Introduction
As a public health problem worldwide, ~71 million
people are chronically infected with hepatitis C virus (HCV),
accounting for ~1% of the global population (1–3). HCV
is a small positive-sense single-stranded RNA virus and belongs to
the genus Hepacivirus of the family Flaviviridae (4,5),
which includes certain well-known neurotropic viruses, including
yellow fever, dengue, zika and tickborne encephalitis viruses.
Human hepatocytes are the main target cells for HCV
infection, which can cause life-threatening liver diseases,
including chronic hepatitis, fibrosis and hepatocellular carcinoma
(6–9). In addition, extra-hepatic
manifestations, including abnormalities in the central nervous
system (CNS), are also observed in HCV-infected patients. Previous
studies have reported that 50–60% of individuals
chronically-infected with HCV had increased incidence of nervous
system disorders, including chronic fatigue syndrome, depression
and cognitive dysfunction, which may persist (10–13),
even after spontaneous or treatment induced peripheral virus
clearance. Furthermore, two studies (9,14)
have identified that HCV can target microglia cells and astrocytes
in the CNS, as HCV RNA and proteins were detected in these two
types of CNS cells using laser capture microscopy, but not detected
in oligodendrocytes or neurons. In addition, alteration of striatal
dopaminergic neurotransmission has been reported in HCV-infected
subjects (15). Taken together,
emerging evidence suggests that the CNS appears vulnerable to HCV
infection.
HCV entry involves interactions between the viral
surface-resident glycoproteins (E1 and E2) and several host factors
(16–19): Scavenger receptor class B type I
(SR-B1), CD81, claudin-1 and occluding. Many brain-derived cell
lines are known to express the above receptors (20,21).
Previous studies have reported the cell culture-derived HCV
replication in primary astrocytes, human SVG astrocyte cell line
and SKNMC peripheral neuroblastoma cell line (22,23).
However, there is no report on HCV infection in glioblastoma cells.
The origin of glioblastoma is not clear, as neural stem cells,
glial progenitors (including oligodendrocyte progenitor cells) and
astrocytes may all be associated with glioma development (24). In the current study, it was
demonstrated that serum-derived HCV could transiently replicate in
the human glioblastoma cell line SF268 and activated the AKT
serine/threonine kinase/glycogen synthase kinase 3β (GSK3β)
pathway.
Materials and methods
Cell culture
Human glioblastoma cell line SF268 (25–27)
was used for current study (a gift from Professor Bao Zhang,
Nanfang Hospital, Southern Medical University, Nanfang, China; the
cell line was authenticated by STR profiling). SF268 cells
originate from human neuromuscular cells and developed as
glioblastoma cell line/anaplastic astrocytoma (non-epithelial) cell
line (28–30). SF268 were cultured at 37°C in a
humidified incubator with 5% CO2 and maintained in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 100 U/ml
penicillin G and 100 U/ml streptomycin (Thermo Fisher Scientific,
Inc.) and 10% fetal bovine serum.
Serum samples
A total of 128 chronic HCV patients (aged 24–59;
63.3% were male) and 14 healthy donors (age 26–51; 57.1% were male)
were enrolled from January 2016 to December 2017 in Nanfang
Hospital (Southern Medical University, Guangzhou, China). Blood
specimens were collected and serum samples were stored at −80°C
prior to further analysis. Laboratory results were recorded from
the patient database; demographics and commercial blood donation
history were obtained using a standardized questionnaire. Potential
HCV infection routes were identified for each patient, including
blood transfusion, intravenous drug use and procedures using shared
medical equipment, such as dental treatment, hemodialysis,
non-sterile tattooing or piercing, unsafe acupuncture, etc.
(Table I).
 | Table I.Demographics and risk factors of
patients associated with hepatitis C virus subtypes. |
Table I.
Demographics and risk factors of
patients associated with hepatitis C virus subtypes.
| Patient ID | No. | Age | Gender | Viral RNA
(IU/ml) | Viral genotype | Core | Risk factor |
|---|
| JX17ZCF | 1 | 43 | Female |
1.23×106 | 2a | + | Othera |
| JX17LXG | 2 | 38 | Male |
1.77×106 | 3a | + | IDUb |
| JX17HLY | 3 | 57 | Male |
2.23×106 | 1b | + | Bloodc |
| JX17LSB | 4 | 29 | Male |
3.20×106 | 6a | + | IDU |
| JX17LMS | 5 | 62 | Male |
7.39×106 | 3a | + | Blood |
| JX16GSQ | 6 | 43 | Male |
1.29×107 | 1b | + | Blood |
| JX16WSJ | 7 | 39 | Male |
3.47×106 | 6a | + | IDU |
Diagnosis of chronic HCV infection was based on
detection of HCV RNA in serum or plasma by using the Cobas
AmpliPrep/Cobas TaqMan HCV test, version 2.0 (lower limit of
detection, 15 IU/ml; Roche Diagnostics GmbH, Mannheim, Germany),
which was used according to the manufacturer's protocols (31) and guidelines (3,32,33).
Samples were determined to be HCV positive via a HCV assay, which
was conducted as described (34).
Anti-HCV antibodies were detected using the Architect HCV assay
(Abbott Japan Co., Ltd., Tokyo, Japan). Exclusion criteria were as
follows: Coinfection with HBV or HIV, renal transplantation,
discontinued treatment, incomplete data, or loss in follow-up
during 12 weeks post-treatment.
The use of patient serum samples were both approved
by the ethical Committee of Nanfang Hospital, Southern Medical
University (no. NFEC-2014-079) and the Medical Ethics Committee at
the Zhongshan School of Medicine, Sun Yat-sen University (no.
2014-072). The experiments were performed in accordance with the
approved guidelines and informed consent was obtained from all
subjects.
HCV infection
Serum was filtered through 0.22-µm polarized filters
prior to incubation with the cells. SF268 cells were seeded in
6-well plates at a density of 4×105 cells/well for 24 h prior to
inoculation. After washing three time with 1X PBS, 200 µl
HCV-positive serum and 1 ml serum-free culture media were added to
each well. The control cells were mock-infected with the
HCV-negative serum from a healthy donor. At 12 h post-infection,
the medium was replaced with 2–3 ml fresh growth medium. The cells
were maintained and harvested at the indicated time points (day 2,
4, 6, 8 and 10 post-infection). The supernatants were collected
every other day and stored at −80°C prior to analysis. Cells were
passaged every 2 days.
