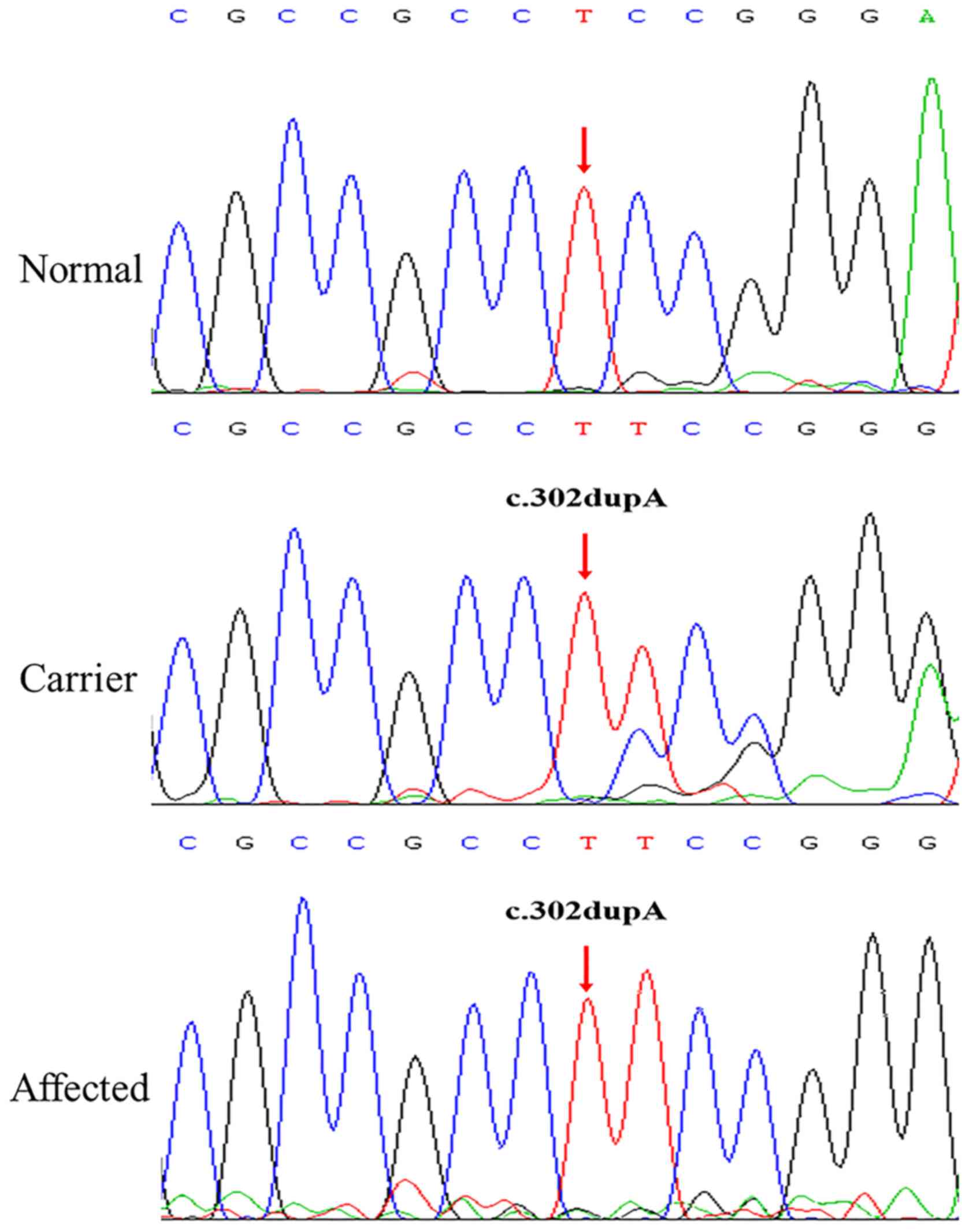
The NHS frameshift mutation (c.302dupA;
p.Ala102fs) identified in the Chinese family examined in the
present study was absent in 1000Genomes, ESP6500 and ExAC
databases, suggesting that it is a novel mutation. Sanger
sequencing verified the presence of the NHS mutation in the
proband (III:2). Subsequent co-segregation analysis among the 5
available individuals in this family detected c.302dupA in the 4
members with congenital cataracts (I:2, II:1, II:3 and III:2), but
not in the unaffected member (II:2) (Fig. 3). The three female members (I:2,
II:1 and II:3) were all heterozygous for this mutation, and
exhibited similar but milder clinical symptoms to the affected male
(III:2). These phenotypic discrepancies and segregation pattern
conform to the disease features of NHS. Therefore, the NHS
frameshift mutation, which altered the open reading frame and
resulted in a premature truncated protein, was the pathogenic
mutation of this family.
