Introduction
Atherosclerosis is the most common cause of
cardiovascular diseases (1). The
processes involved in atherosclerosis are complex and include
endothelial cell activation, phenotypic switching in vascular
smooth muscle cells (VSMCs) and leukocyte infiltration (2). However, the role of VSMCs in
atherosclerosis remains unclear. Mature VSMCs in the medial layer
of arteries exhibit a contractile phenotype and express multiple
contractile proteins (3). In the
context of atherosclerosis, medial VSMCs undergo a phenotypic
switch to a synthetic state (3).
VSMCs may lose contractile markers and acquire the ability to
proliferate and synthesize various components involved in
extracellular matrix deposition, which is involved in the growth of
atherosclerotic plaques following the migration of VSMCs in the
neointima (4). Under stress
conditions, including organ transplant rejection, synthetic VSMCs
are involved in extracellular matrix deposition and express a
variety of chemokines that recruit alloreactive T cells to the
vascular wall (5–7). Therefore, medial VSMCs may accelerate
the atherosclerotic process by releasing chemokines and promoting
leukocyte infiltration.
The phenotypic plasticity of VSMCs is regulated by a
number of signaling pathways (8).
Fibroblast growth factor (FGF) is one of the growth factors
released by VSMCs and exhibits the ability to induce VSMC
dedifferentiation and proliferation. FGF receptor substrate 2
(FRS2) is a scaffold protein interacting with FGF receptors (FGFRs)
that mediates multiple signaling pathways to the activated FGFR
(9). Following the activation of
the FGF signaling pathway, VSMCs undergo a phenotypic switch from a
contractile to a synthetic form (10). Activated FGF signaling inhibits the
expression level of transforming growth factor β receptor (TGFBR) 1
expression via the microRNA let-7 (11–13).
Following activation of the FGF signaling pathway, the
phosphorylation of SMAD2/3 decreases and transforming growth factor
β (TGFβ) signaling is repressed (14,15).
TGFβ was previously demonstrated to maintain VSMCs in a quiescent
state and contractile phenotype (16). In the present study, FGF signaling
was hypothesized to promote a secretory phenotype in VSMC,
promoting the expression of factors involved in extracellular
matrix deposition and pro-inflammatory factors.
In the present study, human VSMCs, and mouse and
human atherosclerotic samples were used to investigate the role of
FGF signaling in VSMCs. FGF was identified to promote a phenotypic
transition in VSMCs, inducing VSMCs to secrete various chemokines
previously identified to be involved in atherosclerosis, including
C-C motif chemokine ligand 2 (CCL2), C-X-C motif chemokine ligand
(CXCL) 9, CXCL10 and CXCL11 (5–7). In
the present study, medial VSMCs were identified to secrete various
chemokines in mouse and human atherosclerotic samples, and
exhibited an increase in FGF signaling. Collectively, the present
results suggested that FGF signaling induced the release of
chemokines by VSMC that may be involved in leukocyte infiltration
and plaque formation in the atherosclerotic process.
Materials and methods
Cells and short hairpin RNA
(shRNA)
Primary human aortic smooth muscle cells (HASMCs)
were purchased from Lonza Group, Ltd. (Basel, Switzerland) and
cultured in Smooth Muscle Cell Growth Medium-2 supplemented with 5%
FBS (Lonza Group, Ltd.). Following 5–8 passages, HASMCs were used
for further experiments. For reverse transcription-quantitative PCR
(RT-qPCR) analysis, cells were serum-deprived in 0.5% FBS for 48 h
prior to treatment with FGF1 (50 ng/ml, BioLegend, Inc., San Diego,
CA, USA). Untreated cells were used as control. For FGF signaling
inhibition, HASMCs were washed with Hank's balanced salt solution
and transfected with a lentivirus encoding an shRNA
(5′-CTCTAAATGGCTACCATAATA-3′; Open Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) targeting FRS2 for 6 h
according to the manufacturer's protocol. To construct FRS2α shRNA
lentivirus, 10 µg pLVX–IRES-puro (Addgene, Inc.) carrying the FRS2α
cDNA expression cassette, 2.5 µg of RSV-REV plasmid (Addgene,
Inc.), 5 µg pMDLg/PRRE vector and 3 µg pMD.2G vector (Addgene,
Inc.) were cotransfected into 293T cells using FuGENE 6 (Promega
Corporation, Madison, WI, USA). The medium was harvested 48 h later
and HASMCs were treated with the resulting medium. An empty
pLVX–IRES-puro vector was used as control. HASMCs were grown at
37°C and 5% CO2 in smooth muscle cell growth medium-2
(Lonza Group, Ltd., Basel, Switzerland). 48 h after shRNA
transfection, HASMCs were used for subsequent experiments.
RNA extraction and analysis
The total RNA was extracted from cultured cells
using RNeasy Mini Kit (Qiagen GmbH, Hilden, Germany) according to
the manufacturer's protocol. Tri Reagent was used (Molecular
Research Center, Inc., Cincinnati, OH, USA) to homogenize the
cells. RNA samples were treated with deoxyribonuclease I (DNase I;
Ambion; Thermo Fisher Scientific, Inc.) and were
reverse-transcribed using random hexamers and oligo-dT using the
Multiscribe RT system (TaqMan RT reagents; Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The RT reaction was performed at 25°C for 5 min, followed
by incubations at 46°C for 20 min and at 95°C for 1 min. The
RT-qPCR was performed in 20 µl total volume in duplicate using cDNA
samples derived from total RNA. TaqMan Universal PCR Master Mix,
primers and TaqMan fluorescent probes were purchased from the
Assays-by-Design service (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Thermocycling conditions were as follows:
Uracil-DNA glycosylase activation at 50°C for 2 min, initial
denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for
15 sec and 60°C for 1 min. Using the AmpliTaq Gold polymerase
(Thermo Fisher Scientific, Inc.). Samples were analyzed using CFX
manager 3.1 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Non-retrotranscribed RNA samples were used as negative control. The
expression levels of the target genes were normalized to GAPDH and
quantified using the 2−ΔΔCq method (17). The primers used in the present
study were: Actin α 2, smooth muscle (ACTA2; cat. no.
Hs05005341_m1), calponin 1 (CNN1; cat. no. Hs00154543_m1), TGFβR2
(cat. no. Hs00234253_m1), CCL2 (cat. nos. Hs00234140_m1 and
Mm00441242_m1), CXCL9 (cat. nos. Hs00171065_m1 and Mm00434946_m1),
CXCL10 (cat. nos. Hs00171042_m1 and Mm00445235_m1), CXCL11 (cat.
nos. Hs00171138_m1 and Mm00444662_m1) and GAPDH (cat. nos.
Hs02786624_g1 and Mm99999915_g1).
ELISA
Chemokine concentrations of human CCL2 (cat. no.
DCP00), CXCL9 (cat. no. DCX900), CXCL10 (cat. no. DIP100) and
CXCL11 (cat. no. DCX110) from cultured VSMCs were determined by
ELISA assay (R&D Systems, Inc., Minneapolis, MN, USA) according
to the manufacturer's protocol. Samples and standards were added to
the wells and plates were incubated for 2 h at room temperature.
Detection solution was added to each well and the plates were
further incubated for 2 h at room temperature. Substrate solution
was added to each well and plates were incubated for 30 min at room
temperature. Stop solution was added to each well and the optical
density was detected using a Fusion® plate reader
(PerkinElmer, Inc., Waltham, MA, USA) after 30 min.