Immunofluorescence analysis
Infected cells (6×103 cells/well) in 96-well plate
were fixed with 3.7% paraformaldehyde for ~10 min at 37°C,
permeabilized with 0.2% Triton X-100 in PBS for 10–15 min, and
blocked with 1% bovine serum albumin for 40–45 min at room
temperature, followed by overnight incubation with anti-HCV core
monoclonal antibody C7-50 (1:400; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA; sc-57800) at 4°C. The cells were washed three
times with PBS, incubated with Alexa Fluor® 488 goat
anti-mouse IgG (1:250; H+L; Thermo Fisher Scientific, Inc; A28175)
and DAPI (10 µg/ml) at room temperature for 2 h. The percentage of
HCV infected cells in the wells was estimated using a laser
scanning confocal microscope (LSM 710; Leica Microsystems GmbH,
Wetzlar, Germany).
Reverse transcription-polymerase chain
reaction (RT-PCR) and quantitative detection of HCV RNA
Serum HCV RNA was extracted from plasma (140 µl)
using using the QIAamp Viral RNA mini Kit (Qiagen GmbH, Hilden,
Germany) according to the manufacturer's protocol. cDNA was
synthesized from 20 µl RNA with Superscript III-First-Strand
Synthesis System (Thermo Fisher Scientific, Inc.) with the
following conditions: 25°C for 10 min, 50°C for 50 min, then a
final cycle at 85°C for 5 min. Primers that contained partial E1
region (reference strain H77 nucleotide positions 729–1,322) for
nested-PCR were described previously (23): Outer forward (OF1),
5′-TTGGGTAAGGTCATCGATACCC-3′; outer reverse (OR2),
5′-TGATGTGCCAACTGCCGTTGGT-3′; inner forward (IF3),
5′-TTCGCCGACCTCATGGGGTACAT-3′; and inner reverse (IR4),
5′-GGACCAGTTCATCATCATATCCCA-3′. LA Taq (RR002A; Takara Bio, Inc.,
Otsu, Japan) was used for nested PCR. First round PCR (primers OF1
and OR2) was performed with the following conditions: 94°C for 2
min, 35 cycles of 94°C for 30 sec, 58°C for 1 min and 72°C for 40
sec, then a final cycle at 72°C for 7 min. Second round PCR
(primers IF3 and IR4) was executed with the same conditions with
the annealing temperature was changed to 56°C for 35 sec. The
products were separated on a 1.0% agarose gel dyed with 2.5 µl
Gel-red (1:10,000; cat. no. 41000; Biotium, Inc., Fremont, CA,
USA), and the positive samples (PCR strip located at 594 bp) were
subjected to sequencing (Shanghai Invitrogen Biotechnology Co.,
Ltd., Shanghai, China). To avoid potential contamination, all
experimental procedures were all performed in parallel with the
appropriate positive and negative controls. Sequencing was
performed with IF3 and IR4 primers in both directions using ABI
Prism Big Dye 3.0 terminators on an ABI Prism 3500 genetic analyzer
(Applied Biosystems; Thermo Fisher Scientific, Inc.). HCV RNA in
the cell culture supernatant was detected quantitatively as
aforementioned.
Western blotting and antibodies
Western blotting analysis was performed as
previously described with minor modifications (35). At the indicated time points after
infection, cells were washed with PBS and incubated with lysis
buffer (A8261, Promega Corporation, Madison, WI, USA) for 45 min on
ice and boiled for 10 min. The protein concentration was determined
using the Bicinchoninic Acid Protein Assay Kit (cat. no. 2634,
Bio-Rad Laboratories, Inc., Hercules, CA, USA) and equal amounts of
protein samples (40 µg) were run through 10% Bis-Tris
SDS-polyacrylamide precasted gels for 1 h 30 min at 150 V.
Subsequently, separated proteins were transferred to the
polyvinylidene difluoride membrane (Bio-Rad Laboratories, Inc.) by
wet electroblotting (XCell SureLock Mini-Cell; Invitrogen; Thermo
Fisher Scientific, Inc.) at constant current for 1 h. Membranes
were then washed with PBS plus 1% Tween-20, and blocked with PBS
plus 1% Tween-20 and 3% bovine serum albumin (cat. no. 11021029,
Gibco; Thermo Fisher Scientific, Inc.) for 1 h at room temperature.
The membranes were incubated overnight at 4°C with anti-HCV core
C7-50 (1:200; sc-57800, Santa Cruz Biotechnology, Inc.) or AKT
(1:1,000; cat. no. 4691, Cell Signaling Technology, Inc., Danvers,
MA, USA), phosphorylated (p)AKT (1:1,000; cat. no. 4060; Cell
Signaling Technology, Inc.), GSK3β (1:1,000; cat. no. 9315; Cell
Signaling Technology, Inc.) and pGSK3β (1:1,000; cat. no. 5558;
Cell Signaling Technology, Inc.), or anti β-tubulin (1:1,000; cat.
no. 86298; Cell Signaling Technology, Inc.) with agitation. Goat
anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody,
Alexa Fluor Plus 680 (1:40,000; a32729; Thermo Fisher Scientific,
Inc.) or Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary
Antibody, Alexa Fluor Plus 680 (1:40,000; a32734; Thermo Fisher
Scientific, Inc.) were added to the membrane and incubated for 1 h
at room temperature prior to treatment with an enhanced
chemiluminescence kit (GE Healthcare, Chicago, IL, USA). Finally,
protein bands were visualized using the Odyssey infrared imaging
system (LI-COR Biosciences, Lincoln, NE, USA), while the
quantification of immunodetected protein bands was performed using
ImageJ software (Version: k 1.45; National Institutes of Health,
Bethesda, MD, USA).
Statistical analysis
Statistical analysis was performed with the SPSS
software, version 22.0 (IBM Corp., Armonk, NY, USA). At least three
independent experiments were performed. For descriptive purposes;
quantitative variables are presented as either the mean ± standard
deviation or medians and ranges, as appropriate. Categorical
variables are presented as number and percentages. Statistical
analysis was performed with two-tailed unpaired Student's t-test.
P<0.05 was considered to indicated a statistically significant
difference.
