Introduction
Multidrug-resistant bacterial infections, especially
those caused by Gram-negative pathogens, have become a significant
global public health threat, and they result in considerable
patient mortality and morbidity and cause great economic and
production losses in the community (1). Serratia marcescens (S.
marcescens), a Gram-negative bacillus, is an important
nosocomial pathogen that can cause an array of infections, such as
bloodstream infections, pneumonia, urinary tract infections,
central nervous system infections, and conjunctivitis (2). The development of novel antibiotics,
especially those used to treat multidrug-resistant pathogens, has
stagnated over the last half century. Therefore, gaining more
insights into the genetic mechanisms responsible for the
antimicrobial resistance of pathogens is both urgent and
necessary.
The accumulation of evidence has led to the
identification of a number of genes that are responsible for
intrinsic resistance to different classes of antibiotics, including
β-lactams, aminoglycosides, and fluoroquinolones (3). Intrinsic mechanisms underlying
bacterial antibiotic resistance include naturally occurring genes
found in the chromosome of the host, such as the multiple
multidrug-resistant efflux systems of β-lactamase of Gram-negative
bacteria (4). A recent study
reported an isolate of S. marcescens harboring the 16S rRNA
methyltransferase gene rmtB, together with various
β-lactamase genes and quinolone resistance genes (5). Another study revealed that
imipenem-resistance in S. marcescens may be mediated by the
plasmid expression of Klebsiella pneumoniae carbapenemase-2
(KPC-2) (6). In addition, evidence
has demonstrated that Gram-negative bacteria can employ several
strategies to protect themselves from polymyxin antibiotics,
including a variety of lipopolysaccharide (LPS) modifications in
addition to the formation of capsules, use of efflux pumps, and
overexpression of the outer membrane protein OprH (7). Although many studies aimed at
elucidating the underlying mechanisms of antibiotic resistance have
been performed, much remains largely unknown, especially the
molecular mechanisms of the multi-drug resistance of S.
marcescens, and awaits discovery.
The development of next-generation sequencing
technologies has provided valuable resources for genetic research
and other scientific disciplines (8,9). In
this study, the parental S. marcescens strain and S.
marcescens strains exhibiting multidrug-resistance were
analyzed with high-throughput RNA sequencing to identify variations
at the transcriptome level. Differentially expressed genes (DEGs)
between the parental strain and the multidrug-resistant S.
marcescens strains were screened, followed by functional
enrichment analysis, protein-protein interaction (PPI) network
construction, and module extraction. The results provide additional
molecular clues that will aid in elucidating the mechanisms and
metabolic pathways related to multidrug-resistance in S.
marcescens.
Materials and methods
Bacterial strains and culture
conditions
Bacterial isolation, identification, and culture
were performed according to conventional methods. The strains were
derived from sputum, blood, lavage fluid, urine, and throat swab
samples; transferred onto Columbia agar supplemented with 5% sheep
blood (bioMérieux, Marcy l'Etoile, France) and MacConkey's agar
plates; and incubated at 35°C for 24 h. The oxidase-negative
Gram-negative bacilli was identified using the VITEK® 2
GN card (bioMérieux) (10). The
drug sensitivity AST-GN16 card was used to test the antimicrobial
agent susceptibility of the isolated strains. In this study, a
total of three drug-susceptible S. marcescens strains (named
MYQT1, MYQT2, and MYQT3) and three multidrug-resistant S.
marcescens strains (named MYQT4, MYQT5, and MYQT6) were
obtained from six different patients and used for the follow-up
analysis.
Total RNA extraction
The cultures were centrifuged at 8,000 × g to
precipitate bacterial cells. Total RNA was extracted using the hot
phenol method as previously described with modifications (11). Subsequently, the bacterial cells
were washed two times with RNAse-free saline or phosphate-buffered
saline (PBS; cat. no. E607016-0500; BBI solutions, Cardiff, UK).
Then, 400–600 µl TES solution was added according to the
precipitation amount, and the bacterial cells were resuspended. The
same amount of phenol-water (Sinopharm Chemical Reagent Co., Ltd.,
Shanghai, China) was added followed by violent mixing. Centrifuge
tubes containing a mixture of each sample, TES, and phenol-water
were agitated at 65°C for 30–60 min in a Thermomixer Compact 5350
(Eppendorf, Hamburg, Germany), and then, the tubes were placed on
ice and allowed to stand for 5 min. Then, the mixtures were
centrifuged at 11,000 × g for 10 min at 4°C. The upper aqueous
phase was selected and transferred to a new tube. Subsequently, a
1/2 volume of TRK-1002 lysis-solution and 2/3 volume of 95% ethyl
alcohol was added to the upper aqueous phase, followed by vortex
blending. Total RNA was then extracted using a TRK-1002
Purification kit (LC Sciences, Houston TX, USA), following the
manufacturer's instructions. RNA quality was evaluated using an
Agilent Bioanalyser (Agilent Technologies, Inc., Santa Clara, CA,
USA).