Animal studies
In total, 16 male C57BL/6J mice (WT; age, 8–12
weeks; body weight, 25 g) and apolipoprotein E (ApoE)-deficient
mice (ApoE−/−; age, 8–12 weeks; body weight, 25 g) were
purchased from The Nanjing Biomedical Research Institute of Nanjing
University (Nanjing, China). Mice were housed under a 12-h
light/dark cycle and maintained at a temperature of 30–34°C with
30–70% relative humidity. The WT mice were fed with standard rodent
chow and the ApoE−/− mice were fed with a high-fat diet
(normal chow supplemented with 1.25% cholesterol and 40% kcal fat)
for 4 months. Each group consisted of eight mice. To harvest the
aortic arteries, mice were anesthetized and perfused through the
left ventricle sequentially with 0.9% sodium chloride. Mouse
brachiocephalic arteries were removed under a dissection microscope
and preserved in 10% paraformaldehyde at 4°C prior to embedding in
optimum cutting temperature (OCT) compound. Samples were cut into
5-µm thick transverse sections for immunofluorescence, and 10-µm
thick transverse sections were cut for laser-capture
microdissection and RT-qPCR analysis. Animal experiments were
performed according to published guidelines (5), and the experimental procedures were
approved by The Ethics Committee of The First Hospital of China
Medical University (Shenyang, China).
Artery donors
The Ethics Committee of The First Hospital of China
Medical University approved the research protocols and the
experiments involving human subjects. All donors provided written
informed consent. The families of the patients provided informed
consent for the use of samples from deceased organ donors. Clinical
characteristics are listed in Table
I. Only patients >18 years old were enrolled in the present
study. Patients with cardiomyopathy and aortic aneurism were
excluded from the present study. In total, six patients (age,
62.2±12.6 years; 1 female, 5 males) with coronary atherosclerosis
assessed by histological examination and six healthy subjects (age,
38.9±4.6 years; 3 females, 3 males) were enrolled in the present
study. Human coronary arteries were collected from explanted hearts
of transplant recipients or deceased organ donors. The samples were
collected in The First Hospital of China Medical University between
January 2008 and December 2018.
 | Table I.Clinical characteristics of patients
with atherosclerosis and healthy subjects. |
Table I.
Clinical characteristics of patients
with atherosclerosis and healthy subjects.
| Characteristic | Healthy subjects
(n=6) | Patients with
atherosclerosis (n=6) |
|---|
| Age (years) | 38.9±4.6 | 62.2±12.6 |
| Female to male
ratio | 1:1 | 1:5 |
| Hyperlipidemia | 0 (0%) | 5 (83.3%) |
| Current smoker | 0 (0%) | 2 (33.3%) |
Immunofluorescence
Human coronary arteries or mouse brachiocephalic
arteries were flushed with PBS, fixed in 10% paraformaldehyde at
4°C overnight, embedded in OCT blocks, and cut into 5-µm thick
sections at −20°C. Sections were incubated with primary antibodies
at 4°C overnight, washed with TBS three times, and incubated with
Alexa Fluor®-conjugated secondary antibodies at room
temperature for 1 h. The following primary antibodies were used:
Mouse anti-smooth muscle actin (SMA; 1:200; cat. no. ab7817; Abcam,
Cambridge, UK), rabbit anti-human/mouse phosphorylated (p-)FGFR1
(1:100; cat. no. ab59194; Abcam) and rabbit anti-human/mouse TGFβR2
(1:100; cat. no. ab216483; Abcam). The following secondary
antibodies were used: Alexa Fluor® 488 donkey anti-mouse
IgG (1:200; cat. no. A21202; Invitrogen; Thermo Fisher Scientific,
Inc.) and Alexa Fluor® 594 donkey anti-rabbit IgG
(1:200; cat. no. A21207; Thermo Fisher Scientific, Inc.). Five
sections from each graft and four fields of view for each section
were analyzed. The nuclei of positive cells were counted using an
epifluorescence microscope under high magnification (magnification,
×400).
Laser-capture microdissection
Human coronary arteries or mouse aortic arteries
were flushed with PBS without fixation, embedded in OCT blocks, cut
into 10-µm thick sections at −20°C and placed on nuclease-free,
membrane-covered frame slides. The medial layer of the aortic wall,
primarily consisting of VSMCs, was cut using a Pixcell
laser-capture epifluorescence microscope (magnification, 100×;
Arcturus; Thermo Fisher Scientific, Inc.) with a laser power
setting of 38 mW, pulse duration of 1 msec, and a 10 µm spot size.
The cellular material was captured using CapSure LCM caps (Thermo
Fisher Scientific, Inc.) and digested in Nanoprep lysis buffer
(Agilent Technologies, Inc., Santa Clara, CA, USA). The DNA content
in each sample was quantified using the Picogreen fluorometric
assay system (Molecular Probes; Thermo Fisher Scientific, Inc.).
The total RNA was isolated using the PicoPure RNA isolation kit
(Thermo Fisher Scientific, Inc.). RNA samples were treated with
DNase I (Ambion; Thermo Fisher Scientific, Inc.) and were
reverse-transcribed using random hexamers and oligo-dT with the
Multiscribe RT system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The RT
reaction was performed at 25°C for 5 min, followed by incubations
at 46°C for 20 min and at 95°C for 1 min. An iCycler and its system
interface software (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
were used to perform qPCR. Amplification was performed as follows:
Uracil-DNA glycosylase activation at 50°C for 2 min, initial
denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for
15 sec and 60°C for 1 min. Using the AmpliTaq Gold polymerase
(Thermo Fisher Scientific, Inc.). Non-retrotranscribed RNA samples
were used as negative control. The expression levels of the target
genes were normalized to GAPDH and quantified using the
2−ΔΔCq method (17).
Standard curves for PCR were constructed to identify the gene copy
number in each sample. The EC marker CD31 (cat. nos. Mm00476702_m1
and Hs00169777_m1; Thermo Fisher Scientific, Inc.) was undetectable
by RT-qPCR, suggesting that only medial VSMCs were isolated without
contamination of other layers (data not shown).
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). Data are
presented as the mean ± standard error. Unpaired two-tailed
Student's t-test was performed to compare two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
FGF signaling induces a synthetic
phenotype in VSMCs and promotes inflammation
To investigate the role of FGF in VSMCs phenotypic
switching and the expression levels of inflammatory molecules,
HASMCs were cultured with or without FGF1 at a concentration of 50
ng/ml. RT-qPCR and ELISA were used to examine the phenotype of
cultured VSMCs. The mRNA expression level of TGFβR2, involved in
VSMC differentiation (16), was
decreased in cells treated with FGF compared with untreated
controls. Consistently with the decreased expression level of
TGFβR2, the mRNA expression levels of genes encoding contractile
proteins, including CNN1 and ACTA2, were decreased following
treatment with FGF (Fig. 1A).
Treatment with FGF significantly increased the protein and mRNA
expression levels of the inflammatory chemokines CCL2, CXCL9,
CXCL10 and CXCL11 (Fig. 1B and C).
The present data suggested that treatment with FGF induced TGFβR2
inhibition and loss of contractile markers. In addition, FGF
activation induced VSMCs to express various chemokines.