Results
Demographic and clinical
characteristics
Of the seven samples tested, six (85.7%) were from
male patients (Table I). Their
ages ranged from 29 to 62 years with a mean (± standard deviation)
of 44±11.4 years. HCV RNA levels (IU/ml) varied between 1.23×106
and 1.29×107. HCV genotypes included 1b, 2a, 3a and 6a. The
transmission routes are listed in Table I.
HCV replication in human glioblastoma
cell line SF268
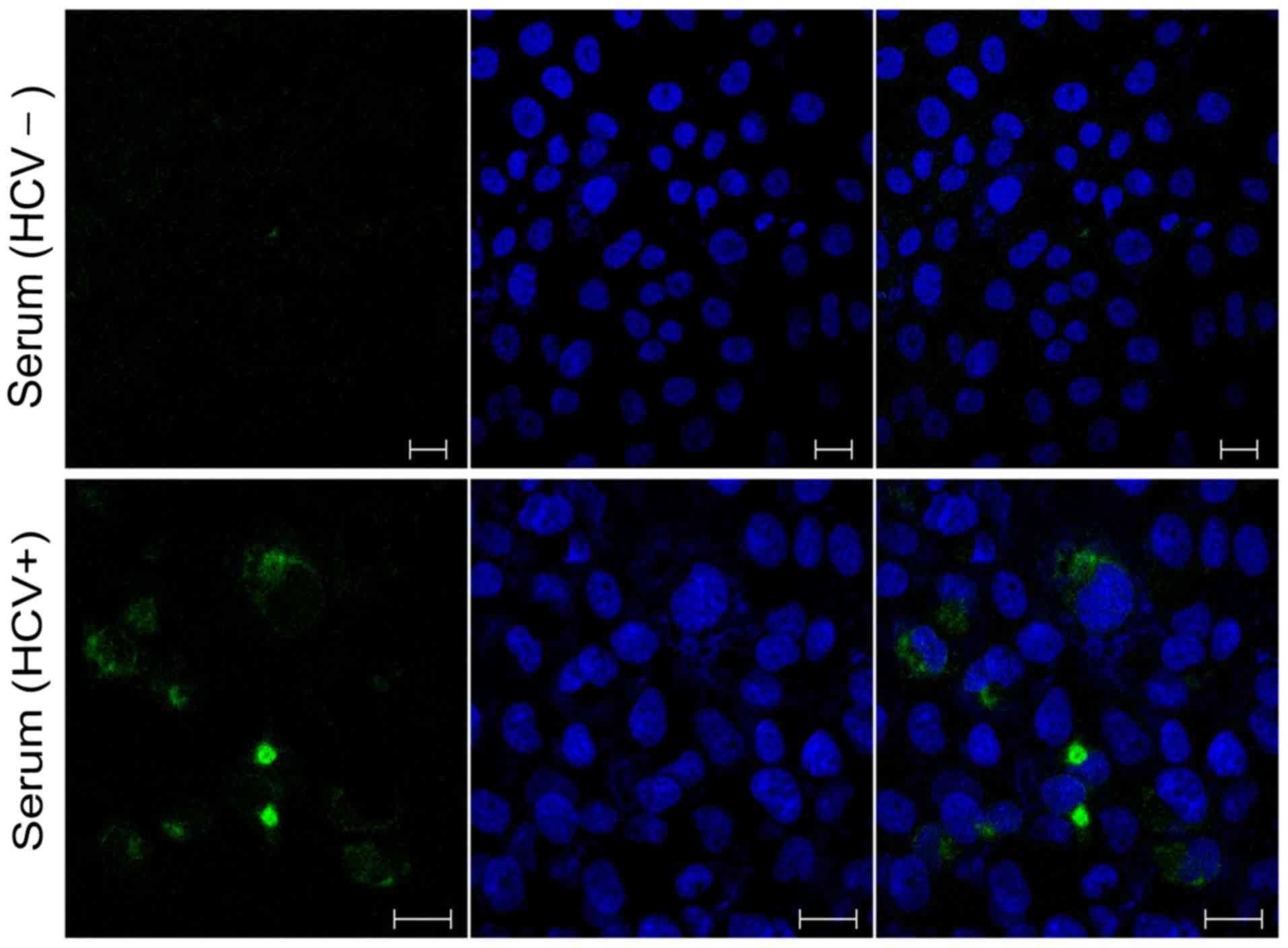
HCV core protein was detected in cells infected with
HCV-positive serum on the day 4 post-infection (~1% of the cells
exhibited efficient HCV infection; Fig. 1). Total RNA was extracted from
SF268 cells exposed to with HCV-positive or -negative serum, and
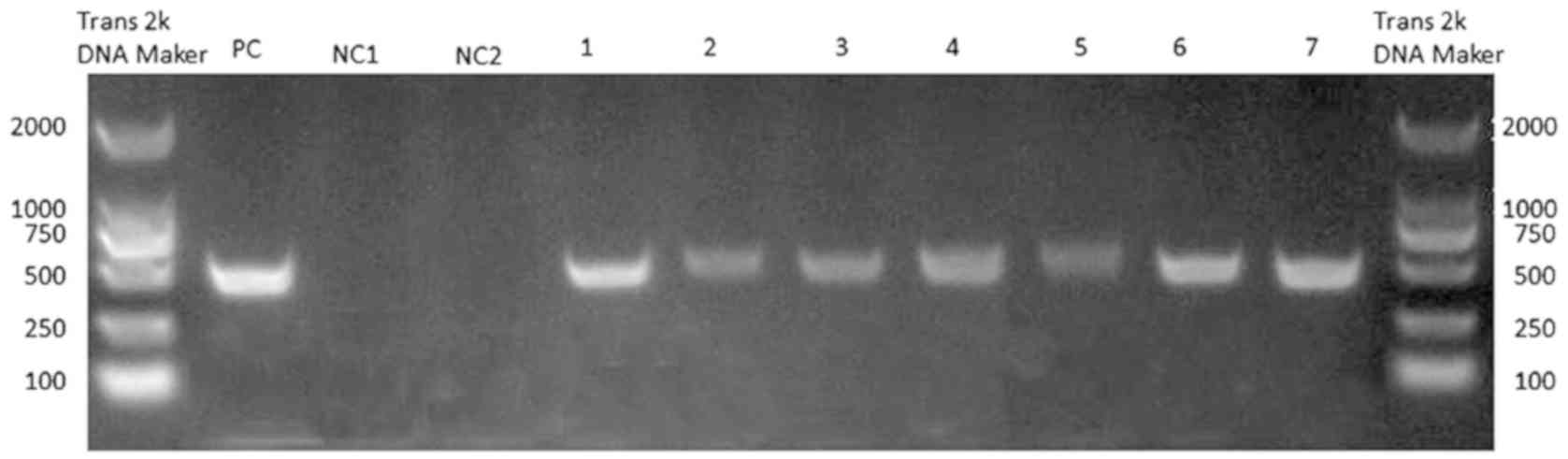
reverse transcribed to cDNA. HCV-negative strand-specific PCR was
performed to detect the virus replicative intermediate in the
SF268. Each of seven HCV-positive serum infected SF268 cells was
HCV RNA positive on day 4, while it remained negative in the
HCV-negative serum-infected SF268 (Fig. 2). qPCR was performed in cell
culture supernatant-derived RNA samples in order to quantify viral
RNA load and affirm RNA replication. The mean RNA levels (IU/ml,
log10) on day 4 post-infection were between 2.68 and 5.00 (Fig. 3) in seven medium samples. HCV RNA
and core protein were undetectable when the culture was extended to
day 6, 8 and 10 (data not shown). These data indicate that human
SF268 glioblastoma cell line can be infected with serum-derived
HCV, but the infection was transient.