Library preparation and Illumina
sequencing
To remove ribosomal RNA, we used a Ribo-Zero™
Magnetic kit (Bacteria) (cat. no. MRZB12424; Illumina, Inc., San
Diego, CA, USA) according to the manufacturer's protocol. RNA
samples were subjected to further purification using a Zymo RNA
Clean and Concentrator kit (cat. no. R1015; Zymo Research, Irvine,
CA, USA) to enrich the mRNA according to the manufacturer's
instructions. Each mRNA sample was suspended in 10 µl of RNase-free
water, and the concentration of the obtained RNA was determined.
Bacterial mRNA was fragmented and stranded, and paired-end
libraries of total RNA were generated using Illumina TruSeq
Stranded Total RNA HT Sample Preparation kits (cat. no.
RS-122-2203, Illumina, Inc.). All the samples were sequenced using
an Illumina HiSeq X10 sequencer (Illumina, Inc.).
Mapping of reads and differential
expression analysis
RNA-seq datasets were obtained from six samples from
two experiment settings. The original RNA-seq datasets were MYQT1,
MYQT2, MYQT3, MYQT4, MYQT5, and MYQT6 with 9748744, 9669644,
9765080, 9638041, 9742165, and 9750956 read pairs. All RNA-seq
reads were cleaned with Trimmomatic (12), and then, the read qualities were
ascertained with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
In order to determine the appropriate reference genome to use for
read mapping, all cleaned RNA-seq reads we first used to perform
BLAST (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/)
searches against the NCBI nt database. The BLAST results indicated
that S. marcescens subsp. marcescens Db11 was the closest
reference genome. The six cleaned RNA-seq datasets to S.
marcescens subsp. marcescens Db11 were then mapped using Bowtie
2 (13). In addition, the genomic
viewer Integrative Genomics Viewer (IGV) (14) was used to evaluate the mapping
quality. BEDtools (15) was used
to calculate the read count of each gene over the six samples.
Correlation analysis was performed to ensure that the read count
qualities were stable within samples (across three different
batches).
edgeR (16) uses
the calcNormFactors function to normalize for RNA composition by
finding a set of scaling factors for the library sizes that
minimize the log-fold changes (FC) between the samples for most
genes (16). In this study, edgeR
(16) was used to perform
differential gene expression analysis. edgeR uses the Cox-Reid
profile-adjusted likelihood (CR) method for estimating dispersions
(16). The screening criteria were
|log2FC|>1 and a P-value <0.05. Additionally, IGV
was used to zoom in on some significant DEGs.
Functional enrichment of DEGs
The functional enrichment tool DAVID (17) was applied to perform enrichment
analysis. In the analysis, functional annotations of gene ontology
(GO), including the ontology of cellular component (CC), biological
process (BP), and molecular function (MF) were mostly focused on
(18). The GO terms with a gene
count >2 and a P-value <0.05 were considered statistically
significant.
PPI construction and analysis
The STRING database (19) contains known and predicted
protein-protein associations that are integrated and transferred
across organisms. Since S. marcescens in this study was not
included in the STRING database, the Serratia odorifera
4Rx13 strain was used in the database which has a high degree
of homology with S. marcescens. The protein sequences of the
DEGs were downloaded from the NCBI database and blasted in the
STRING database, and the interactions between the proteins were
predicted. Required Confidence (combined score) >0.4 was
selected as the threshold for predicting PPIs.
Cytoscape 3.4.0 (https://cytoscape.org/) is an open source software
project for biological network visualization and data integration.
According to the network connectivity, important nodes in the PPI
network could be identified (20).
In the present study, three calculation methods for determining
network topology properties were combined, including degree
centrality (21), betweenness
centrality (22), and closeness
centrality (23), and the
importance of nodes was analyzed in the network. The Cytoscape
plugin CytoNCA (24) was used for
the calculation of network topology properties (parameter setting:
Network, without weight). In CytoNCA output, the higher the node
score is, the more important the position in the network, and the
more likely it is to be the key node.
Module selection and analysis
In PPI networks, similar functional proteins tend to
cluster together and co-occur in central network locations
(25). Therefore, studying the
protein complex of a PPI network or functional clustering module
can help determine the unknown functions of proteins. In the
present study, the MCODE (26)
tool was applied to extract significant modules from the PPI
network. The default parameters were set as Degree Cutoff: 2; Node
Score Cutoff: 0.2; K-Core: 2; and Max. Depth: 100. Moreover, GO
enrichment analysis was performed for genes in the selected modules
with a threshold of a gene count >2 and a P-value <0.05.
Results
RNA-seq analysis of the bacterial
samples
After read cleaning, there were 8088448, 7929058,
8070351, 8229630, 8240862, and 8193913 read pairs in MYQT1, MYQT2,
MYQT3, MYQT4, MYQT5, and MYQT6, respectively. The overall alignment
rates to the closest reference genome were 72, 81, 93, 77, 82, and
82% for MYQT1, MYQT2, MYQT3, MYQT4, MYQT5, and MYQT6, respectively.