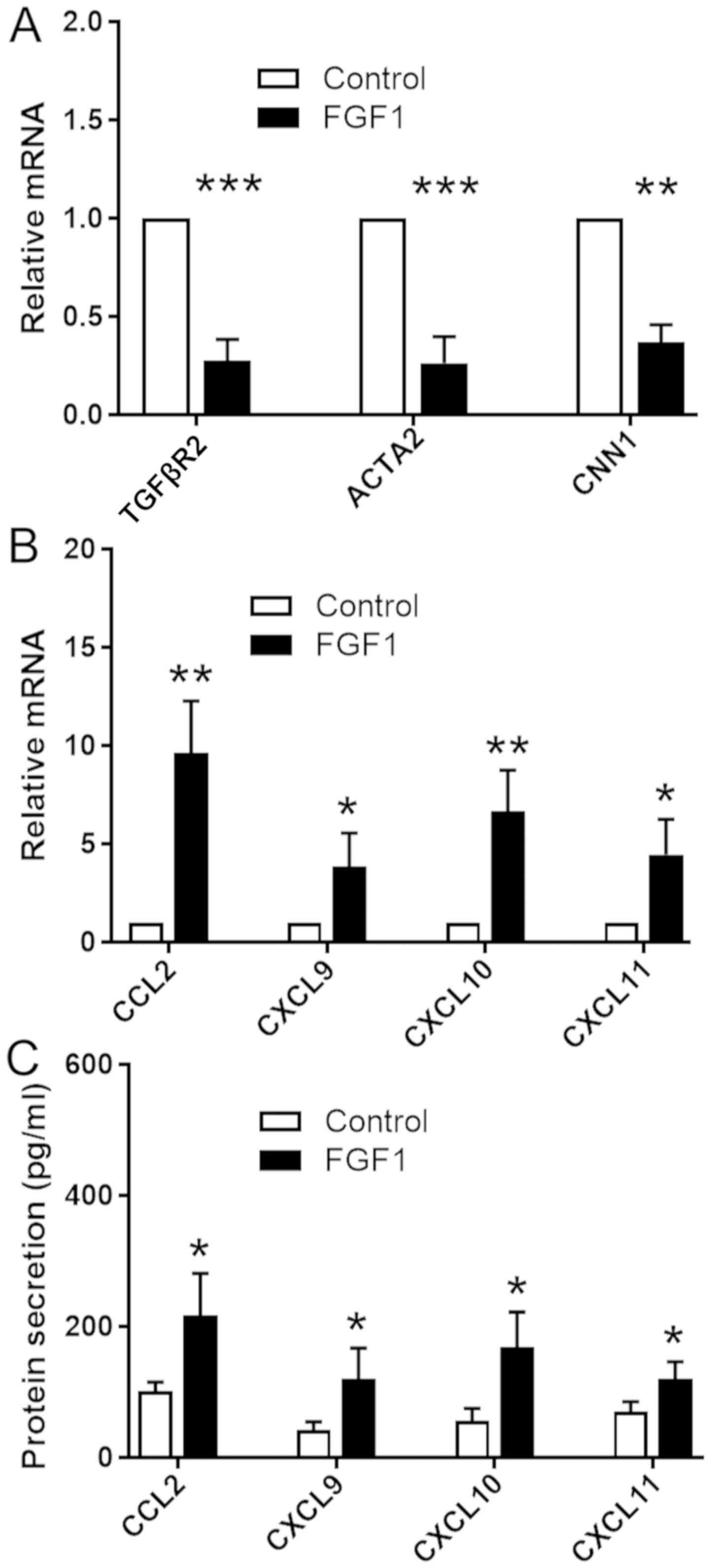 | Figure 1.FGF signaling downregulates the
expression level of TGFβR2 and upregulates the expression levels of
various chemokines in VSMCs. Human aortic smooth muscle cells were
treated with or without FGF1 (50 ng/ml). Reverse
transcription-quantitative polymerase chain reaction was used to
assess the mRNA expression levels of (A) TGFβR2, ACTA2, CNN1 and
(B) various chemokines. GAPDH was used as the internal control.
Data are presented as fold changes in treated cells relative to
untreated controls. (C) Quantification of chemokines levels
secreted by VSMCs by ELISA. Experiments were repeated three times.
Data are presented as the mean ± standard error. Statistical
significance was determined by unpaired two-tailed Student's
t-test. *P<0.05, **P<0.01, ***P<0.001 vs.
untreated cells. VSMCs, vascular smooth muscle cells; FGF1,
fibroblast growth factor 1; TGFβR2, transforming growth factor β
receptor 2; ACTA2, actin α 2, smooth muscle; CNN1, calponin 1;
CCL2, C-C motif chemokine ligand 2; CXCL, C-X-C motif chemokine
ligand. |
Inhibition of FGF signaling in VSMCs
maintains their contractile phenotype and suppresses
inflammation
To further investigate the role of FGF signaling in
the phenotypic transition of VSMCs and in the synthesis of
inflammatory cytokines, FRS2 shRNA was used to knockdown the
expression level of FRS2 in HASMCs. RT-qPCR and ELISA were
performed to examine the expression levels of various chemokines in
cultured VSMCs. A significant decrease in the expression level of
FRS2 was detected in VSMCs transfected with FRS2 shRNA (Fig. 2A). Additionally, the expression
levels of genes encoding the contractile proteins CNN1 and SMA
(encoded by ACTA2) were significantly increased following FRS2
knockdown. In addition, the expression level of TGFβR2 was
upregulated following FRS2 knockdown cells compared with control
cells (Fig. 2B). The present
results further suggested that FGF signaling may inhibit TGFβ
signaling and influence the phenotype of VSMCs. Furthermore, the
expression levels of genes encoding chemokines was significantly
decreased following FRS2 knockdown (Fig. 2C). Additionally, the secretion
levels of CXCL9, CXCL10 and CXCL11 were undetectable in the FRS2
knockdown group (Fig. 2D). The
present results suggested that activation of the FGF signaling in
VSMCs caused a significant decrease in the expression levels of
contractile markers and an increase in inflammatory markers.
Collectively, the present results suggested that FGF signaling may
serve a role in the phenotypic switching of VSMCs by increasing the
secretion of chemokines by VSMCs. In addition, inhibition of FGF
signaling was sufficient to significantly decrease inflammation in
VSMCs, indicated by the changes in chemokines.
