Introduction
Hepatocellular carcinoma (HCC) is the most common
type of primary liver cancer in adults, accounting for the highest
mortality rate in patients with cirrhosis (1). HCC is typically associated with
hepatitis virus infection [hepatitis B virus (HBV) or hepatitis C
virus (HCV)] or exposure to aflatoxin and alcohol; ~75% of HCC
cases are induced by HBV infection (2,3).
Patients with HCC are characterized by the presentation of yellow
skin, weight loss, abdominal swelling, nausea, loss of appetite,
vomiting, abdominal pain or fatigue (4). The stages of disease progression in
newly diagnosed patients can greatly affect the prognosis of HCC
(5). Patient outcome is typically
poor, with only 10–20% of HCC cases fully recovering following
surgery (6). HCC commonly occurs
in males aged 30–50 years; annually, 662,000 cases of
HCC-associated mortality are reported worldwide (7). Therefore, the pathogenesis of
HBV-induced HCC requires further investigation to improve the
diagnosis and treatment of this disease.
Long noncoding RNAs (lncRNAs) serve important roles
in various cellular activities, including gene expression
regulation, tumor growth, apoptosis, autophagy and cell
differentiation (8,9). Via regulation of lncRNAs, such as
zinc finger E-box binding homeobox 2 antisense RNA 1, HBV X (HBx)
promotes the metastasis of HCC cells via the induction of
epithelial-mesenchymal transition (10). The expression of lncRNA
downregulated expression by HBx is reduced in HBV-associated HCC
samples, and exhibits an inverse correlation with HBx expression
and functions as a tumor suppressor in HBV-associated
hepatocarcinogenesis (11). The
lncRNA Unc-51 like kinase 4 pseudogene 2 is upregulated in
HBV-associated HCC tissues and may be involved in mediating disease
pathogenesis by associating with enhancer of zeste homolog 2
(12). The expression of lncRNA
LINC00152 can be enhanced by HBx, and its suppression is a
potential therapeutic strategy for the treatment of HCC (13,14).
The serum expression levels of lncRNAs AX800134 and uc001ncr were
identified as potential diagnostic markers for HBV-associated HCC
(15). The lncRNAs uc003wbd and
AF085935 are dysregulated in the serum of patients with HBV or HCC,
and may be potential targets for the screening of HBV and HCC
(16). The lncRNA DBH antisense
RNA 1 contributes to cell proliferation and survival via the
Ras/mitogen activated protein kinase signaling pathway, and serves
a carcinogenic role in HBV-associated HCC (17). Therefore, identifying the lncRNAs
associated with HBV-induced HCC is important for understanding the
underlying mechanisms and identifying novel therapies for the
treatment of this disease.
Bioinformatics methods are extensively used for
analyzing gene expression profiles to investigate the mechanisms of
human diseases (18). Wang et
al (19) analyzed the RNA-Seq
data of patients in The Cancer Genome Atlas (TCGA), and used four
independent prognostic lncRNAs identified by univariate Cox
proportional hazards (Cox-PH) regression analysis to construct a
risk score model. Zheng et al (20) sorted the samples downloaded from
TCGA into four cohorts, based on their clinical history of viral
hepatitis infection and alcohol consumption. Then, the lncRNAs
dysregulated in normal samples versus three tumor sample cohorts,
based on HBV infection, HCV infection and history of alcohol
consumption, were identified to further select for
disease-associated lncRNAs; however, a risk score model was not
generated and further investigation is required. Yuan et al
(21) collected samples from HCC
patients, patients with HBV-positive chronic hepatitis and
cancer-free controls, and subsequently conducted reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis of 10 candidate lncRNAs to identify differentially
expressed lncRNAs in HCC patients compared with patients with
chronic hepatitis or healthy controls. Risk score analysis revealed
that the combination of three lncRNAs with α-fetoprotein could
distinguish patients with HCC from those with chronic hepatitis or
healthy controls. In the present study, the RNA-Seq data of
patients in TCGA and three other datasets of HBV infection were
downloaded. The RNA-Seq data from TCGA, GSE55092 and GSE19665 were
integrated together to determine differentially expressed RNAs
(DE-RNAs). Subsequently, prognosis-associated lncRNAs were selected
by univariate Cox-PH regression analysis. The risk score system
based on these lncRNAs was supported by the validation dataset,
GSE10186. The constructed risk score system in the present study
differs from those in the three aforementioned studies, and may
provide a novel basis for predicting the prognosis of patients with
HBV-induced HCC.
Materials and methods
Expression profile data
The mRNA-sequencing data of HCC (platform: Illumina
HiSeq 2000 RNA Sequencing; extracted on 11th February 2018) were
extracted from TCGA (https://cancergenome.nih.gov/) database, which
included 100 HCC and 26 normal samples.
Additionally, microarray data in the Gene Expression
Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database were
identified using ‘hepatocellular carcinoma’ as the key word.
Relevant databases were selected based on the following criteria:
i) The database contained gene expression profile data; ii) the
samples were solid tumor tissues from patients with HCC; iii) the
database contained HBV infection information; and iv) the database
contained human expression profiles. A total of three databases
[including GSE55092 (22),
GSE19665 (23) and GSE10186
(24,25)] were selected. GSE55092 (including
39 HCC samples and 81 normal samples) and GSE19665 (including 5 HCC
samples and 5 normal samples) were based on the Affymetrix-GPL570
platform (Affymetrix; Thermo Fisher Scientific, Inc., Waltham, MA,
USA); the databases contained no prognosis information, and were
used for screening prognosis-associated lncRNAs and constructing
the risk score system. GSE10186 (including 118 HCC samples;
platform: Affymetrix-GPL5474; Affymetrix; Thermo Fisher Scientific,
Inc.) contained prognosis information and used for validating the
risk score system. Among the 118 HCC samples, there were 79 samples
with HBV infection status and prognosis information (including 19
HBV positive samples and 60 HBV negative samples; 48 alive samples
and 31 dead samples, mean survival time=88.62±45.04 months)
(Table I).
 | Table I.Clinical information of samples in
TCGA, GSE19665 and GSE10186. |
Table I.
Clinical information of samples in
TCGA, GSE19665 and GSE10186.
|
Characteristics | TCGA | GSE19965 | GSE10186 |
|---|
| Tumor samples | 100 | 5 | 79 |
| Control
samples | 26 | 5 | 0 |
| Age (mean ± SD,
years) | 61.64±14.70 | 64.30±8.23 | NA |
| Sex
(male/female) | 60/40 | 9/1 | NA |
| Neoplasm
histological grade (G1/G2/G3/G4/NA) | 12/51/35/1/1 | NA | NA |
| Pathologic stage
(I/II/III/IV/NA) | 38/33/23/3/3 | NA | NA |
| Satellite lesions
(positive/negative/NA) | NA | NA | 2/59/18 |
| Pathology
differentiated (moderately/poorly/moderately-poorly) | NA | 7/1/2 | NA |
| Microvascular
invasion (positive/negative/NA) | 36/52/12 | NA | 16/45/18 |
| Alcohol status
(Yes/No/NA) | NA | NA | 46/30/3 |
| HBV infection
(positive/negative/NA) | 57/43 | 5/5 | 19/60 |
| Live status
(dead/alive) | 42/58 | NA | 48/31 |
| Overall survival
time (mean ± SD, months) | 31.22±29.53 | NA | 88.62±45.04 |
Data preprocessing
The datasets were preprocessed by the following two
methods according to their differences in testing platforms. For
TCGA, the preprocessCore package (version 1.40.0, http://bioconductor.org/packages/release/bioc/html/preprocessCore.html)
(26) in R was applied for data
normalization. For the CEL files based on Affy platform, format
conversion, the supplement of missing values, background correction
and data standardization were conducted with the oligo package
(version 1.41.1, http://www.bioconductor.org/packages/release/bioc/html/oligo.html)
(27) in R.
Then, lncRNAs were annotated with the Ref_seq and
Transcript_ID provided by annotation platforms. The detection
sequences in the platforms were aligned with the human reference
genome GRCh38 by Clustal 2 software (http://www.clustal.org/clustal2/) (28). By combining the annotation and
alignment results, lncRNAs and relevant expression information were
finally obtained (29,30).