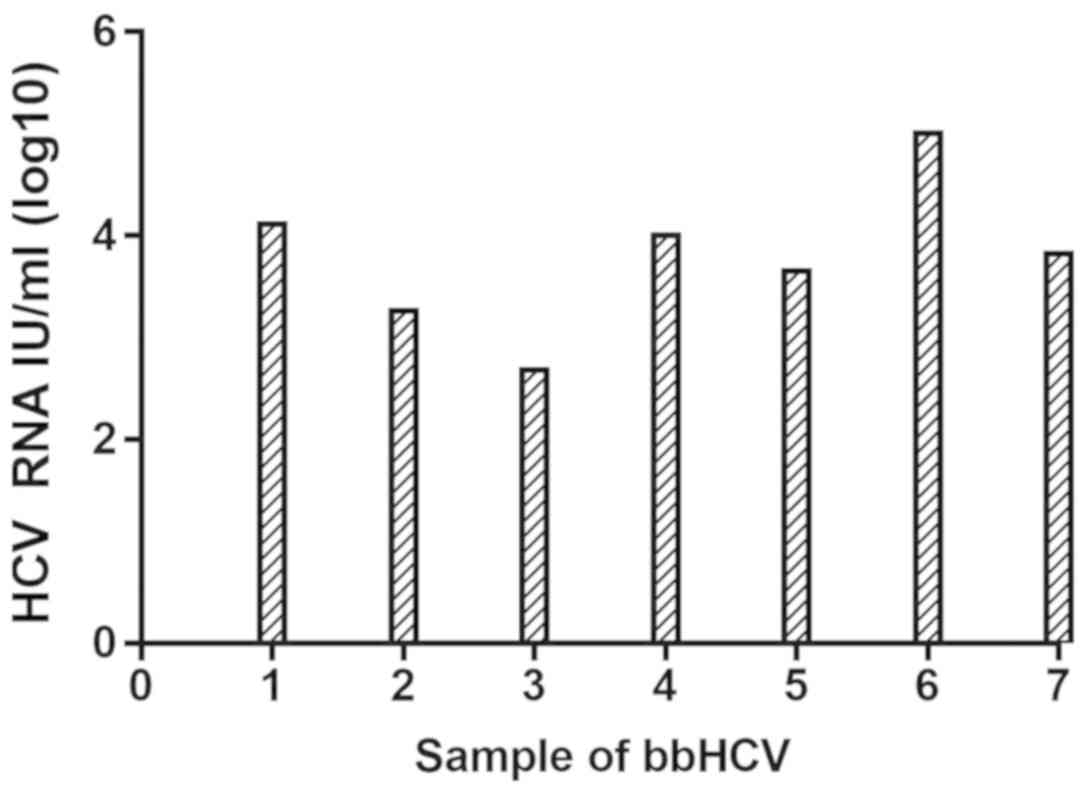 | Figure 3.qPCR for HCV RNA. On day 4
post-infection, cell supernatant was collected and subjected to
qPCR for the detection of HCV RNA. The mean HCV RNA levels (IU/ml,
log10) on day 4 post-infection were 4.11 for JX17ZCF (patient 1),
3.26 for JX17LXG (patient 2), 2.68 for JX17HLY (patient 3), 4.01
for JX17LSB (patient 4), 3.65 for JX17LMS (patient 5), 5.00 for
JX16GSQ (patient 6) and 3.82 for JX16WSJ (patient 7). bbHCV,
blood-borne HCV; qPCR, quantitative polymerase chain reaction; HCV,
hepatitis C virus. |
Activation of phosphoinositide
3-kinase (PI3K)-AKT signaling
Previous studies reported that HCV utilizes the
PI3K/AKT pathway to facilitate viral entry and replication in human
hepatocytes (10,36). However, little is known about the
effect of HCV infection on the AKT signaling pathway in neuronal
cell lines. To explore the effect of HCV infection on AKT pathway
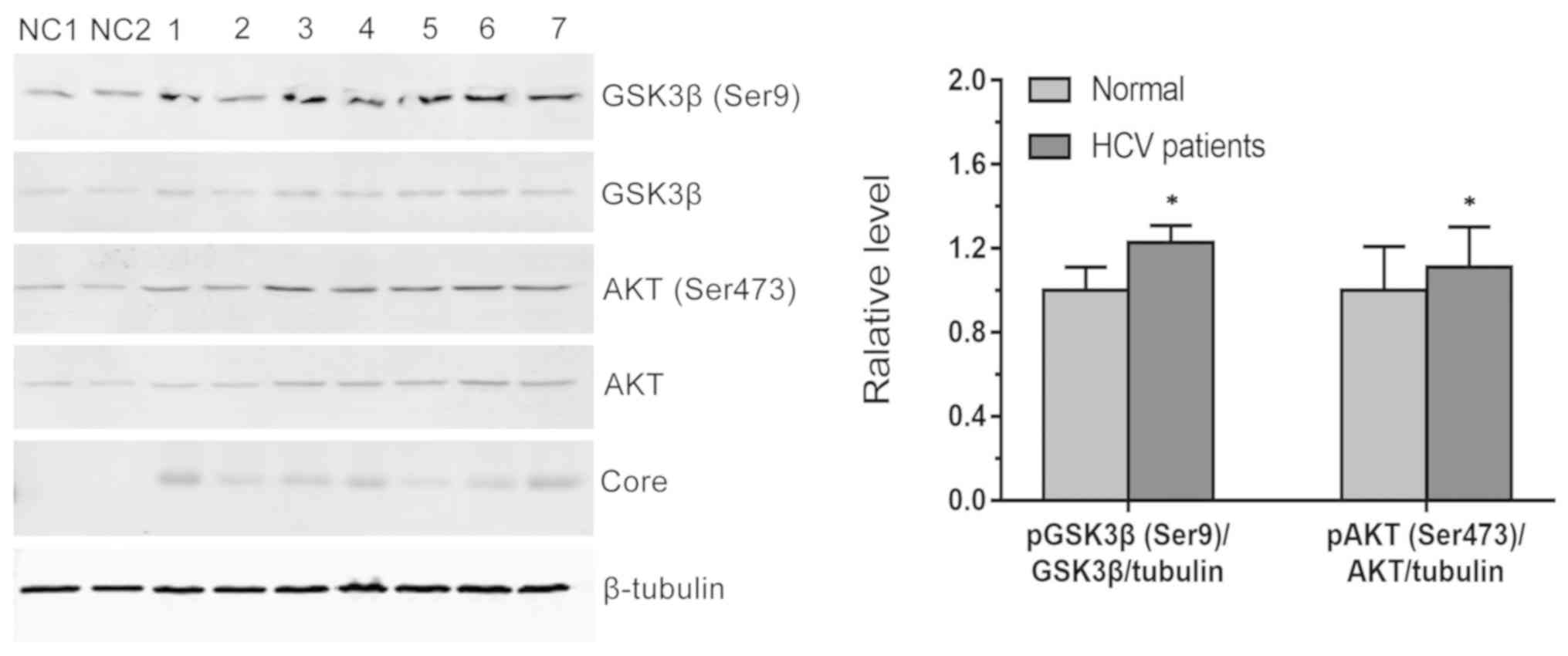
in neuronal cell lines, AKT and pAKT (Ser473) protein levels were
assessed at day 4 post-infection. As shown in Fig. 4, AKT (Ser473) phosphorylation was
increased cells expressing HCV core proteins. Phosphorylation of
GSK3β, a downstream target of AKT, was also activated by HCV
infection. These data demonstrate that HCV infection of
glioblastoma cells activated the AKT pathway and downstream
signaling.
Discussion
To the best our knowledge, this is the first study
demonstrating that the human glioblastoma cell line SF268 can be
infected by HCV, and to detect activation of the AKT/GSK3β pathway
following HCV infection.
Glioblastoma (World Health Organization grade IV
glioma) is the most lethal primary brain tumor, with increasing
prevalence. It was previously reported that four human glioblastoma
multiform tumor cell lines, U373MG, T98G, DBTRG and U87MG, could
not be infected with cell culture-derived HCV, even though these
cell lines express certain key factors involved in HCV entry
(scavenger receptor class B type 1 and Claudin-1) (23). In the current study, HCV-inoculated
SF268 cells were positive for HCV RNA on day 4 post-infection;
however, the HCV RNA titers were lower than the input viral RNA
levels by ~3 logs, suggesting two possible scenarios: i) Residual
HCV inoculum was not completely removed; ii) cells were infected,
but with low replication. To distinguish the two possibilities, HCV
core protein expression was also assessed in the HCV-infected SF268
on day 4, 6, 8 and 10 post-infection. Supporting with the PCR
amplification results, HCV core protein was detectable on day 4
post-infection, suggesting that there was established HCV
replication; however, only ~1% of the cells exhibited efficient HCV
infection and both HCV RNA and core protein expression became
undetectable beyond day 4, indicating that HCV infection in SF268
was transient and the replication was low. Overexpression of