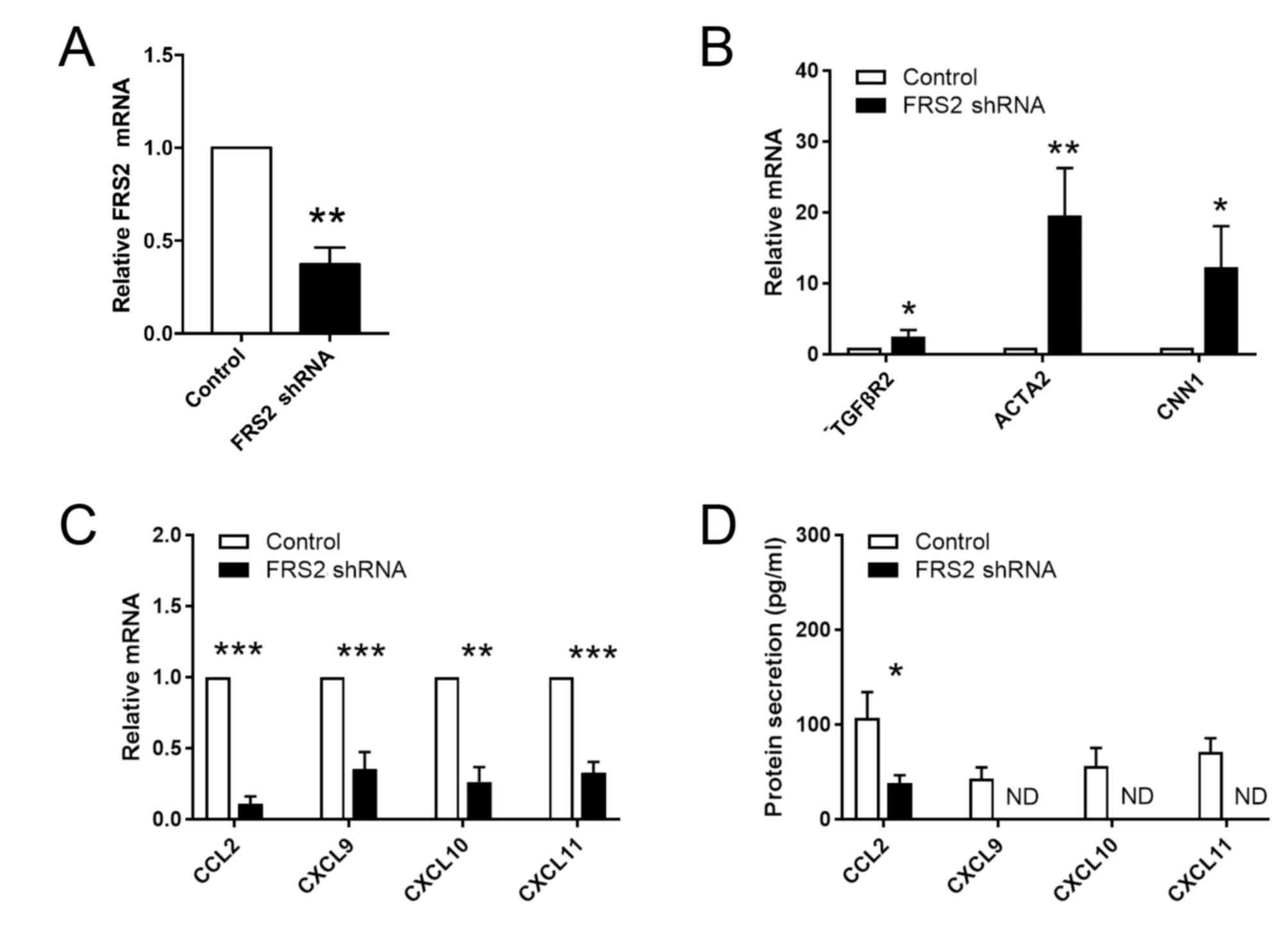 | Figure 2.Inhibition of FGF signaling
upregulates TGFβR2 and downregulates chemokines in VSMCs. Human
artery smooth muscle cells were transfected with FRS2 to inhibit
FGF signaling. (A) Efficiency of shRNA transfection in VSMCs was
measured by RT-qPCR. RT-qPCR was used to assess the expression
levels of (B) TGFβR2 and (C) various chemokines. GAPDH was used as
the internal control. Data are presented as fold changes in
transfected cells relative to untreated controls. (D) Protein
levels of chemokines secreted by VSMCs were determined by ELISA.
Experiments were repeated three times. Data are presented as the
mean ± standard error. Statistical significance was determined by
unpaired two-tailed Student's t-test. *P<0.05, **P<0.01,
***P<0.001 vs. empty vector control. FGF, fibroblast growth
factor; VSMCs, vascular smooth muscle cells; RT-qPCR, reverse
transcription-quantitative PCR; FRS2, fibroblast growth factor
receptor substrate 2; shRNA, short hairpin RNA; TGFβR2,
transforming growth factor β receptor 2; ACTA2, Actin α 2, smooth
muscle; CNN1, calponin 1; CCL2, C-C motif chemokine ligand 2; CXCL,
C-X-C motif chemokine ligand; ND, not detectable. |
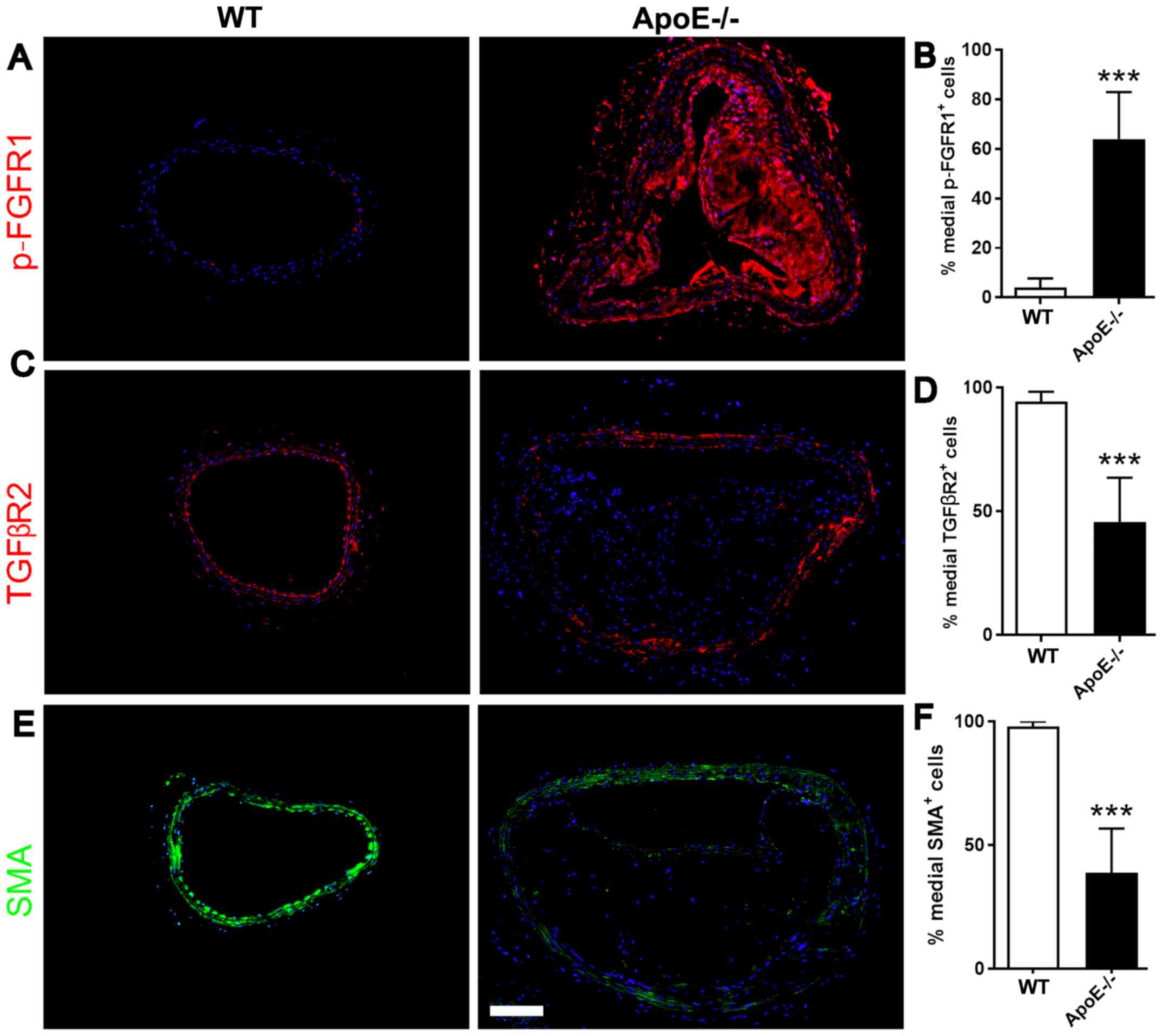
Increased FGF signaling in aortic
VSMCs of ApoE−/− mice
FGF signaling was previously demonstrated to be
involved in atherosclerosis (15).