Weighted gene co-expression network
analysis (WGCNA)
WGCNA is an algorithm for the construction of a
co-expression network and the identification of disease-associated
modules (31). With TCGA as the
training dataset, and GSE55092 and GSE19665 as the validation
datasets, the R package WGCNA (version 1.61, http://cran.r-project.org/web/packages/WGCNA/index.html)
(31) was used to build a
co-expression network and screen the stable modules associated with
HCC. The processes of WGCNA included calculating correlations in
expression between the datasets, and determining adjacent function
and module partition (each module contained ≥200 RNA,
cutHeight=0.99). Additionally, functional annotation for the stable
modules was conducted via the userListEnrichment function in the
WGCNA package (31).
Differential expression analysis
For TCGA, GSE55092 and GSE19665, the DE-RNAs between
HCC and normal samples were analyzed via the MetaDE.ES algorithm in
the MetaDE package (version 1.0.5, http://cran.r-project.org/web/packages/MetaDE/)
(32,33). The RNAs with Qpval >0.05,
tau2=0, and P<0.05 and false discovery rate <0.05
were defined as consensus DE-RNAs. In particular, this study
focused on the differential expression of lncRNAs in stable
modules.
Construction and validation of risk
score system
Univariate Cox regression analysis in survival
package (version 2.4, http://cran.r-project.org/web/packages/survival/index.html)
(34) was performed using TCGA to
select for prognosis-associated lncRNAs from the lncRNAs in stable
modules. The lncRNAs with P<0.05 were considered to be
prognosis-associated lncRNAs.
Subsequently, the optimal lncRNA combinations were
screened by the Cox-PH model in penalized package (http://bioconductor.org/packages/penalized/) (35). The parameter ‘lambda’ in the Cox-PH
model was acquired via 1,000× calculation based on a
cross-validation likelihood (cvl) algorithm (36). The risk score system was
constructed via weighting the expression level
(exprlncRNA) of each lncRNA in the optimal lncRNA
combination using the corresponding regression coefficient (β). The
formula of the risk score system was as follows:
Risk score = βlncRNA1 ×
exprlncRNA1 + βlncRNA2 ×
exprlncRNA2 + … + βlncRNAn ×
exprlncRNAn.
Additionally, the robustness of the risk score
system in prognosis prediction was evaluated using GSE10186 as the
validation dataset, with Kaplan-Meier (KM) survival curves and
receiver operating characteristic (ROC) curve analysis.
Analysis of lncRNA-associated
pathways
Gene sets were extracted from stable modules
involving the optimal lncRNAs. Using Gene Set Enrichment Analysis
(http://software.broadinstitute.org/gsea/index.jsp)
(37), pathway enrichment analysis
was performed to identify lncRNA-associated pathways. The cut-off
criterion was set as P<0.05.
Results
WGCNA is able to select for stable
modules
There were 15,988 mRNAs and 851 lncRNAs shared by
GSE55092, GSE19665 and TCGA. The modules significantly associated
with HCC were selected by WGCNA. The consistency of the expression
values of the common RNAs was analyzed to ensure the comparability
of RNA expression in the three datasets. The expression
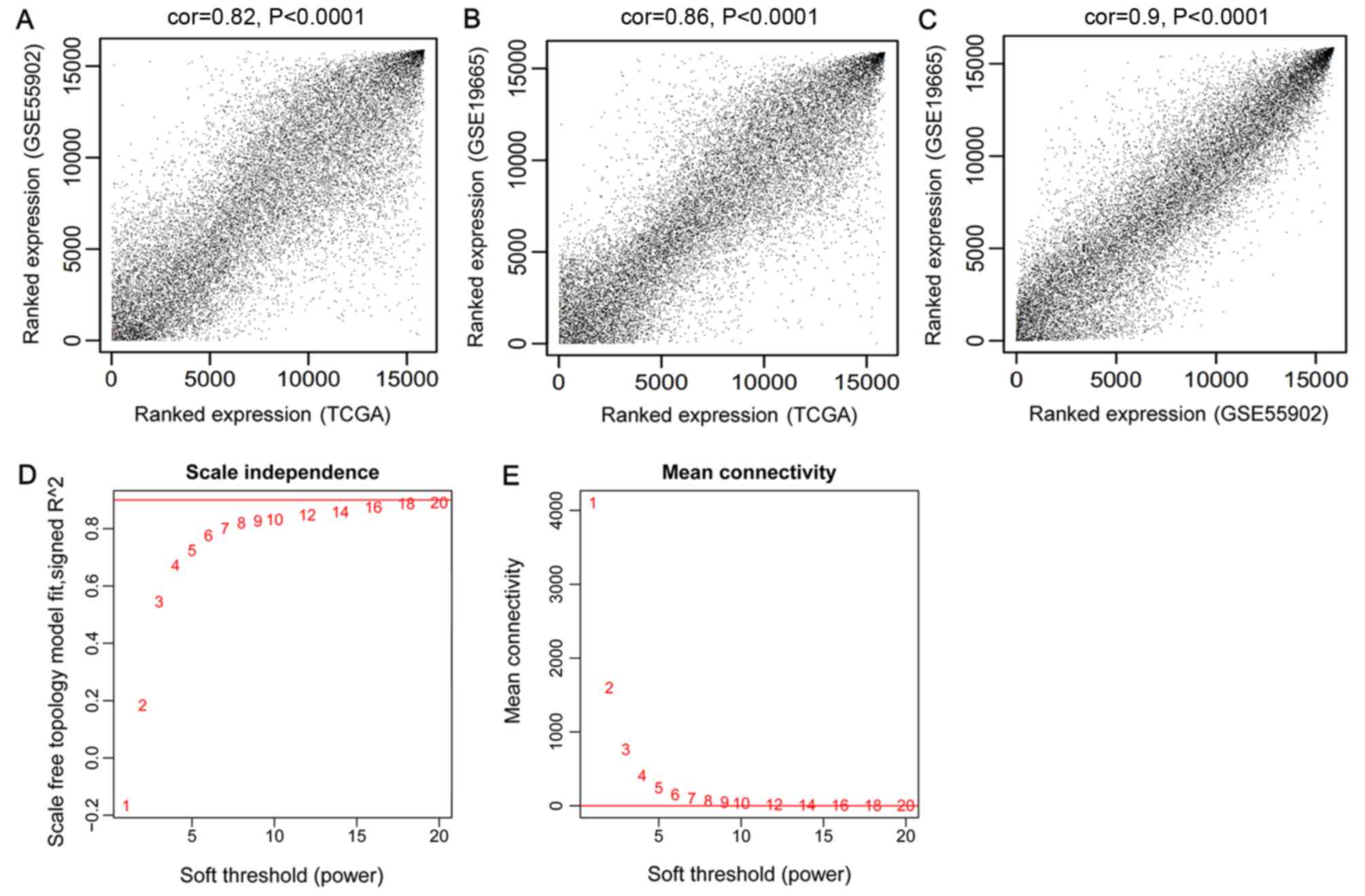
correlations were all >0.80 and P<1×10−200.
Therefore, the three datasets exhibited significant and positive
correlations (Fig. 1A-C).
An appropriate adjacency matrix weighting parameter
β (power) was selected to enable the co-expression network to
approach a scale-free network distribution. The squares of the
correlation coefficients between log(k) and log[p(k)] were acquired
to select parameter β. A higher square value indicated that the
co-expression network was closer to scale-free network distribution
(Fig. 1D). The corresponding
parameter β was selected when the square value first reached 0.9,
namely β=8. The mean connectivity degree of the RNAs in the
co-expression network was 8 when β=8, which was in accordance with
small world architecture (Fig.
1E).