microRNA-122 (miR-122) in SF268 cells may serve to improve
transfection efficiency in future studies, as miR-122 has been
reported to stimulate HCV replication (37). In contrast with the study by Bürgel
et al (23), the data of
the current study support that SF268 glioblastoma cells can be
infected by serum-derived HCV. This discrepancy could be due to the
difference between serum-derived HCV and cell culture-derived HCV
interactions with cells, the concentration of viral particles used
in our study was much lower and there have been no previous studies
using HCV with the SF268 cell line. However, the current data
indicate that human SF268 glioblastoma cell line supports HCV
replication, and this response may fluctuate with respect to
different patient samples, as the presence of various known and
unknown inflammatory factors such as tumor necrosis factor-α
(TNF-α) in the patient serum may promote viral entry. Other studies
have reported HCV replication in primary astrocytes inoculated with
serum-derived HCV (38).
Additionally, other studies have indicated that HCV RNA sequences
isolated from brain and cerebrospinal fluid samples were different
to the serum-derived viruses, suggesting that HCV could replicate
in the brain and adapt in a tissue-specific manner (39–41).
HCV infection has been associated with the incidence of Parkinson's
disease in a number of epidemiological studies (42–44).
Together, these findings suggest that HCV may spread across the
brain-blood barrier and infect neurological cells, which may have
important implications for viral neurological pathogenesis.
It has been reported that HCV infection activates
AKT at the early stage of infection to enhance its entry, and this
activation is mediated by interaction between the HCV E2 envelope
protein and its co-receptors, Claudin-1 and CD81 (36). In line with this observation, the
PI3K-AKT signaling pathway was activated in HCV-infected SF268
cells in the current study. Other viruses, such as paramyxoviruses
has also been reported to regulate the pathway to promote their
replication (45,46). Influenza A virus also activates
this pathway to enhance viral replication at a post-entry step, and
its NS1 protein can also activate PI3K to suppress apoptosis
(47). Human cytomegalovirus
(48), coxsackievirus B3 (49), and varicella-zoster virus (50) also require PI3K/AKT activation for
efficient infection. It has been reported that the HCV NS5A protein
binds to PI3K and activates the signaling pathway. In addition, the
HCV core protein can induce the AKT Ser473 phosphorylation, but
with no apparent effect on Thr308 phosphorylation, and also impair
the insulin signaling pathway (51). The data of the current study
revealed that AKT phosphorylation in SF268 cells occurred at day 4
post-infection in parallel with detection core protein, indicating
an association between HCV infection and activation of AKT.