Discussion
In the Chinese family examined in the present study,
the proband (III:2), his mother (II:3), maternal aunt (II:1) and
maternal grandmother (I:2) exhibited congenital cataracts with
other ocular abnormalities, but no marked craniofacial or dental
anomalies. Therefore, all congenital cataracts-associated genes
were initially screened. Subsequent to exclusion of high-frequency
variants, genes with recessive inheritance patterns and those
causing syndromic cataracts, a novel NHS frameshift mutation
(c.302dupA; p.Ala102fs) was identified, which altered the open
reading frame and resulted in a premature truncated protein. Sanger
sequencing demonstrated that the mutation co-segregated with the
primary phenotype; congenital cataracts.
NHS, located on Xp22.13, contains 10 exons
and encodes at least 5 isoforms resulting from alternative splicing
(16,17). NHS-A and NHS-1A are the two primary
isoforms, encoding proteins of 1,630 and 1,651 amino acids,
respectively, and are transcribed from exon 1. These 2 isoforms
differ in that NHS-1A contains exon 3a and NHS-A does not (17). NHS-B is transcribed from exon 1b,
is translated from exon 4, and encodes a protein 1,335 amino acids
in length. NHS-C is transcribed and translated from exon 1a and
encodes a protein containing 1,453 amino acids (6–8).
Null mutations in NHS have been identified as causative of
NHS (5,11). At present, ≥40 different NHS
mutations have been described, originating in India, China,
Australia, the United Kingdom, Turkey, Canada and the United States
of America (http://cat-map.wustl.edu/). A number
of these are nonsense mutations or indels, while a few are splice
site mutations or copy number variations.
NHS is conserved in humans and other
vertebrates, including mice (18).
It is expressed in multiple tissues that regulate morphology and
development, including the lens, craniofacial mesenchyme, dental
primordia and brain (19). NHS
isoforms exhibit spatial differences in expression. Isoforms
containing exon 1 are expressed in the epithelia and are localized
to the cell periphery, while isoforms lacking exon 1 are expressed
in non-epithelial tissue and localized to the cytoplasm (7). Therefore, the pleiotropic features of
NHS may be attributed to differences in subcellular localization
and NHS protein isoforms. The N-termini of NHS-A and NHS-1A have a
functional Wiskott-Aldrich syndrome protein-family
verprolin-homologous protein homology domain, which serves an
important role in the regulation of actin remodeling and the
maintenance of cell morphology by interacting with Abl interactor
proteins (17). NHS
mutations in exon 1 were demonstrated to only disrupt the NHS-A and
NHS-1A isoforms, while mutations in exons 3, 5 and 6 appear to
affect ≥1 isoform type (20,21).
The frameshift mutation identified in exon 1 (c.302dupA;
p.Ala102fs) in the present study may cause the NHS disease
phenotype by disrupting NHS-A and NHS-1A function.
In summary, the present study described the clinical
manifestations of an affected male and carrier females in a Chinese
family with NHS, and identified a novel frameshift mutation
(c.302dupA; p.Ala102fs) in the NHS exon 1. The results of
the present study broadens the known pathogenic mutation spectrum
of NHS and will aid genetic counseling for patients with
NHS. The identification of atypical NHS in the present study, with
congenital cataracts and other ocular abnormalities but no marked
craniofacial or dental anomalies, caused by an NHS mutation
suggests that diagnosis may require a combination of clinical
assessment and genetic analysis. There are certain limitations in
the present study. Firstly, a frameshift insertion variant of
NHS that was completely co-segregated with the disorder was
identified. This provided only genetic evidence to the
pathogenicity of the variant. Therefore, future experiments may be
required to confirm these data. For example, whether the insertion
affects the mRNA production, for example nonsense-mediated mRNA
decay, may be verified by reverse transcription polymerase chain
reaction. Concomitantly, the variant produced a truncated protein,
which requires additional confirmation by performing western blot
analysis. Finally, whether this frameshift variant is specific to
this family or common in the Chinese NHS population was not
determined. Future studies will require the analysis of more
patients, to include a range of NHS variants.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science and Technology Basic Work (grant no. 2014FY130100),
National Infrastructure of Chinese Genetic Resources (grant no.
YCZYPT [2017] 01), the National Key Research and Development
Program of China (grant no. 2016YFC1000307-6), and Guangxi Science
and Technology Project (grant no. ZY18164008).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MW, AQ, XM and BW were involved in the conception
and design of the study. HM, and KW were responsible for the
acquisition of data. MW, AQ, HM and KW were responsible for
analysis and interpretation of the data. MW and AQ drafted the
manuscript, and XM and BW critically revised the manuscript for
important intellectual content. All authors read and approved the
final version to be published.
Ethics approval and consent to
participate
Approval of the present study was obtained from the
Ethics Committee of the National Research Institute for Family
Planning. All procedures adhered to the guidelines of the
Declaration of Helsinki and the Association for Research in Vision
and Ophthalmology statement on human subjects. Written informed
consent was obtained from the patients.
Patient consent for publication
Written informed consent for publication of clinical
details and clinical images was obtained from the patients.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NHS
|
Nance-Horan Syndrome
|
|
WES
|
whole exome sequencing
|
References
|
1
|
Nance WE, Warburg M, Bixler D and
Helveston EM: Congenital X-linked cataract, dental anomalies and
brachymetacarpalia. Birth Defects Orig Artic Ser. 10:285–291.