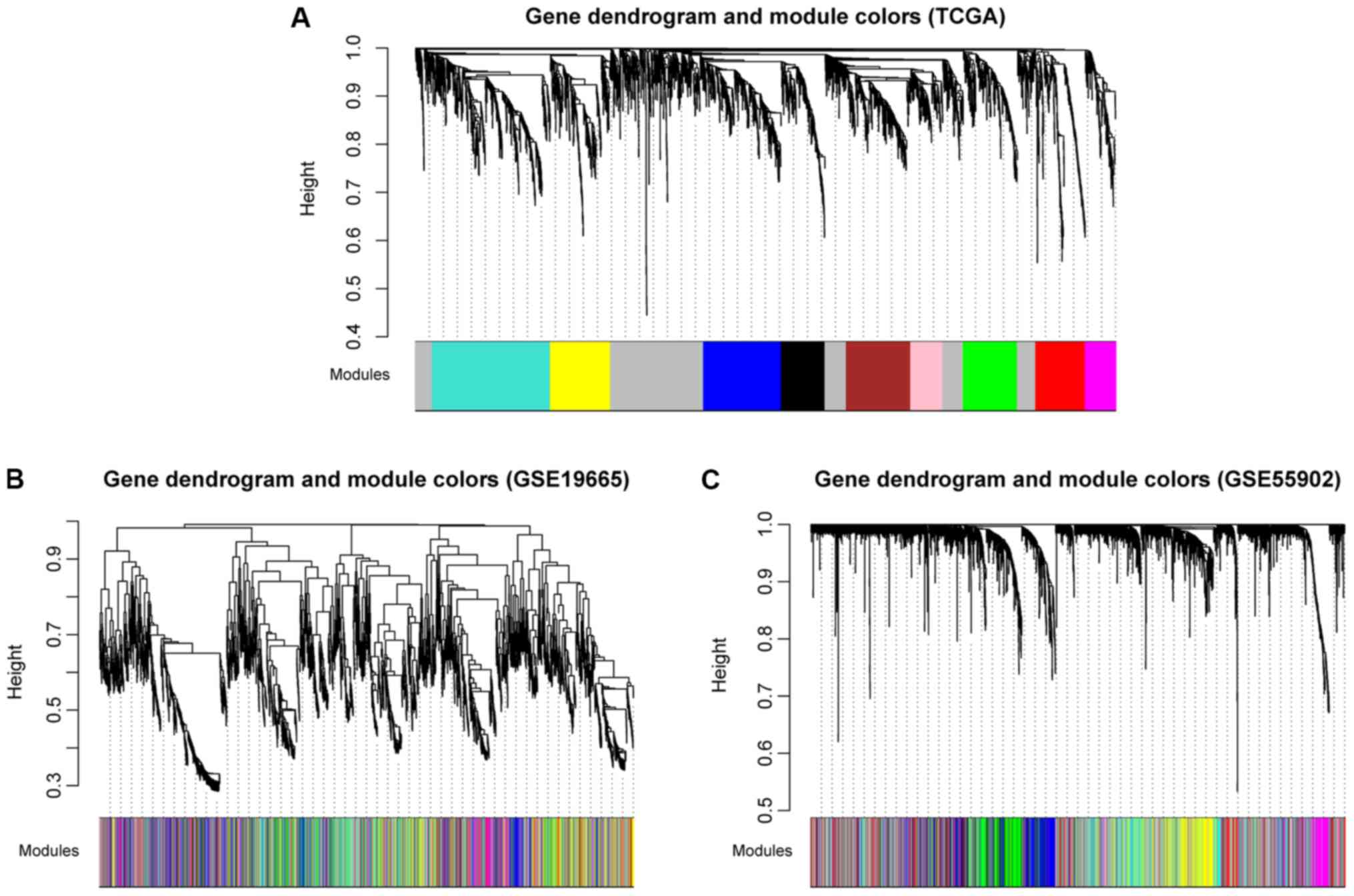
Using TCGA as the training dataset, a total of 10
modules were identified by constructing RNA adjacent matrices and
system clustering trees (Fig. 2A).
According to the modules of TCGA and the RNAs in each module,
corresponding module partitioning was performed with GSE19665
(Fig. 2B) and GSE55092 (Fig. 2C) to determine the stabilities of
the modules of TCGA. Module partitions and correlations for TCGA
were presented in Fig. 3A and B,
respectively. The results suggested that RNAs within the same
module were gathered together, thus possessing similar expression
(Fig. 3A). Additionally, the
clustering results of GSE55092 (Fig.
3C) and GSE19665 (Fig. 3D)
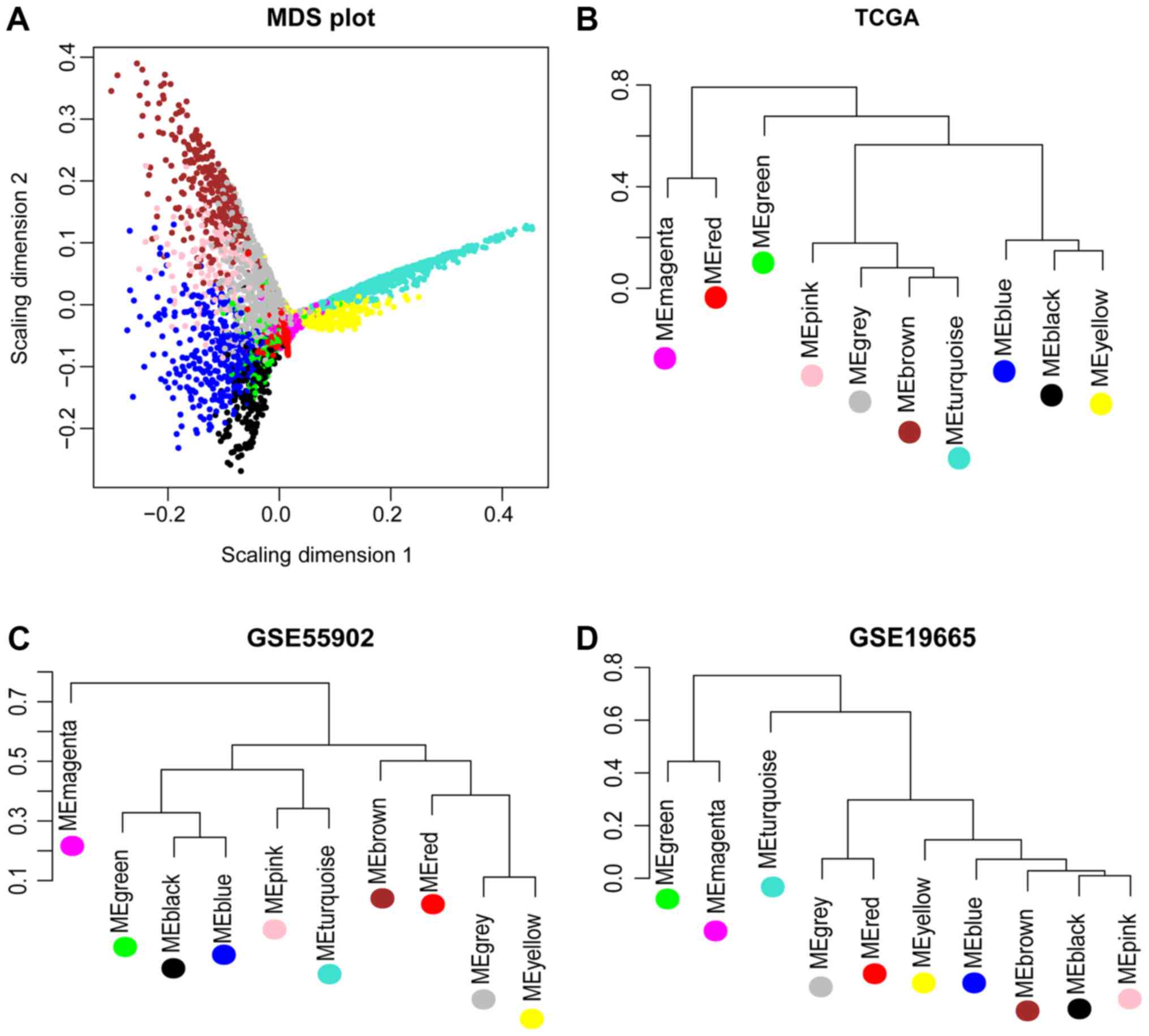
indicated that magenta, blue, yellow and green modules were
characterized by independent branches; four modules (blue, magenta,
yellow and green) were revealed to be stable modules (preservation
Z score >10). Additionally, functional annotation demonstrated
that the lncRNAs in blue, magenta, yellow and green modules
respectively associated with ‘inflammatory responses’, ‘cell
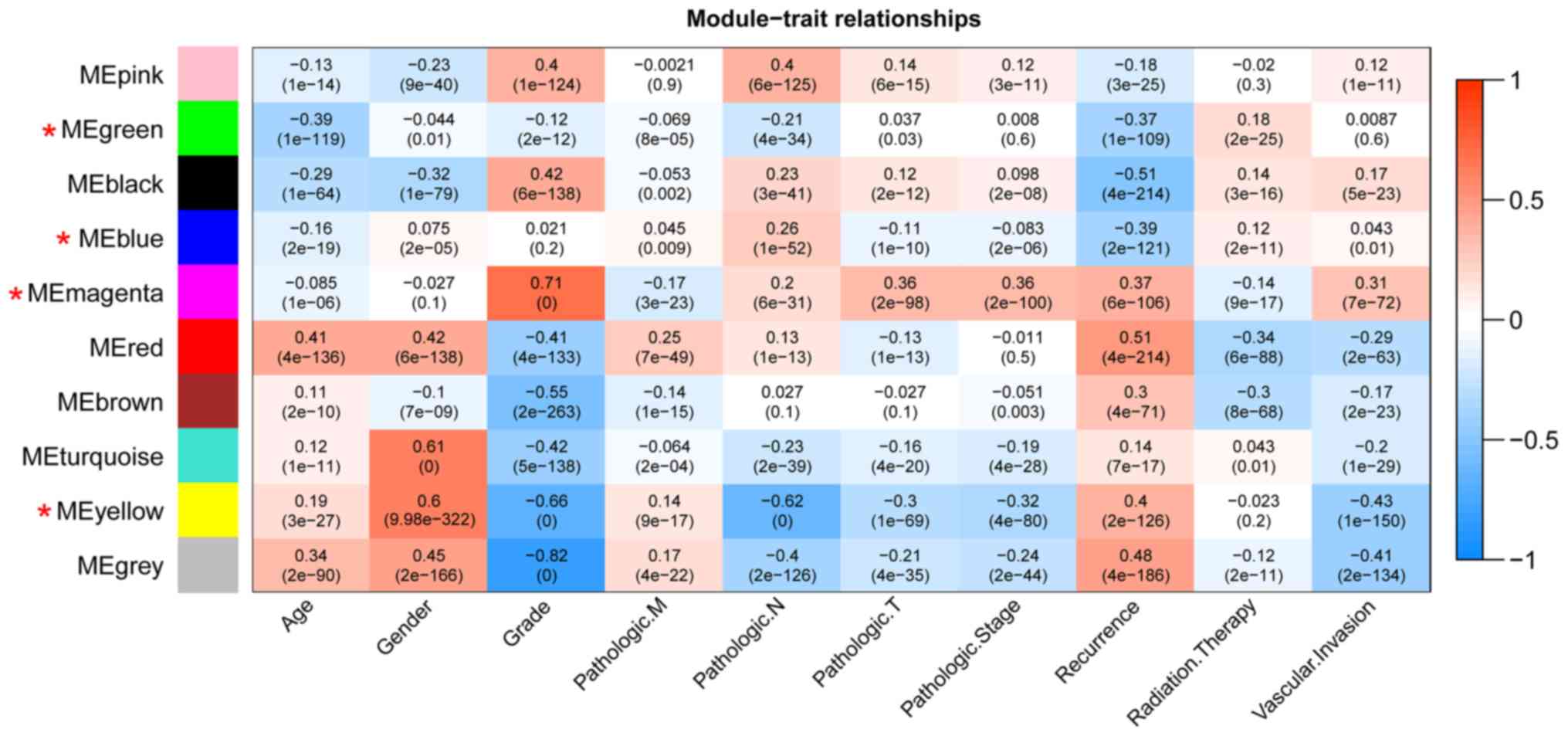
cycle’, ‘blood coagulation’ and ‘cell adhesion’ (Table II). Furthermore, the clinical
information [including age, gender, grade, tumor, node and
metastasis (TNM) stage, pathological stage, recurrence, radiation
therapy and vascular invasion] of the samples in TCGA were
integrated to calculate the correlation between the RNAs in each
module and clinical factor. The results revealed that the four