The activation of the PI3K/AKT pathway may also lead
to CNS immune responses and subsequently cause neuronal injury. A
previous study investigated the effects of HCV core protein on
microglia, neurons and astrocytes (38), indicating that HCV core protein
triggered immune activation of glial cells and was neurovirulent by
inducing the expression of pro-inflammatory cytokines [including
interleukin (IL)-1β, IL-6 and TNF-α] and chemokines (including
C-X-C motif chemokine 10 and interleukin-8). The current study
suggests that the PI3K/AKT pathway was activated by HCV infection
in SF268 cells, a human glioblastoma cell lines derived from glial
cells. This serum-derived HCV infection of SF268 cells may provide
a novel culture system for the study of HCV-induced neuro-immune
activation and potential neuronal injury.
To the best of our knowledge, no previous studies
have investigated the role of HCV in glioblastoma tumorigenesis;
however, the findings of the present study suggest that HCV may
have a role in the carcinogenesis of glioblastoma by inducing a
chronic inflammatory state, which creates a pro-carcinogenic
environment. Long-term infection with HCV creates an oncogenic
environment through a combination of viral protein expression,
persistent inflammation, oxidative stress and chronically
deregulated signaling events, which accumulate to cause genetic
instability. According to the study by Bokemeyer et al
(52), HCV infection may induce
neuroinflammation and carcinogenesis as choline, creatine and
myo-inositol, which are usually indicate glial activation and
macrophage infiltration in chronic inflammation, were all
significantly higher in HCV-infected patients than in controls.
Additionally, compared with HCV-negative controls, HCV-positive
patients were demonstrated to exhibit significantly higher levels
of pro-inflammatory cytokines, including IL-1α, IL-1β, TNF-α, IL-12
and IL-18 (53). Furthermore,
post-mortem studies of HCV quasispecies and replicative
intermediates also indicated that microglia may be an infection
locus, leading to neuroinflammatory activity (54,55).
Finally, HCV core protein has been reported to trigger activation
of the extracellular signal-related kinase/signal transducer and
activator of transcription 3 system via Toll-like receptor 2 in the
CNS (56), which may have a role
in neurodegeneration. In summary, HCV may have a role in
glioblastoma carcinogenesis by inducing a chronic inflammatory
state, and this may be an important research direction.
There are certain limitations in the present study.
Firstly, the sample size was relatively small as this was a single
center study, though HCV infection of SF268 cells was clearly
demonstrated. Additionally, identification of cellular factors that
restricted long-term HCV infection in SF268 cells was not performed
and will be a focus of further studies. Furthermore, additional
studies are required investigate HCV infection of neurons, as
glioblastoma cell lines are not neuronal cells, although the SF268
cell line appears to be a useful tool for studying HCV infection in
the CNS. The current study suggests novel methods for establishing
a HCV infection system to investigate HCV neuropathogenesis.
In summary, to the best of our knowledge, the
current study is the first to demonstrate transient HCV infection
in a human glioblastoma cell line. The results provide evidence
that HCV is able to enter and spread in a non-liver cancer cell
line in vitro and may explain extra-hepatic symptoms in
patients with chronic HCV infection. Additionally, the PI3K-AKT
signaling pathway was activated in HCV-infected SF268 cells,
suggesting that SF268 cells may be used as a model for
investigating HCV-nerve cells interactions.
Acknowledgements
Not applicable.
Funding
This study was supported by the grants from the
National Natural Sciences Foundation of China (grant nos. 81470856,
81772923 and 31700150). The funding agencies had no role in study
design, data collection and analysis, decision to publish, or
preparation of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors contributions
GY, ZZ, YYZ, YPL and YZ conceived and designed the
research. GY, JL and CH collected the samples. GY, LR, ZZ and JL
performed the experiments. GY, LR, ZZ, LM and MC analyzed
experimental results. GY and ZZ wrote the manuscript. YZ, YYZ and
YPL supervised the project and edited the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
ORCID: Guosheng Yuan, orcid.org/0000-0002-2118-4553; Yuanping Zhou.
orcid.org/0000-0002-2493-396X.
References
|
1
|
European Association for the Study of the
Liver. Electronic address, . easloffice@easloffice.eu: EASL
recommendations on treatment of hepatitis C 2016. J Hepatol.
66:153–194. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mohd Hanafiah K, Groeger J, Flaxman AD and
Wiersma ST: Global epidemiology of hepatitis C virus infection: New
estimates of age-specific antibody to HCV seroprevalence.
Hepatology. 57:1333–1342. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
AASLD/IDSA HCV Guidance Panel: Hepatitis C
guidance: AASLD-IDSA recommendations for testing, managing, and
treating adults infected with hepatitis C virus. Hepatology.
62:932–954. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dorner M, Horwitz JA, Donovan BM, Labitt
RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, et
al: Completion of the entire hepatitis C virus life cycle in
genetically humanized mice. Nature. 501:237–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lorenz IC, Marcotrigiano J, Dentzer TG and
Rice CM: Structure of the catalytic domain of the hepatitis C virus
NS2-3 protease. Nature. 442:831–835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ahmad J, Eng FJ and Branch AD: HCV and
HCC: Clinical update and a review of HCC-associated viral mutations
in the core gene. Semin Liver Dis. 31:347–355. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vandenbulcke H, Moreno C, Colle I, Knebel
JF, Francque S, Sersté T, George C, de Galocsy C, Laleman W,
Delwaide J, et al: Alcohol intake increases the risk of HCC in
hepatitis C virus-related compensated cirrhosis: A prospective
study. J Hepatol. 65:543–551. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang XF and Korangy F: Intrahepatic
landscape of regulatory T-cell subsets in chronically HCV-infected
patients with cirrhosis and HCC. Hepatology. 60:1461–1462. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wilkinson J, Radkowski M and Laskus T:
Hepatitis C virus neuroinvasion: Identification of infected cells.