1974.PubMed/NCBI
|
|
2
|
Walpole I, Hockey A and Nicoll A: The
Nance-Horan syndrome. J Med Genet. 27:632–634. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li H, Yang L, Sun Z, Yuan Z, Wu S and Sui
R: A novel small deletion in the NHS gene associated with
Nance-Horan syndrome. Sci Rep. 8:23982018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chograni M, Rejeb I, Jemaa LB, Châabouni M
and Bouhamed HC: The first missense mutation of NHS gene in a
Tunisian family with clinical features of NHS syndrome including
cardiac anomaly. Eur J Hum Genet. 19:851–856. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brooks SP, Ebenezer ND, Poopalasundaram S,
Lehmann OJ, Moore AT and Hardcastle AJ: Identification of the gene
for Nance-Horan syndrome (NHS). J Med Genet. 41:768–771. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Coccia M, Brooks SP, Webb TR,
Christodoulou K, Wozniak IO, Murday V, Balicki M, Yee HA,
Wangensteen T, Riise R, et al: X-linked cataract and Nance-Horan
syndrome are allelic disorders. Hum Mol Genet. 18:2643–2655. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li A, Li B, Wu L, Yang L, Chen N and Ma Z:
Identification of a novel NHS mutation in a Chinese family with
Nance-Horan syndrome. Curr Eye Res. 40:434–438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tug E, Dilek NF, Javadiyan S, Burdon KP
and Percin FE: A Turkish family with Nance-Horan Syndrome due to a
novel mutation. Gene. 525:141–145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lewis RA, Nussbaum RL and Stambolian D:
Mapping X-linked ophthalmic diseases. IV. Provisional assignment of
the locus for X-linked congenital cataracts and microcornea (the
Nance-Horan syndrome) to Xp22.2-p22.3. Ophthalmology. 97:110–121.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bixler D, Higgins M and Hartsfield J Jr:
The Nance-Horan syndrome: A rare X-linked ocular-dental trait with
expression in heterozygous females. Clin Genet. 26:30–35. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burdon KP, McKay JD, Sale MM,
Russell-Eggitt IM, Mackey DA, Wirth MG, Elder JE, Nicoll A, Clarke
MP, FitzGerald LM, et al: Mutations in a novel gene, NHS, cause the
pleiotropic effects of Nance-Horan syndrome, including severe
congenital cataract, dental anomalies, and mental retardation. Am J
Hum Genet. 73:1120–1130. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Messina-Baas O and Cuevas-Covarrubias S:
Inherited congenital cataract: A guide to suspect the genetic
etiology in the cataract genesis. Mol Syndromol. 8:58–78. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nozaki F, Kusunoki T, Okamoto N, Yamamoto
Y, Miya F, Tsunoda T, Kosaki K, Kumada T, Shibata M and Fujii T:
ALDH18A1-related cutis laxa syndrome with cyclic vomiting. Brain
Dev. 38:678–684. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bhola PT, Hartley T, Bareke E; Care4Rare
Canada Consortium, ; Boycott KM, Nikkel SM and Dyment DA: Autosomal
dominant cutis laxa with progeroid features due to a novel, de novo
mutation in ALDH18A1. J Hum Genet. 62:661–663. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gershoni-Baruch R, Baruch Y, Viener A and
Lichtig C: Fechtner syndrome: Clinical and genetic aspects. Am J
Med Genet. 31:357–367. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun W, Xiao X, Li S, Guo X and Zhang Q:
Exome sequencing of 18 Chinese families with congenital cataracts:
A new sight of the NHS gene. PLoS One. 9:e1004552014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brooks SP, Coccia M, Tang HR, Kanuga N,
Machesky LM, Bailly M, Cheetham ME and Hardcastle AJ: The
Nance-Horan syndrome protein encodes a functional WAVE homology
domain (WHD) and is important for co-ordinating actin remodelling
and maintaining cell morphology. Hum Mol Genet. 19:2421–2432. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding X, Patel M, Herzlich AA, Sieving PC
and Chan CC: Ophthalmic pathology of Nance-Horan syndrome: Case
report and review of the literature. Ophthalmic Genet. 30:127–135.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shoshany N, Avni I, Morad Y, Weiner C,
Einan-Lifshitz A and Pras E: NHS gene mutations in ashkenazi jewish
families with Nance-Horan Syndrome. Curr Eye Res. 42:1240–1244.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramprasad VL, Thool A, Murugan S,
Nancarrow D, Vyas P, Rao SK, Vidhya A, Ravishankar K and
Kumaramanickavel G: Truncating mutation in the NHS gene: Phenotypic
heterogeneity of Nance-Horan syndrome in an Asian Indian family.
Invest Ophthalmol Vis Sci. 46:17–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sharma S, Ang SL, Shaw M, Mackey DA, Gécz
J, McAvoy JW and Craig JE: Nance-Horan syndrome protein, NHS,
associates with epithelial cell junctions. Hum Mol Genet.
15:1972–1983. 2006. View Article : Google Scholar : PubMed/NCBI
|