stable modules were significantly correlated to grade, TNM stage,
pathologic stage, recurrence and radiation therapy (Fig. 4). Thus, the lncRNAs in the four
stable modules were examined for subsequent analysis.
| Table II.Stabilities of the 10 modules
identified in TCGA and the biological functions enriched for the
lncRNAs in the modules. |
Table II.
Stabilities of the 10 modules
identified in TCGA and the biological functions enriched for the
lncRNAs in the modules.
| TCGA | Color | Module size | mRNA | LncRNA | Preservation
Z-score | Module
annotation |
|---|
| Module 1 | black | 206 | 206 | 0 | 5.6804 | Chemotaxis |
| Module 2 | blue | 371 | 364 | 7 | 18.9870 | Inflammatory
response |
| Module 3 | brown | 303 | 302 | 1 | 0.7094 | Oxidation-reduction
process |
| Module 4 | green | 264 | 255 | 9 | 26.5495 | Cell adhesion |
| Module 5 | grey | 796 | 794 | 2 | 0.8546 | Response to
nutrient levels |
| Module 6 | magenta | 147 | 143 | 4 | 26.2491 | Cell cycle |
| Module 7 | pink | 150 | 150 | 0 | 8.0652 | Regulation of cell
proliferation |
| Module 8 | red | 233 | 232 | 1 | 0.3724 | Synaptic
transmission |
| Module 9 | turquoise | 555 | 552 | 3 | 6.3217 | Ion transport |
| Module 10 | yellow | 286 | 283 | 3 | 25.7553 | Blood
coagulation |
Differential expression analysis
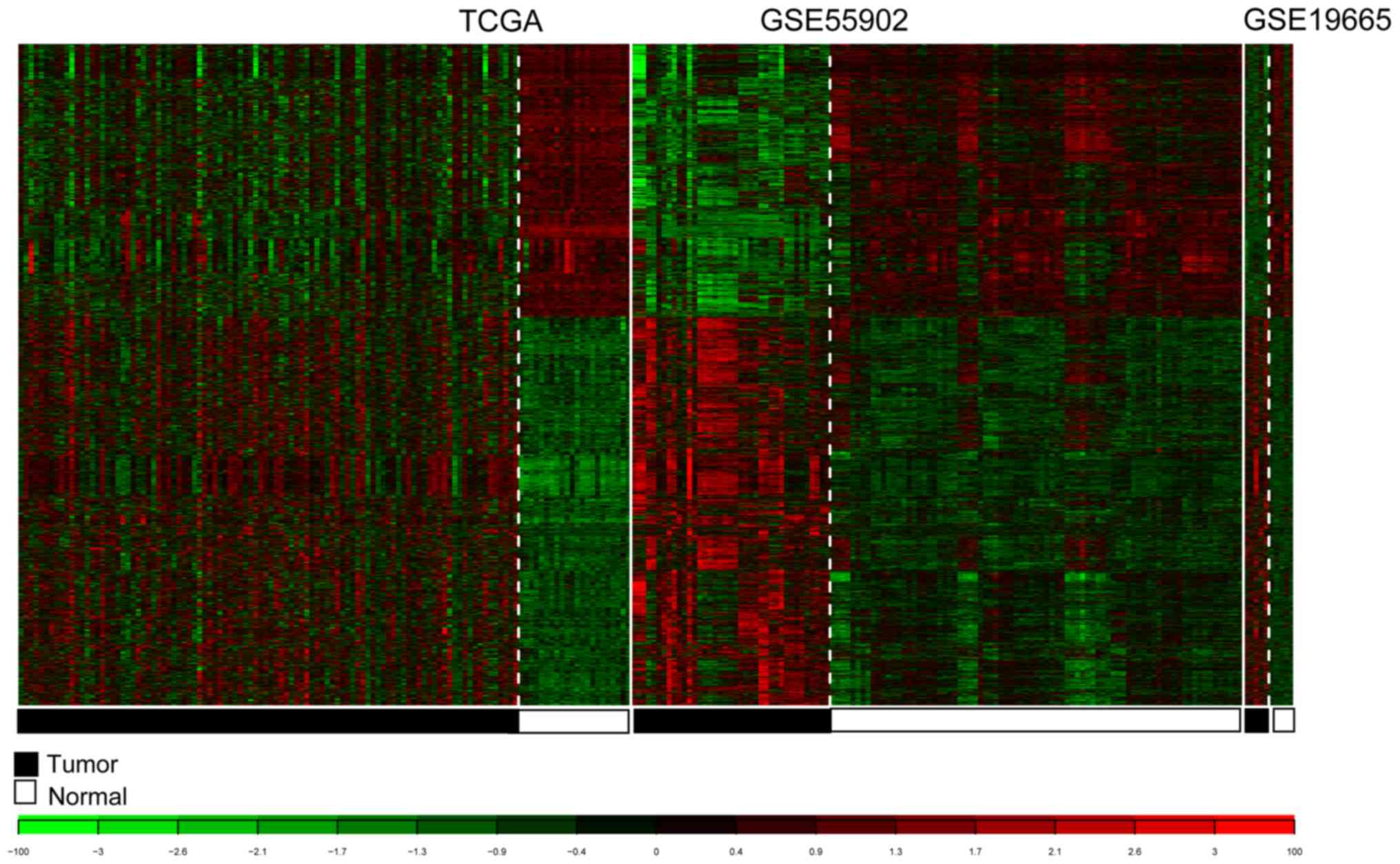
For TCGA, GSE55092 and GSE19665, 3,051 consensus
DE-RNAs were reported. The 3,051 DE-RNAs included 10 lncRNAs and
3,041 mRNAs. The clustering heatmaps for the consensus DE-RNAs in
the three datasets are presented in Fig. 5.