J Virol. 83:1312–1319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mannová P and Beretta L: Activation of the
N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: Control of cell
survival and viral replication. J Virol. 79:8742–8749. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mariotto S, Ferrari S and Monaco S:
HCV-related central and peripheral nervous system demyelinating
disorders. Inflamm Allergy Drug Targets. 13:299–304. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pham TN, MacParland SA, Mulrooney PM,
Cooksley H, Naoumov NV and Michalak TI: Hepatitis C virus
persistence after spontaneous or treatment-induced resolution of
hepatitis C. J Virol. 78:5867–5874. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Radkowski M, Gallegos-Orozco JF, Jablonska
J, Colby TV, Walewska-Zielecka B, Kubicka J, Wilkinson J, Adair D,
Rakela J and Laskus T: Persistence of hepatitis C virus in patients
successfully treated for chronic hepatitis C. Hepatology.
41:106–114. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Letendre S, Paulino AD, Rockenstein E,
Adame A, Crews L, Cherner M, Heaton R, Ellis R, Everall IP, Grant
I, et al: Pathogenesis of hepatitis C virus coinfection in the
brains of patients infected with HIV. J Infect Dis. 196:361–370.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weissenborn K, Ennen JC, Bokemeyer M, Ahl
B, Wurster U, Tillmann H, Trebst C, Hecker H and Berding G:
Monoaminergic neurotransmission is altered in hepatitis C virus
infected patients with chronic fatigue and cognitive impairment.
Gut. 55:1624–1630. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bankwitz D, Steinmann E, Bitzegeio J,
Ciesek S, Friesland M, Herrmann E, Zeisel MB, Baumert TF, Keck ZY,
Foung SK, et al: Hepatitis C virus hypervariable region 1 modulates
receptor interactions, conceals the CD81 binding site, and protects
conserved neutralizing epitopes. J Virol. 84:5751–5763. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grigorov B, Reungoat E, Gentil Dit Maurin
A, Varbanov M, Blaising J, Michelet M, Manuel R, Parent R, Bartosch
B, Zoulim F, et al: Hepatitis C virus infection propagates through
interactions between Syndecan-1 and CD81 and impacts the hepatocyte
glycocalyx. Cell Microbiol. 19:2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lavie M, Sarrazin S, Montserret R,
Descamps V, Baumert TF, Duverlie G, Séron K, Penin F and Dubuisson
J: Identification of conserved residues in hepatitis C virus
envelope glycoprotein E2 that modulate virus dependence on CD81 and
SRB1 entry factors. J Virol. 88:10584–10597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tong Y, Zhu Y, Xia X, Liu Y, Feng Y, Hua
X, Chen Z, Ding H, Gao L, Wang Y, et al: Tupaia CD81, SR-BI,
claudin-1, and occludin support hepatitis C virus infection. J
Virol. 85:2793–2802. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Duffy HS, John GR, Lee SC, Brosnan CF and
Spray DC: Reciprocal regulation of the junctional proteins
claudin-1 and connexin43 by interleukin-1beta in primary human
fetal astrocytes. J Neurosci. 20:RC1142000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Husemann J and Silverstein SC: Expression
of scavenger receptor class B, type I, by astrocytes and vascular
smooth muscle cells in normal adult mouse and human brain and in
Alzheimer's disease brain. Am J Pathol. 158:825–832. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rajalakshmy AR, Malathi J and Madhavan HN:
Serum-derived hepatitis C virus 1a infection of human astrocyte
cell line SVG. J Viral Hepat. 23:211–216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bürgel B, Friesland M, Koch A, Manns MP,
Wedemeyer H, Weissenborn K, Schulz-Schaeffer WJ, Pietschmann T,
Steinmann E and Ciesek S: Hepatitis C virus enters human peripheral
neuroblastoma cells-evidence for extra-hepatic cells sustaining
hepatitis C virus penetration. J Viral Hepat. 18:562–570. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zong H, Verhaak RG and Canoll P: The
cellular origin for malignant glioma and prospects for clinical
advancements. Expert Rev Mol Diagn. 12:383–394. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Al Hassan M, Fakhoury I, El Masri Z,
Ghazale N, Dennaoui R, El Atat O, Kanaan A and El-Sibai M:
Metformin treatment inhibits motility and invasion of glioblastoma
cancer cells. Anal Cell Pathol (Amst). 2018:59174702018.PubMed/NCBI
|
|
26
|
Shih SR, Weng KF, Stollar V and Li ML:
Viral protein synthesis is required for Enterovirus 71 to induce
apoptosis in human glioblastoma cells. J Neurovirol. 14:53–61.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stein S, Zhao R, Haeno H, Vivanco I and
Michor F: Mathematical modeling identifies optimum lapatinib dosing
schedules for the treatment of glioblastoma patients. PLoS Comput
Biol. 14:e10059242018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chan JF, Yip CC, Tsang JO, Tee KM, Cai JP,
Chik KK, Zhu Z, Chan CC, Choi GK, Sridhar S, et al: Differential
cell line susceptibility to the emerging Zika virus: Implications
for disease pathogenesis, non-vector-borne human transmission and
animal reservoirs. Emerg Microbes Infect. 5:e932016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Perazzoli G, Prados J, Ortiz R, Caba O,
Cabeza L, Berdasco M, Gónzalez B and Melguizo C: Temozolomide
resistance in glioblastoma cell lines: Implication of MGMT, MMR,
P-Glycoprotein and CD133 expression. Plos One. 10:e1401312015.
View Article : Google Scholar
|
|
30
|
Westphal M, Harsh GR IV, Rosenblum ML and
Hammonds RJ Jr: Epidermal growth factor receptors in the human
glioblastoma cell line SF268 differ from those in epidermoid
carcinoma cell line A431. Biochem Biophys Res Commun. 132:284–289.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pas S, Molenkamp R, Schinkel J, Rebers S,
Copra C, Seven-Deniz S, Thamke D, de Knegt RJ, Haagmans BL and
Schutten M: Performance evaluation of the new Roche cobas
AmpliPrep/cobas TaqMan HCV test, version 2.0, for detection and
quantification of hepatitis C virus RNA. J Clin Microbiol.
51:238–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
European Association for the Study of the
Liver. Electronic address, . simpleeasloffice@easloffice.eu;
European Association for the Study of the Liver: EASL
recommendations on treatment of hepatitis C 2018. J Hepatol.