The ApoE−/− mouse model is the most common murine model
used to investigate atherosclerosis. To examine the role of FGF and
TGFβ signaling in the phenotype of VSMCs in vivo,
ApoE−/− mice and WT C57BL/6J mice were used to compare
the protein expression level of p-FGFR1 and TGFβR2 in medial VSMCs.
Immunofluorescence staining was performed to investigate the
protein expression levels of p-FGFR1, TGFβR2 and ACTA2 in VSMCs.
The medial VSMCs in ApoE−/− mice exhibited an increased
protein expression level of p-FGFR1, whereas SMA (encoded by ACTA2)
was decreased (Fig. 3A and B). The
protein expression levels of TGFβR2 and SMA were decreased in VSMCs
of ApoE−/− mice compared with WT mice (Fig. 3C-F). The present results suggested
that FGF signaling was activated in the medial VSMCs of aortic
arteries exhibiting atherosclerotic plaques, as assessed by SMA
staining. Additionally, activation of FGF signaling inhibited the
expression level of TGFβR2 and contractile proteins, suggesting the
initiation of a phenotypic switch in medial VSMCs.
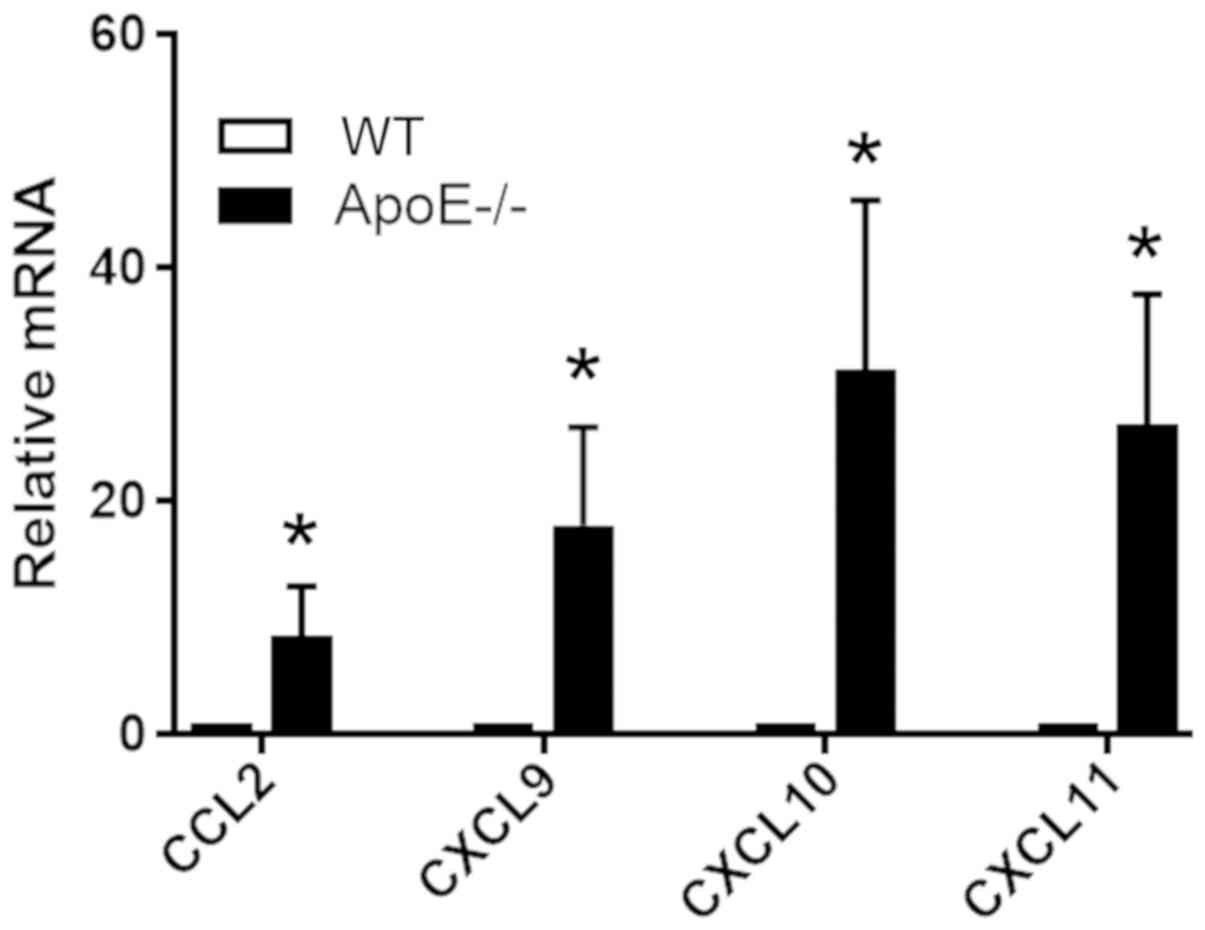
Aortic VSMCs express chemokines in
ApoE−/− mice
To examine the protein expression levels of
inflammatory molecules in medial VSMCs in vivo,
ApoE−/− mice and WT mice were used to investigate the
expression levels of various genes encoding chemokines in the
medial VSMCs of aortic arteries. To detect the mRNA expression
levels of genes associated with inflammation in medial VSMCs and
not in surrounding cells in the intima and adventitia,
laser-capture microdissection was used to collect medial VSMCs.
qPCR analysis of dissected medial tissues suggested that VSMCs in
WT mice exhibited significant decreased expression levels of CCL2,
CXCL9, CXCL10 and CXCL11, compared with ApoE−/− mice
(Fig. 4). In addition, VSMCs in
ApoE−/− mice exhibited increased p-FGFR1 and decreased
expression of contractile proteins. Collectively, the present data
were consistent with the aforementioned in vitro results and
demonstrated that active FGF signaling altered the phenotype of
VSMCs by inducing the expression of various inflammatory
molecules.