Construction and validation of the
risk score system
The expression levels of the lncRNAs in stable
modules were extracted from TCGA, and then 14 prognosis-associated
lncRNAs were selected based on univariate Cox regression analysis.
Using the Cox-PH model, the optimal lncRNA combination was selected
from the 14 prognosis-associated lncRNAs. Finally, a 9-lncRNA
optimal combination was obtained, involving: DiGeorge syndrome
critical region gene 9 (DGCR9); glucosidase, β, acid 3
(GBA3); HLA complex group 4 (HCG4);
N-acetyltransferase 8B (NAT8B); neighbor of breast cancer 1
gene 2 (NBR2); prostate androgen-regulated transcript 1
(PART1); ret finger protein like 1 antisense RNA 1
(RFPL1S); solute carrier family 22 member 18 antisense
(SLC22A18AS) and T-cell leukemia/lymphoma 6 (TCL6;
Table III). The formula for the
risk score system based on the optimal lncRNA combination was:
| Table III.LncRNAs in the optimal lncRNA
combination. |
Table III.
LncRNAs in the optimal lncRNA
combination.
| LncRNA | Coefa | Hazard ratio | P-value | Module color |
|---|
| DGCR9 | −0.0308 | 0.90 | 0.0230 | blue |
| GBA3 | 0.2033 | 1.07 | 0.0240 | magenta |
| HCG4 | 0.4416 | 1.11 | 0.0170 | magenta |
| NAT8B | 0.7662 | 1.11 | 0.0120 | magenta |
| NBR2 | −0.5517 | 0.72 | 0.0068 | yellow |
| PART1 | 0.3786 | 1.04 | 0.0490 | green |
| RFPL1S | 0.0590 | 1.09 | 0.0340 | green |
|
SLC22A18AS | 0.0427 | 1.11 | 0.0200 | green |
| TCL6 | 1.4731 | 1.23 | 0.0004 | green |
Risk score = (−0.03084) × ExpDGCR9 +
(0.203324) × ExpGBA3 + (0.441589) × ExpHCG4 +
(0.766193) × ExpNAT8B + (−0.5517) × ExpNBR2 +
(0.378576) × ExpPART1 + (0.058961) ×
ExpRFPL1S + (0.042655) × ExpSLC22A18AS +
(1.473117) × ExpTCL6.
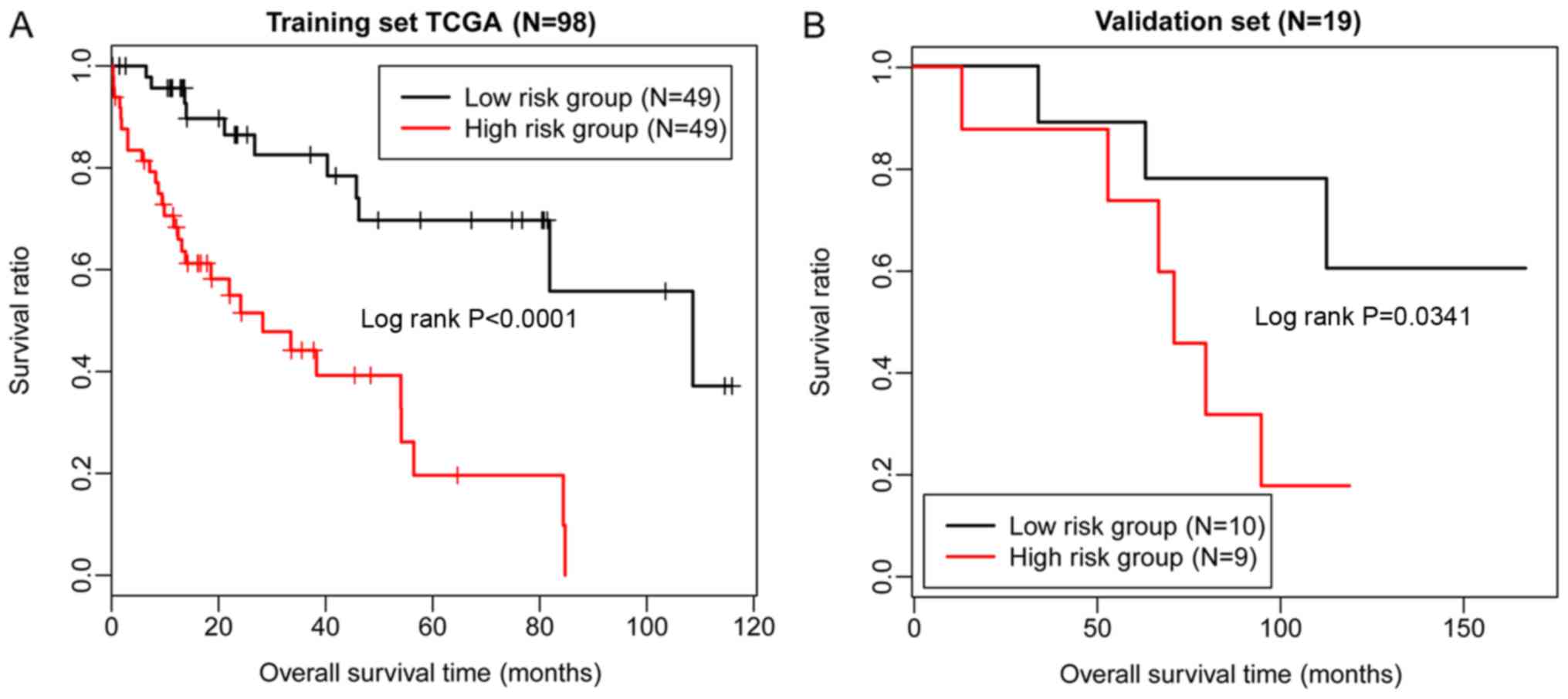
Risk scores were calculated for the samples in the
dataset from TCGA using the risk score system. Based on the median
of risk scores, the samples in TCGA were classified into high- and
low-risk groups. Then, the difference between the survival times of
individuals within the two groups was characterized by KM survival
curves. The results indicated that the risk score system could
effectively distinguish the high- and low-risk groups (P<0.01;
Fig. 6A). Subsequently, the risk
score system was applied to the validation dataset GSE10186,
demonstrating that the high- and low-risk groups could also be
differentiated (P=0.0341; Fig.