69:461–511. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Omata M, Kanda T, Wei L, Yu ML, Chuang WL,
Ibrahim A, Lesmana CR, Sollano J, Kumar M, Jindal A, et al: APASL
consensus statements and recommendations for hepatitis C
prevention, epidemiology, and laboratory testing. Hepatol Int.
10:681–701. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang H, Yuan G, Du Y, Cai X, Liu J, Hu C,
Liang B, Hu G, Tang X and Zhou Y: Effects of preventive therapy for
latent tuberculosis infection and factors associated with treatment
abandonment: A cross-sectional study. J Thorac Dis. 10:4377–4386.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang Z, Liu L, Jiang X, Zhai S and Xing
D: The essential Role of Drp1 and its regulation by S-nitrosylation
of parkin in dopaminergic neurodegeneration: Implications for
parkinson's disease. Antioxid Redox Signal. 25:609–622. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Z, Tian Y, Machida K, Lai MM, Luo G,
Foung SK and Ou JH: Transient activation of the PI3K-AKT pathway by
hepatitis C virus to enhance viral entry. J Biol Chem.
287:41922–41930. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li YP, Gottwein JM, Scheel TK, Jensen TB
and Bukh J: MicroRNA-122 antagonism against hepatitis C virus
genotypes 1–6 and reduced efficacy by host RNA insertion or
mutations in the HCV 5′ UTR. Proc Natl Acad Sci USA. 108:4991–4996.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vivithanaporn P, Maingat F, Lin LT, Na H,
Richardson CD, Agrawal B, Cohen EA, Jhamandas JH and Power C:
Hepatitis C virus core protein induces neuroimmune activation and
potentiates human immunodeficiency Virus-1 neurotoxicity. PLoS One.
5:e128562010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fishman SL, Murray JM, Eng FJ, Walewski
JL, Morgello S and Branch AD: Molecular and bioinformatic evidence
of hepatitis C virus evolution in brain. J Infect Dis. 197:597–607.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Laskus T, Radkowski M, Bednarska A,
Wilkinson J, Adair D, Nowicki M, Nikolopoulou GB, Vargas H and
Rakela J: Detection and analysis of hepatitis C virus sequences in
cerebrospinal fluid. J Virol. 76:10064–10068. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Radkowski M, Wilkinson J, Nowicki M, Adair
D, Vargas H, Ingui C, Rakela J and Laskus T: Search for hepatitis C
virus negative-strand RNA sequences and analysis of viral sequences
in the central nervous system: Evidence of replication. J Virol.
76:600–608. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Boyd JT, Wangensteen KJ, Krawitt EL,
Hamill RW, Kao CH and Tsai HH: Hepatitis C virus infection as a
risk factor for Parkinson disease: A nationwide cohort study.
Neurology. 87:3422016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tsai HH, Liou HH, Muo CH, Lee CZ, Yen RF
and Kao CH: Hepatitis C virus infection as a risk factor for
Parkinson disease: A nationwide cohort study. Neurology.
86:840–846. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen HH, Liu PF, Tsai HH, Yen RF and Liou
HH: Re: Wangensteen et al. of a letter on ‘Hepatitis C virus
infection: A risk factor for Parkinson's disease’. J Viral Hepat.
23:5602016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sun M, Fuentes SM, Timani K, Sun D, Murphy
C, Lin Y, August A, Teng MN and He B: Akt plays a critical role in
replication of nonsegmented negative-stranded RNA viruses. J Virol.
82:105–114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhirnov OP: Biochemical variations in
cytolytic activity of ortho- and paramyxoviruses in human lung
tumor cell culture. Biochemistry (Mosc). 82:1048–1054. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Preusse M, Schughart K and Pessler F: Host
genetic background strongly affects pulmonary microRNA expression
before and during influenza A virus infection. Front Immunol.
8:2462017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Peppenelli MA, Arend KC, Cojohari O,
Moorman NJ and Chan GC: Human cytomegalovirus stimulates the
synthesis of select Akt-dependent antiapoptotic proteins during
viral entry to promote survival of infected monocytes. J Virol.
90:3138–3147. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen Z, Yang L, Liu Y, Tang A, Li X, Zhang
J and Yang Z: LY294002 and Rapamycin promote coxsackievirus-induced
cytopathic effect and apoptosis via inhibition of PI3K/AKT/mTOR
signaling pathway. Mol Cell Biochem. 385:169–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu X and Cohen JI: Varicella-zoster virus
ORF12 protein activates the phosphatidylinositol 3-kinase/Akt
pathway to regulate cell cycle progression. J Virol. 87:1842–1848.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Banerjee S, Saito K, Ait-Goughoulte M,
Meyer K, Ray RB and Ray R: Hepatitis C virus core protein
upregulates serine phosphorylation of insulin receptor substrate-1
and impairs the downstream akt/protein kinase B signaling pathway
for insulin resistance. J Virol. 82:2606–2612. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bokemeyer M, Ding XQ, Goldbecker A, Raab
P, Heeren M, Arvanitis D, Tillmann HL, Lanfermann H and Weissenborn
K: Evidence for neuroinflammation and neuroprotection in HCV
infection-associated encephalopathy. Gut. 60:370–377. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wilkinson J, Radkowski M, Eschbacher JM
and Laskus T: Activation of brain macrophages/microglia cells in
hepatitis C infection. Gut. 59:1394–1400. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Grover VP, Pavese N, Koh SB, Wylezinska M,
Saxby BK, Gerhard A, Forton DM, Brooks DJ, Thomas HC and
Taylor-Robinson SD: Cerebral microglial activation in patients with
hepatitis C: In vivo evidence of neuroinflammation. J Viral Hepat.
19:e89–e96. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Forton DM, Hamilton G, Allsop JM, Grover
VP, Wesnes K, O'Sullivan C, Thomas HC and Taylor-Robinson SD:
Cerebral immune activation in chronic hepatitis C infection: A
magnetic resonance spectroscopy study. J Hepatol. 49:316–322. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Paulino AD, Ubhi K, Rockenstein E, Adame
A, Crews L, Letendre S, Ellis R, Everall IP, Grant I and Masliah E:
Neurotoxic effects of the HCV core protein are mediated by
sustained activation of ERK via TLR2 signaling. J Neurovirol.
17:327–340. 2011. View Article : Google Scholar : PubMed/NCBI
|