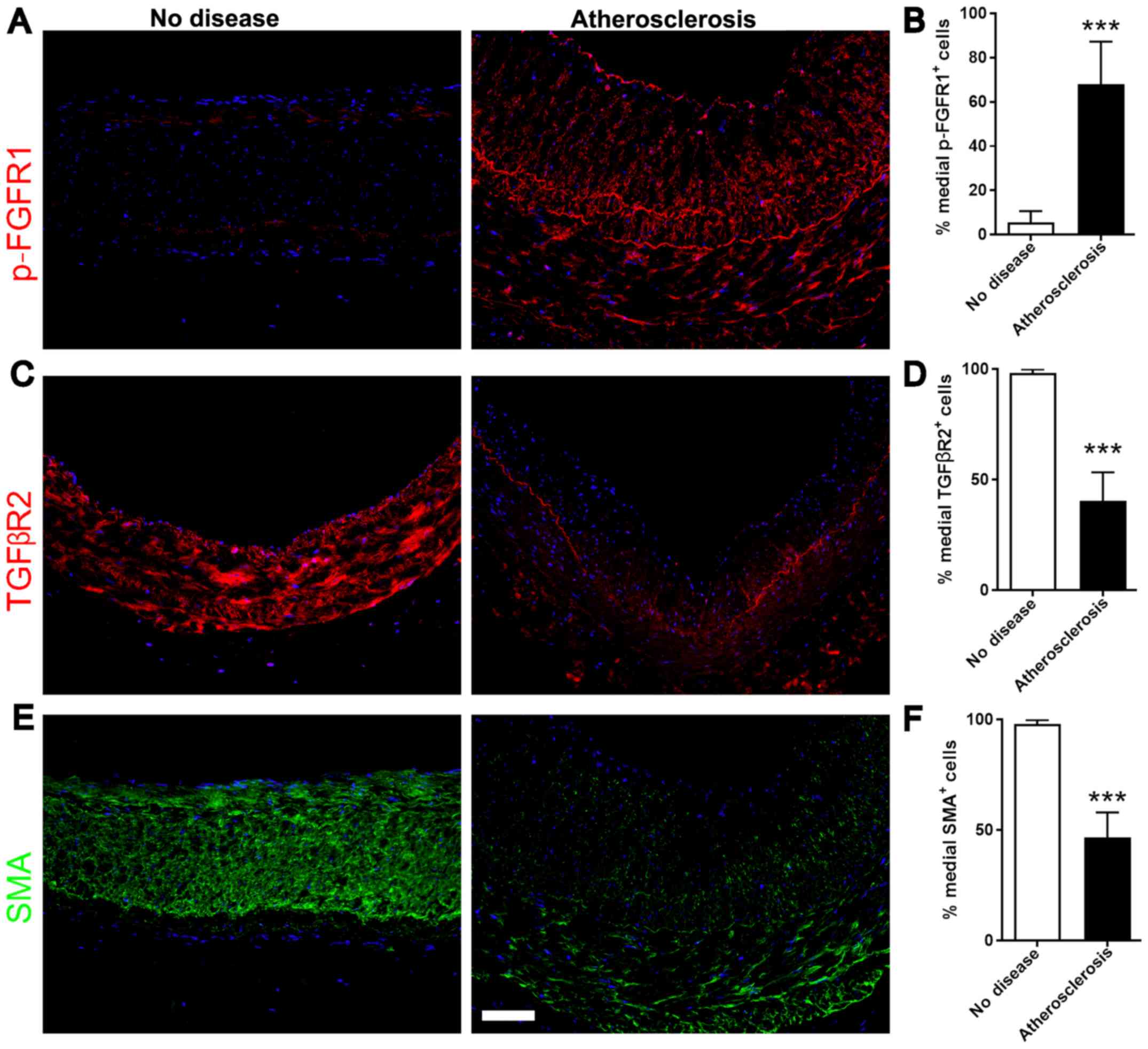
Increased FGF signaling in medial
VSMCs of human coronary arteries
To further investigate the role of FGF and TGFβ
signaling in the phenotype of VSMCs in human, coronary arteries
were collected from patients with atherosclerotic plaques and
healthy donors. Subsequently, the expression levels of p-FGFR1 and
TGFβR2 in medial layer VSMCs were analyzed. Immunofluorescence
staining was used to examine the protein expression levels of
p-FGFR1, TGFβR2 and SMA. Consistent with the aforementioned results
in mouse, the medial VSMCs in patients with atherosclerotic plaques
exhibited upregulated p-FGFR1 and downregulated SMA expression
(Fig. 5A and B). The protein
expression levels of TGFβR2 exhibited the same trend of SMA, with
decreased expression in the medial VSMCs of arteries from patients
with atherosclerosis compared with healthy controls (Fig. 5C-F). The present results suggested
that FGF signaling was activated in the medial VSMCs of coronary
arteries of patients with advanced atherosclerotic plaques.
Collectively, FGF signaling may inhibit the protein expression
level of TGFβR2 and contractile factors, inducing a phenotypic
switch in medial VSMCs.
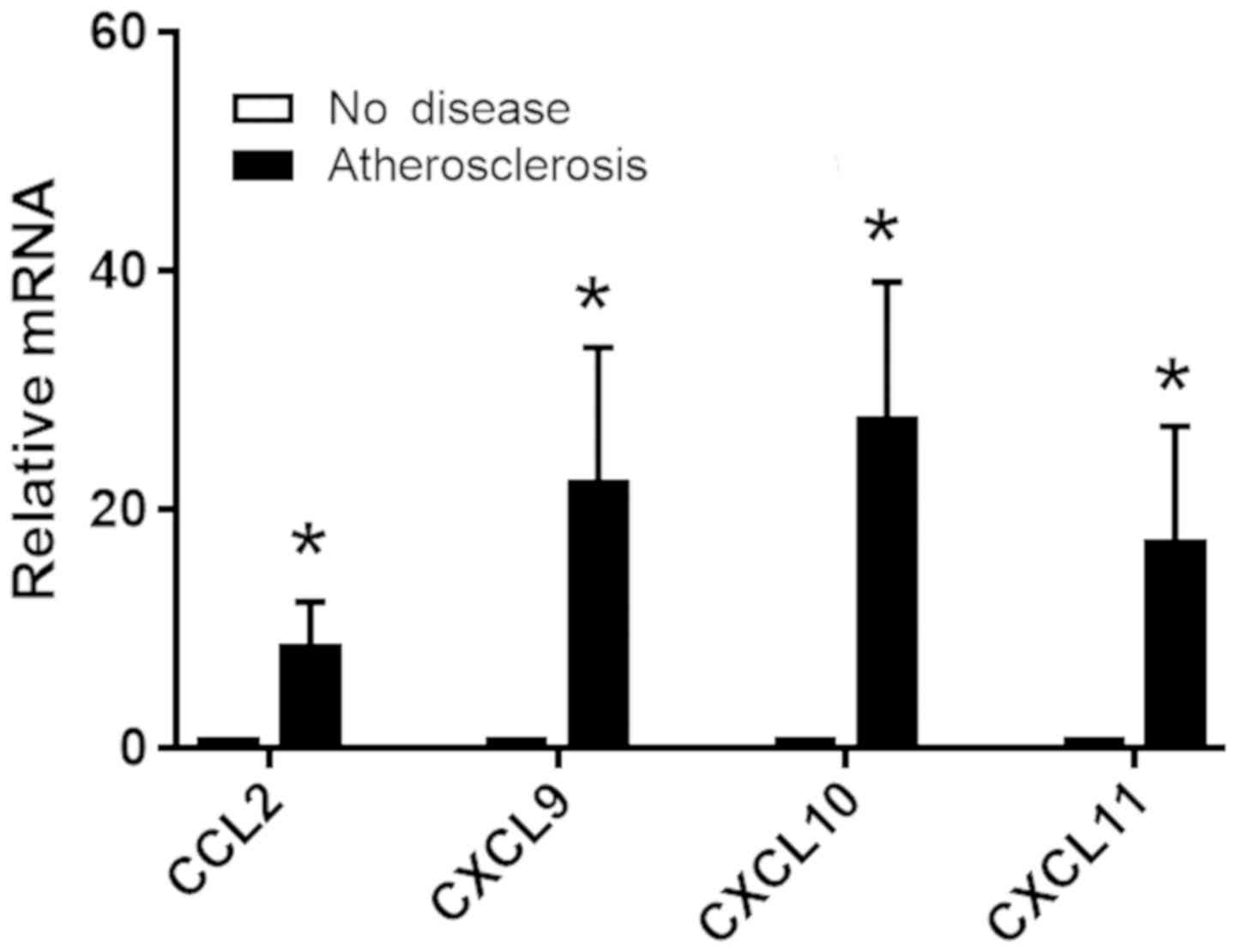
Medial VSMCs release chemokines in
human coronary arteries
To examine the secretion of inflammatory molecules
by medial VSMCs in human, coronary arteries with advanced plaques
and healthy arteries were collected, and the expression levels of
various chemokines in the medial VSMCs of coronary arteries were
investigated. Laser-capture microdissection was used to remove the
medial layer of human coronary arteries and to collect only medial
VSMCs. RT-qPCR was used to measure the mRNA expression levels of
inflammatory cytokines synthesized by medial VSMCs, and not by
additional cells in the intima and adventitia. The mRNA expression
levels of genes encoding the chemokines CCL2, CXCL9, CXCL10 and
CXCL11 were significantly increased in coronary arteries from
patients with atherosclerosis compared with healthy controls
(Fig. 6). The increase in the
expression levels of genes encoding chemokines in human patients
was observed in addition to the activation of FGF signaling and the
decrease in the expression level of SMA. Collectively, the present
results are consistent with the aforementioned analyses in mice and
suggested that upregulation of FGF signaling may induce VSMCs
phenotypic switching by promoting the secretion of inflammatory
molecules by VSMCs.