6B). Therefore, the risk score system exhibited high
robustness, and the nine lncRNAs were significantly associated with
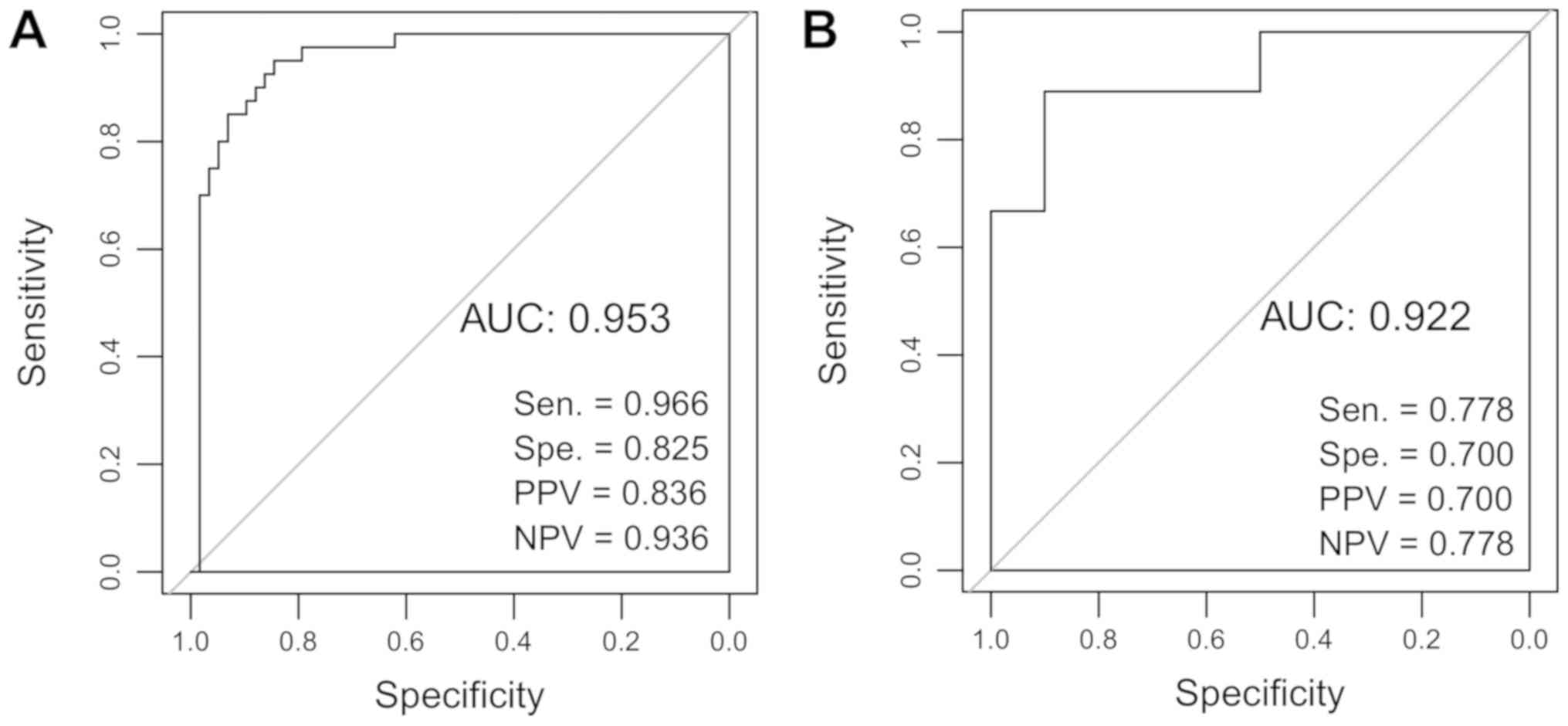
the prognosis of patients with HCC. Furthermore, ROC curve analysis
was applied to evaluate the predictive diagnostic value of the
9-lncRNA risk score system using TCGA and the validation dataset.
The sensitivity, specificity, positive predictive value, negative
predictive value, and the area under the ROC curves (AUC) were
determined. The AUC values of the 9-lncRNA risk score system for
TCGA and GSE10186 were 0.953 and 0.922, respectively (Fig. 7).
Analysis of lncRNA-associated
pathways
mRNAs closely associated with the nine lncRNAs were
selected from the four stable modules, and an lncRNA-mRNA
co-expression network was constructed (Fig. 8). In particular,
phosphoenolpyruvate carboxykinase 2 (PCK2) was positively
regulated by the lncRNA GBA3 in the co-expression network.
The gene sets corresponding to the nine lncRNAs were separately
determined with pathway enrichment analysis. The results revealed
that the mRNAs associated with the nine lncRNAs were mainly
enriched in ‘cell cycle’, ‘drug metabolism’, ‘peroxisome
proliferator-activated receptor (PPAR) signaling pathway’, ‘cell
focal adhesion’, ‘calcium signaling pathways’, and ‘endogenous cell
receptor interactions’.
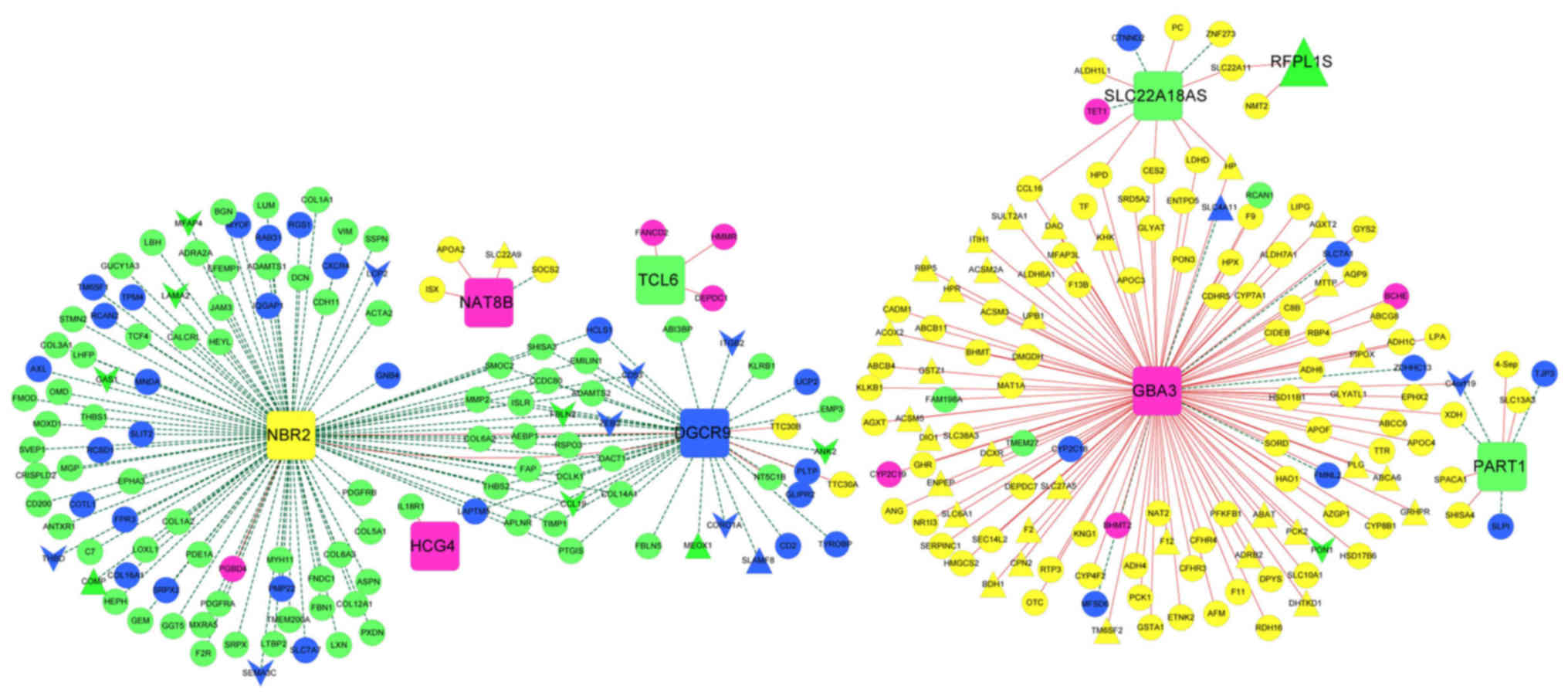 | Figure 8.LncRNA-mRNA co-expression network for
the nine optimal lncRNAs. Squares represent lncRNAs. Regular
triangles and inverted triangles separately represent consensus
upregulated genes and downregulated genes. Circles represent
non-consensus differentially expressed RNAs co-expressed with
lncRNAs. The color of a node indicates the module involved. Red and
green lines represent positive and negative co-expression
associations, respectively. LncRNA, long noncoding RNA; DGCR9,
DiGeorge syndrome critical region gene 9; GBA3, glucosidase, β,
acid 3; HCG4, HLA complex group 4; NAT8B, N-acetyltransferase 8B;
NBR2, neighbor of breast cancer 1 gene 2; PART1, prostate
androgen-regulated transcript 1; RFPL1S, ret finger protein like 1
antisense RNA 1; SLC22A18AS, solute carrier family 22 member 18
antisense; TCL6, T-cell leukemia/lymphoma 6. |
Discussion
In the present study, blue, magenta, yellow and
green modules were screened as four stable modules by WGCNA.
Additionally, the four stable modules were determined to be
significantly associated with certain clinical factors, including
grade, TNM stage, pathologic stage, recurrence and radiation
therapy. For TCGA, GSE55092 and GSE19665, a total of 3,051
consensus DE-RNAs were identified, including 10 lncRNAs and 3,041
mRNAs. Subsequently, 14 prognosis-associated lncRNAs were selected,
and a 9-lncRNA optimal combination, including DGCR9, GBA3, HCG4,
NAT8B, NBR2, PART1, RFPL1S, SLC22A18AS and TCL6 was
identified. A risk score system was built based on the optimal
lncRNA combination, which effectively distinguished high- and
low-risk individuals within the validation dataset GSE10186.
DGCR5 expression was reported to be lower in
HCC serum and tissues (38);
therefore, DGCR5 may function as a valuable diagnostic and
prognostic marker in patients with HCC. There was a significant
correlation reported between NAT2 polymorphism and HCC in
smokers positive for HBV, indicating that NAT2 may be
associated with HBV-associated hepatocarcinogenesis in smokers
(39,40). NAT10 exhibits higher levels
of expression in HCC tissues compared with peritumoral tissues
(41); thus, NAT10 may be
applied in the prognosis and treatment of patients with HCC.
NAT10 overexpression enhances the tumorigenic activity of
mutated p53 via upregulating its expression, and is correlated with
the poor survival of patients, suggesting that NAT10 serves
critical roles in the prognosis and therapy of p53-mutated HCC
(42). Therefore, DGCR9 and
NAT8B may be important in the pathology of HCC.
In the present study, PCK2 was proposed to be
positively regulated by GBA3 in the lncRNA-mRNA
co-expression network. The insulin signaling pathway (involving
PCK2) and the ubiquitin-mediated proteolysis pathway
[involving HECT, UBA and WWE domain containing 1, E3 ubiquitin
protein ligase (HUWE1)] serve critical roles in
hepatocarcinogenesis, and PCK2 and HUWE1 may affect
the proliferation of HCC cells via involvement in the
aforementioned pathways (43). Via
the induction of NBR2 and adenosine
5′-monophosphate-activated protein kinase/PPARα signaling,
microRNA-19a can suppress the autophagy of
D-GalN/lipopolysaccharide-stimulated hepatocytes (44). SLC22A18 is a paternally
imprinted gene that encodes a polyspecific organic cation
transporter, which exhibits gain-of-imprinting in breast cancers
and hepatocarcinomas (45).
SLC22A18 is predominantly expressed in fetal and adult
kidney and liver tissues; additionally, SLC22A18 and
SLC22A18AS exhibit genomic imprinting in adult liver and
breast tissues (46).
Collectively, these studies suggest that GBA3, NBR2 and
SLC22A18AS expression may affect the progression of HCC in
patients.
To the best of our knowledge, no studies have
previously reported associations of PART1 or TCL6
with HCC; however, PART1 and TCL6 have been linked to
the prognosis of other tumors. For example, the lncRNA PART1
correlated with the overall survival and progression-free survival
of patients with oral squamous cell carcinoma (47). PART1 also contributes to cell
proliferation and apoptosis in prostate cancer by suppressing
Toll-like receptor signaling pathways (48); therefore, PART1 may present a
potential therapeutic target. Additionally, the expression of
TCL6 is downregulated in clear cell renal cell carcinoma,
and may be an unfavorable prognostic indicator for the disease
(49). Thus, it is possible that
PART1 and TCL6 may also be involved in the
pathogenesis of HCC.
There are certain limitations to the present study.
The constructed 9-lncRNA risk score system requires the
demonstration of clinical relevance by using clinical samples
obtained from an independent patient cohort. Additionally, platform
differences and data heterogeneities between the downloaded
datasets may affect the accuracy of the risk score system. The
validation dataset, GSE10186, contained the largest number of
samples with HBV infection information among the three GEO
datasets; however, a greater number of samples is required for
rigorous and robust analysis.
In conclusion, four stable modules and 14
prognosis-associated lncRNAs were identified. A risk score system
was established based on the optimal nine lncRNAs, which may be
valuable for predicting the prognosis of patients with HBV-positive
HCC, and improve understanding of the pathology of HCC.
Furthermore, employing the system with a larger independent cohort
of patients is required for further validation.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Project
supported by the Presidential Foundation of the 302 Hospital of the
People's Liberation Army (grant no. YNKT2014027).
Availability of data and materials
The datasets used during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
HL performed data analysis and wrote the manuscript.
PZ, XJ, YZ, YC, TY, JW and LW contributed significantly in the
interpretation and the analysis of data, and in revising the
manuscript. YS conceived and designed the study. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Gastroenterologist. 362:1907–1917.
2003.
|
|
2
|
Nguyen VTT, Law MG and Dore GJ: Hepatitis
B-related hepatocellular carcinoma: Epidemiological characteristics
and disease burden. J Viral Hepat. 16:453–463. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hiotis SP, Rahbari NN, Villanueva GA,
Klegar E, Luan W, Wang Q and Yee HT: Hepatitis B vs. hepatitis C
infection on viral hepatitis-associated hepatocellular carcinoma.
BMC Gastroenterol. 12:642012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Waghray A, Murali AR and Menon KN:
Hepatocellular carcinoma: From diagnosis to treatment. World J
Hepatol. 7:1020–1029. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuen MF, Hou JL and Chutaputti A; Asia
Pacific Working Party on Prevention of Hepatocellular Carcinoma, :
Hepatocellular carcinoma in the Asia pacific region. J
Gastroenterol Hepatol. 24:346–353. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giannini EG, Farinati F, Ciccarese F,
Pecorelli A, Rapaccini GL, Di Marco M, Benvegnù L, Caturelli E,
Zoli M, Borzio F, et al: Prognosis of untreated hepatocellular
carcinoma. Hepatology. 61:184–190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jemal A, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beckedorff FC, Amaral MS,
Deocesano-Pereira C and Verjovski-Almeida S: Long non-coding RNAs
and their implications in cancer epigenetics. Biosci Rep.
33:e000612013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jin Y, Wu D, Yang W, Weng M, Li Y, Wang X,
Zhang X, Jin X and Wang T: Hepatitis B virus × protein induces
epithelial-mesenchymal transition of hepatocellular carcinoma cells
by regulating long non-coding RNA. Virol J. 14:2382017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lv D, Wang Y, Zhang Y, Cui P and Xu Y:
Downregulated long non-coding RNA DREH promotes cell proliferation
in hepatitis B virus-associated hepatocellular carcinoma. Oncol
Lett. 14:2025–2032. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu TT, Xu XM, Hu Y, Deng JJ, Ge W, Han NN
and Zhang MX: Long noncoding RNAs in hepatitis B
virus-relatedhepatocellular carcinoma. World J Gastroenterol.
21:7208–7217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deng X, Zhao XF, Liang XQ, Chen R, Pan YF
and Liang J: Linc00152 promotes cancer progression in hepatitis B
virus-associated hepatocellular carcinoma. Biomed Pharmacother.
90:100–108. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li J, Wang X, Tang J, Jiang R, Zhang W, Ji
J and Sun B: HULC and Linc00152 act as novel biomarkers in
predicting diagnosis of hepatocellular carcinoma. Cell Physiol
Biochem. 37:687–696. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang K, Guo WX, Li N, Gao CF, Shi J, Tang
YF, Shen F, Wu MC, Liu SR and Cheng SQ: Serum LncRNAs profiles
serve as novel potential biomarkers for the diagnosis of
HBV-positive hepatocellular carcinoma. PLoS One. 10:e01449342015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu J, Xie F, Geng L, Shen W, Sui C and
Yang J: Investigation of serum lncRNA-uc003wbd and lncRNA-AF085935
expression profile in patients with hepatocellular carcinoma and
HBV. Tumor Biol. 36:3231–3236. 2015. View Article : Google Scholar
|
|
17
|
Nguyen QT, Lee EJ, Huang MG, Park YI,
Khullar A and Plodkowski RA: Diagnosis and treatment of patients
with thyroid cancer. Am Health Drug Benefits. 8:30–40.