Discussion
The role of the phenotypic switching of VSMCs in the
process of atherosclerosis is not fully understood. In the present
study, FGF signaling was identified to induce phenotypic switching
of cultured VSMCs from a contractile state to a secretory state,
characterized by chemokine production. Suppressing FGF signaling in
VSMCs decreased the secretion of chemokines. Furthermore, analysis
of arteries from ApoE−/− mice and human atherosclerotic
patients suggested that medial VSMCs exhibited an increase in the
activation of FGF signaling and in the expression levels of
pro-inflammatory factors. Collectively, the present results
suggested that FGF signaling may have a significant role in the
regulation of VSMCs dedifferentiation and in the expression of
pro-inflammatory factors. Additionally, the present results
suggested that FGF signaling may be a pro-atherogenic factor, able
to suppress contractile markers and to induce a secretory phenotype
in VSMCs.
The FGF family includes 28 multifunctional ligands
that function as growth factors (18). FGF receptors are a family of
receptor tyrosine kinases consisting of five members (19–22).
In the arterial wall, endothelial cells and VSMCs express FGFs and
FGFRs (23). The FGFRs expressed
at high levels in VSMCs are FGFR1, FGFR2 and FGFR3, which are
activated by FGF1 (14). FGF
signaling was previously demonstrated to induce dedifferentiation
VSMCs from a contractile phenotype to a proliferative and synthetic
state by inhibiting TGFβ signaling (14,15).
In the present study, activated FGF signaling was identified to
increase the secretion of various pro-inflammatory factors. The
present results suggested that FGF signaling may induce an increase
in the protein expression levels of multiple chemokines in VSMCs,
promoting leukocyte trafficking to inflammatory sites, including
atherosclerotic plaques.
VSMCs are the only cell type in the medial layer of
arteries. Arterial VSMCs exhibit contractile functions and certain
synthetic functions, including elastin and collagen deposition.
Under homeostatic conditions, VSMCs maintain a quiescent phenotype
and are involved in extracellular matrix deposition and in the
turnover of collagen and elastin (7). Various stresses, including
inflammatory stimuli from microbial infection and infiltrated
leukocytes, are able to induce a pro-inflammatory response in
VSMCs, initiating phenotype switching, and promoting the release of
numerous cytokines and chemokines (6,24,25).
The expression levels of inflammatory factors expressed by
endothelial cells and synthetic VSMCs are similar (26,27).
Similarly to endothelial cells, VSMCs can secrete large amounts of
pro-inflammatory factors, recruiting macrophages and T cells to the
vessel wall, particularly to the neointima and adventitia (28–30).
Under atherosclerotic conditions, VSMCs in the
medial layer of arteries undergo a phenotypic switch in response to
various stimuli, including hypercholesterolemia, activated
endothelial cells and infiltrated leukocytes (3). In the present study, synthetic VSMCs
were identified to secrete increased levels of pro-inflammatory
molecules compared with contractile VSMCs, and synthetic VSMCs may
serve a significant role in initiating atherosclerosis. Various
chemokines expressed by VSMCs may initiate and promote
atherosclerosis. CCL2 is a potent chemoattractant that is able to
recruit monocytes and macrophages to the vessel wall, and induce
them to infiltrate into the neointima through the arterial lumen
and into the adventitia through the postcapillary venules (31–34).
Macrophages transdifferentiate into foam cells in microenvironments
exhibiting high circulating levels of cholesterol and may promote
plaque development. CXCL9, CXCL10 and CXCL11 are important
chemoattractants that recruit CXC receptor type 3-expressing T
cells into the vessel wall (34–38).
These T cells may induce the release of interferon γ (IFNγ) and
tumor necrosis factor α (TNFα) derived from macrophages and VSMCs,
promoting the progression of atherosclerotic plaques (5–7).
Notably, the level of CXCL10 secreted by synthetic VSMCs is between
10- and 100-fold higher compared with macrophages and T cells in
atherosclerotic arteries, whereas macrophages and T cells are the
principal cell types that secrete IFNγ and TNFα (7). This crosstalk between leukocytes and
VSMCs may induce a positive feedback loop in arteries causing
progression of atherosclerotic plaques.
Collectively, the findings of the present study
suggested that FGF signaling-induced phenotype switching leads to
increased secretion of chemokines by VSMCs, which may be involved
in leukocyte infiltration into the vascular lumen and initiation of
atherosclerotic lesion development. The present results suggested
that FGF signaling may promote atherosclerosis by increasing VSMC
proliferation and by inducing chemokine expression in VSMCs.
Additionally, the present study suggested that modulation of the
FGF signaling pathway may be an effective strategy for the
treatment and prevention of atherosclerosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant no. 81770488) and the
Liaoning Province Clinical Training Project (grant no.
LNCCC-C03-2015).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
MQ and SX designed the study and prepared the
manuscript. MQ performed the experiments. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of The First Hospital of China Medical University
(Shenyang, China). All patients or family members of the organ
donors provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanz J and Fayad ZA: Imaging of
atherosclerotic cardiovascular disease. Nature. 451:953–957. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bennett MR, Sinha S and Owens GK: Vascular
smooth muscle cells in atherosclerosis. Circ Res. 118:692–702.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu R, Leslie KL and Martin KA: Epigenetic
regulation of smooth muscle cell plasticity. Biochim Biophys Acta.
1849:448–453. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burns WR, Wang Y, Tang PC, Ranjbaran H,
Iakimov A, Kim J, Cuffy M, Bai Y, Pober JS and Tellides G:
Recruitment of CXCR3+ and CCR5+ T cells and production of
interferon-gamma-inducible chemokines in rejecting human arteries.
Am J Transplant. 5:1226–1236. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ahmad U, Ali R, Lebastchi AH, Qin L, Lo
SF, Yakimov AO, Khan SF, Choy JC, Geirsson A, Pober JS and Tellides
G: IFN-gamma primes intact human coronary arteries and cultured
coronary smooth muscle cells to double-stranded RNA- and
self-RNA-induced inflammatory responses by upregulating TLR3 and
melanoma differentiation-associated gene 5. J Immunol.
185:1283–1294. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tellides G and Pober JS: Inflammatory and
immune responses in the arterial media. Circ Res. 116:312–322.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kawai-Kowase K and Owens GK: Multiple
repressor pathways contribute to phenotypic switching of vascular
smooth muscle cells. Am J Physiol Cell Physiol. 292:C59–C69. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lindner V and Reidy MA: Proliferation of
smooth muscle cells after vascular injury is inhibited by an
antibody against basic fibroblast growth factor. Proc Natl Acad Sci
USA. 88:3739–3743. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jackson CL and Reidy MA: Basic fibroblast
growth factor: Its role in the control of smooth muscle cell
migration. Am J Pathol. 143:1024–1031. 1993.PubMed/NCBI
|
|
11
|
Chen PY, Qin L, Barnes C, Charisse K, Yi
T, Zhang X, Ali R, Medina PP, Yu J, Slack FJ, et al: FGF regulates
TGF-β signaling and endothelial-to-mesenchymal transition via
control of let-7 miRNA expression. Cell Rep. 2:1684–1696. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen PY, Qin L, Tellides G and Simons M:
Fibroblast growth factor receptor 1 is a key inhibitor of TGFβ
signaling in the endothelium. Sci Signal. 7:ra902014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen PY, Qin L, Baeyens N, Li G, Afolabi
T, Budatha M, Tellides G, Schwartz MA and Simons M:
Endothelial-to-mesenchymal transition drives atherosclerosis
progression. J Clin Invest. 125:4514–4528. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen PY, Qin L, Li G, Tellides G and
Simons M: Fibroblast growth factor (FGF) signaling regulates
transforming growth factor beta (TGFβ)-dependent smooth muscle cell
phenotype modulation. Sci Rep. 6:334072016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen PY, Qin L, Li G, Tellides G and
Simons M: Smooth muscle FGF/TGFβ cross talk regulates
atherosclerosis progression. EMBO Mol Med. 8:712–728. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lebastchi AH, Khan SF, Qin L, Li W, Zhou
J, Hibino N, Yi T, Rao DA, Pober JS and Tellides G: Transforming
growth factor beta expression by human vascular cells inhibits
interferon gamma production and arterial media injury by
alloreactive memory T cells. Am J Transplant. 11:2332–2341. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ornitz DM and Itoh N: The fibroblast
growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol.
4:215–266. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen PY, Simons M and Friesel R: FRS2 via
fibroblast growth factor receptor 1 is required for
platelet-derived growth factor receptor beta-mediated regulation of
vascular smooth muscle marker gene expression. J Biol Chem.
284:15980–15992. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kouhara H, Hadari YR, Spivak-Kroizman T,
Schilling J, Bar-Sagi D, Lax I and Schlessinger J: A lipid-anchored
Grb2-binding protein that links FGF-receptor activation to the
Ras/MAPK signaling pathway. Cell. 89:693–702. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eswarakumar VP, Lax I and Schlessinger J:
Cellular signaling by fibroblast growth factor receptors. Cytokine
Growth Factor Rev. 16:139–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gotoh N: Regulation of growth factor
signaling by FRS2 family docking/scaffold adaptor proteins. Cancer
Sci. 99:1319–1325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hughes SE: Localisation and differential
expression of the fibroblast growth factor receptor (FGFR)
multigene family in normal and atherosclerotic human arteries.
Cardiovasc Res. 32:557–569. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou J, Tang PC, Qin L, Gayed PM, Li W,
Skokos EA, Kyriakides TR, Pober JS and Tellides G: CXCR3-dependent
accumulation and activation of perivascular macrophages is
necessary for homeostatic arterial remodeling to hemodynamic
stresses. J Exp Med. 207:1951–1966. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu L, Qin L, Zhang H, He Y, Chen H, Pober
JS, Tellides G and Min W: AIP1 prevents graft arteriosclerosis by
inhibiting interferon-γ-dependent smooth muscle cell proliferation
and intimal expansion. Circ Res. 109:418–427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rao RM, Yang L, Garcia-Cardena G and
Luscinskas FW: Endothelial-dependent mechanisms of leukocyte
recruitment to the vascular wall. Circ Res. 101:234–247. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qin L, Huang Q, Zhang H, Liu R, Tellides
G, Min W and Yu L: SOCS1 prevents graft arteriosclerosis by
preserving endothelial cell function. J Am Coll Cardiol. 63:21–29.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Charo IF and Taubman MB: Chemokines in the
pathogenesis of vascular disease. Circ Res. 95:858–866. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tabas I and Glass CK: Anti-inflammatory
therapy in chronic disease: Challenges and opportunities. Science.
339:166–172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He C, Medley SC, Hu T, Hinsdale ME, Lupu
F, Virmani R and Olson LE: PDGFRβ signalling regulates local
inflammation and synergizes with hypercholesterolaemia to promote
atherosclerosis. Nat Commun. 6:77702015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boring L, Gosling J, Cleary M and Charo
IF: Decreased lesion formation in CCR2-/- mice reveals a role for
chemokines in the initiation of atherosclerosis. Nature.
394:894–897. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gerszten RE, Garcia-Zepeda EA, Lim YC,
Yoshida M, Ding HA, Gimbrone MA Jr, Luster AD, Luscinskas FW and
Rosenzweig A: MCP-1 and IL-8 trigger firm adhesion of monocytes to
vascular endothelium under flow conditions. Nature. 398:718–723.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng Y, Qin L, Zacarías NV, de Vries H,
Han GW, Gustavsson M, Dabros M, Zhao C, Cherney RJ, Carter P, et
al: Structure of CC chemokine receptor 2 with orthosteric and
allosteric antagonists. Nature. 540:458–461. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Veillard NR, Steffens S, Pelli G, Lu B,
Kwak BR, Gerard C, Charo IF and Mach F: Differential influence of
chemokine receptors CCR2 and CXCR3 in development of
atherosclerosis in vivo. Circulation. 112:870–878. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Heller EA, Liu E, Tager AM, Yuan Q, Lin
AY, Ahluwalia N, Jones K, Koehn SL, Lok VM, Aikawa E, et al:
Chemokine CXCL10 promotes atherogenesis by modulating the local
balance of effector and regulatory T cells. Circulation.
113:2301–2312. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zernecke A, Bot I, Djalali-Talab Y,
Shagdarsuren E, Bidzhekov K, Meiler S, Krohn R, Schober A,
Sperandio M, Soehnlein O, et al: Protective role of CXC receptor
4/CXC ligand 12 unveils the importance of neutrophils in
atherosclerosis. Circ Res. 102:209–217. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schwarz JB, Langwieser N, Langwieser NN,
Bek MJ, Seidl S, Eckstein HH, Lu B, Schömig A, Pavenstädt H and
Zohlnhöfer D: Novel role of the CXC chemokine receptor 3 in
inflammatory response to arterial injury: Involvement of mTORC1.
Circ Res. 104:189–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tavakolian Ferdousie V, Mohammadi M,
Hassanshahi G, Khorramdelazad H, Khanamani Falahati-Pour S, Mirzaei
M, Allah Tavakoli M, Kamiab Z, Ahmadi Z, Vazirinejad R, et al:
Serum CXCL10 and CXCL12 chemokine levels are associated with the
severity of coronary artery disease and coronary artery occlusion.
Int J Cardiol. 233:23–28. 2017. View Article : Google Scholar : PubMed/NCBI
|