2015.PubMed/NCBI
|
|
18
|
Servant N, Roméjon J, Gestraud P, La Rosa
P, Lucotte G, Lair S, Bernard V, Zeitouni B, Coffin F,
Jules-Clément G, et al: Bioinformatics for precision medicine in
oncology: Principles and application to the SHIVA clinical trial.
Front Genet. 5:1522014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Z, Wu Q, Feng S, Zhao Y and Tao C:
Identification of four prognostic LncRNAs for survival prediction
of patients with hepatocellular carcinoma. Peerj. 5:e35752017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng H, Li P, Kwok JG, Korrapati A, Li
WT, Qu Y, Wang XQ, Kisseleva T, Wang-Rodriguez J and Ongkeko WM:
Alcohol and hepatitis virus-dysregulated lncRNAs as potential
biomarkers for hepatocellular carcinoma. Oncotarget. 9:224–235.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yuan W, Sun Y, Liu L, Zhou B, Wang S and
Gu D: Circulating LncRNAs serve as diagnostic markers for
hepatocellular carcinoma. Cell Physiol Biochem. 44:125–132. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Melis M, Diaz G, Kleiner DE, Zamboni F,
Kabat J, Lai J, Mogavero G, Tice A, Engle RE, Becker S, et al:
Viral expression and molecular profiling in liver tissue versus
microdissected hepatocytes in hepatitis B virus-associated
hepatocellular carcinoma. J Transl Med. 12:2302014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Deng YB, Nagae G, Midorikawa Y, Yagi K,
Tsutsumi S, Yamamoto S, Hasegawa K, Kokudo N, Aburatani H and
Kaneda A: Identification of genes preferentially methylated in
hepatitis C virus-related hepatocellular carcinoma. Cancer Sci.
101:1501–1510. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoshida Y, Nijman SM, Kobayashi M, Chan
JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K,
Hashimoto M, et al: Integrative transcriptome analysis reveals
common molecular subclasses of human hepatocellular carcinoma.
Cancer Res. 69:7385–7392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shtraizent N, DeRossi C, Nayar S,
Sachidanandam R, Katz LS, Prince A, Koh AP, Vincek A, Hadas Y,
Hoshida Y, et al: MPI depletion enhances O-GlcNAcylation of p53 and
suppresses the Warburg effect. elife. 6:e224772017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bolstad BM, Irizarry RA, Astrand M and
Speed TP: A comparison of normalization methods for high density
oligonucleotide array data based on variance and bias.
Bioinformatics. 19:185–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Irizarry RA, Bolstad BM, Collin F, Cope
LM, Hobbs B and Speed TP: Summaries of Affymetrix GeneChip probe
level data. Nucleic Acids Res. 31:e152003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Larkin MA, Blackshields G, Brown NP,
Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm
A, Lopez R, et al: Clustal W and Clustal X version 2.0.
Bioinformatics. 23:2947–2948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou M, Guo M, He D, Wang X, Cui Y, Yang
H, Hao D and Sun J: A potential signature of eight long non-coding
RNAs predicts survival in patients with non-small cell lung cancer.
J Transl Med. 13:2312015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou M, Xu W, Yue X, Zhao H, Wang Z, Shi
H, Cheng L and Sun J: Relapse-related long non-coding RNA signature
to improve prognosis prediction of lung adenocarcinoma. Oncotarget.
7:29720–29738. 2016.PubMed/NCBI
|
|
31
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qi C, Hong L, Cheng Z and Yin Q:
Identification of metastasis-associated genes in colorectal cancer
using metaDE and survival analysis. Oncol Lett. 11:568–574. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang X, Kang DD, Shen K, Song C, Lu S,
Chang LC, Liao SG, Huo Z, Tang S, Ding Y, et al: An R package suite
for microarray meta-analysis in quality control, differentially
expressed gene analysis and pathway enrichment detection.
Bioinformatics. 28:2534–2536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang P, Wang Y, Hang B, Zou X and Mao JH:
A novel gene expression-based prognostic scoring system to predict
survival in gastric cancer. Oncotarget. 7:55343–55351.
2016.PubMed/NCBI
|
|
35
|
Goeman JJ: L1 penalized estimation in the
Cox proportional hazards model. Biom J. 52:70–84. 2010.PubMed/NCBI
|
|
36
|
Knafl GJ, Dixon JK, O'Malley JP, Grey M,
Deatrick JA, Gallo A and Knafl KA: Scale development based on
likelihood cross-validation. Stat Methods Med Res. 21:599–619.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tilford CA and Siemers NO: Gene set
enrichment analysis. Methods Mol Biol. 563:99–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang R, Wang X, Zhang W, Zhangyuan G, Jin
K, Yu W, Xie Y, Xu X, Wang H and Sun B: Down-regulation of LncRNA
DGCR5 correlates with poor prognosis in hepatocellular carcinoma.
Cell Physiol Biochem. 40:707–715. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu MW, Yang SY, Yang SY, Hsiao TJ, Chang
HC, Lin SM, Liaw YF, Chen PJ and Chen CJ: Role of
N-acetyltransferase polymorphisms in hepatitis B related
hepatocellular carcinoma: Impact of smoking on risk. Gut.
47:703–709. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang J, Xu F and Ouyang C: Joint effect
of polymorphism in the N-acetyltransferase 2 gene and smoking on
hepatocellular carcinoma. Tumor Biol. 33:1059–1063. 2012.
View Article : Google Scholar
|
|
41
|
Zhang X, Liu J, Yan S, Huang K, Bai Y and
Zheng S: High expression of N-acetyltransferase 10: A novel
independent prognostic marker of worse outcome in patients with
hepatocellular carcinoma. Int J Clin Exp Pathol. 8:14765–14771.
2015.PubMed/NCBI
|
|
42
|
Li Q, Liu X, Jin K, Lu M, Zhang C, Du X
and Xing B: NAT10 is upregulated in hepatocellular carcinoma and
enhances mutant p53 activity. BMC Cancer. 17:6052017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu YX, Zhang SF, Ying-Hua JI, Guo SJ,
Wang GF and Zhang GW: Whole-exome sequencing identifies mutated
PCK2 and HUWE1 associated with carcinoma cell proliferation in a
hepatocellular carcinoma patient. Oncol Lett. 4:847–851. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu YM, Ma JH, Zeng QL, Lv J, Xie XH, Pan
YJ and Yu ZJ: MiR-19a affects hepatocyte autophagy via regulating
lncRNA NBR2 and AMPK/PPARα in D-GalN/lipopolysaccharide--stimulated
hepatocytes. J Cell Biochem. 119:358–365. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ali AM, Bajaj V, Gopinath KS and Kumar A:
Characterization of the human SLC22A18 gene promoter and its
regulation by the transcription factor Sp1. Gene. 429:37–43. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Martin-kleiner I, Radetić M, Grbeša I,
Parazajder D, Kovačić M, Radetić M and Trošelj KG: The analysis of
the SLC22A18 gene and its natural antisense transcripts in human
papillary thyroid tumors. Proceedings of the Congress of the
Croatian Society of Biochemistry and Molecular Biology with
international participation (HDBMB 2008). Croatian Society of
Biochemistry and Molecular Biology; Zagreb: 2008
|
|
47
|
Li S, Chen X, Liu X, Yu Y, Pan H, Haak R,
Schmidt J, Ziebolz D and Schmalz G: Complex integrated analysis of
lncRNAs-miRNAs-mRNAs in oral squamous cell carcinoma. Oral Oncol.
73:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun M, Geng D, Li S, Chen Z and Zhao W:
LncRNA PART1 modulates toll-like receptor pathway to influence cell
proliferation and apoptosis in prostate cancer cells. Biol Chem.
399:387–395. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Su H, Sun T, Wang H, Shi G, Zhang H, Sun F
and Ye D: Decreased TCL6 expression is associated with poor
prognosis in patients with clear cell renal cell carcinoma.
Oncotarget. 8:5789–5799. 2017.PubMed/NCBI
|