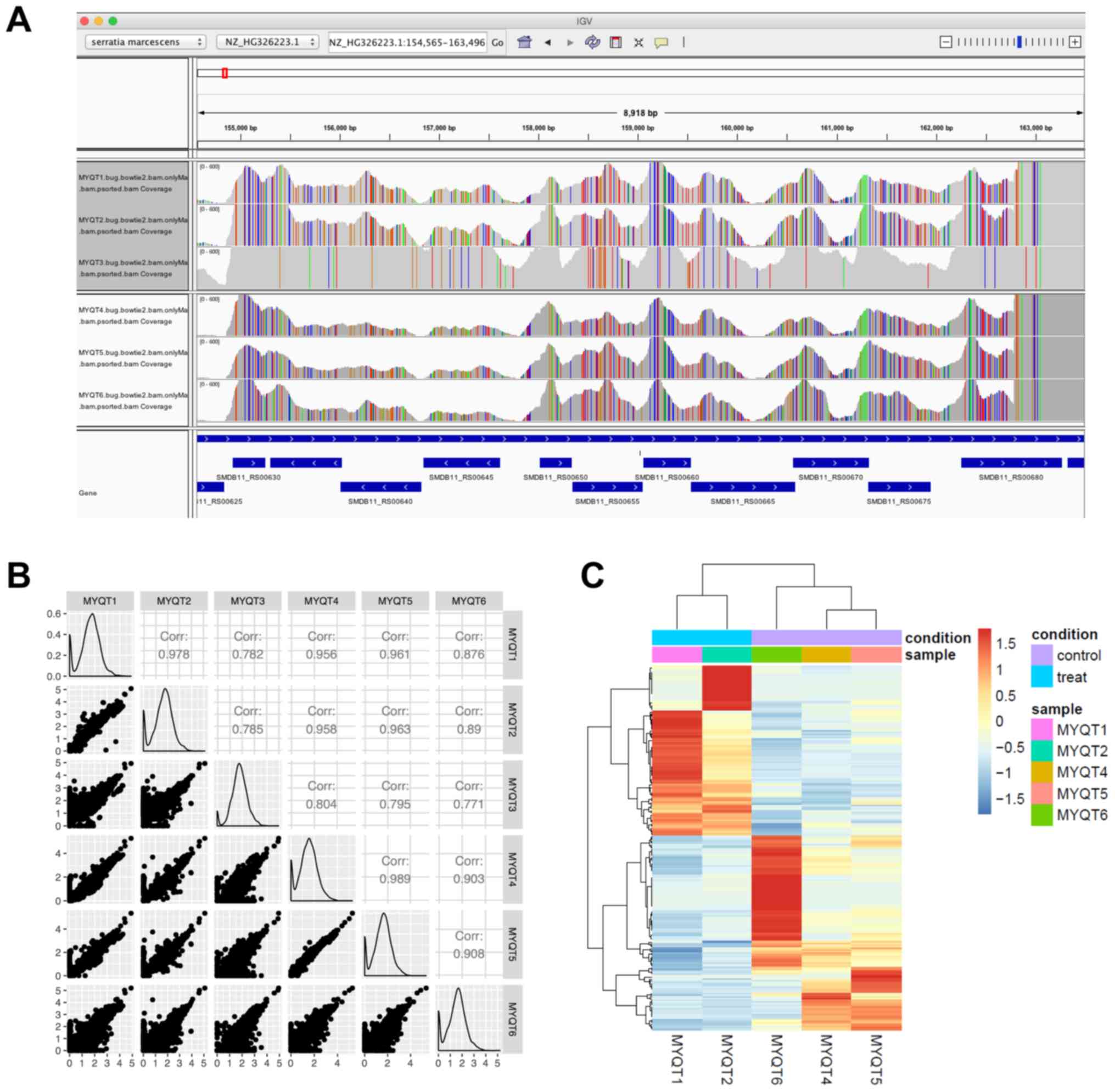
In the IGV screenshot (Fig. 1A),
we present the locus tags from SMDB11_RS00620 to SMDB11_RS00680.
This genomic region was selected picked since most of the samples
had a high read coverage. In the IGV plot, the vertical strips (in
red, green, blue, or orange) indicate where the RNA-seq reads
contained different nucleotides (mutated nucleotides/SNPs). The
five samples excluding MYQT3 had similar SNP patterns, leading us
to question whether the MYQT3 data corresponded to a different
S. marcescens strain. As presented in Fig. 1B, each small scatter plot reveals
the correlation of gene expression [presented and normalized
transcripts per million (TPM)] between two samples. As revealed in
the plot, MYQT3 had very a different expression pattern compared to
MYQT1 and MYQT2, and thus, MYQT3 was excluded from the following
analysis.
Differential expression analysis
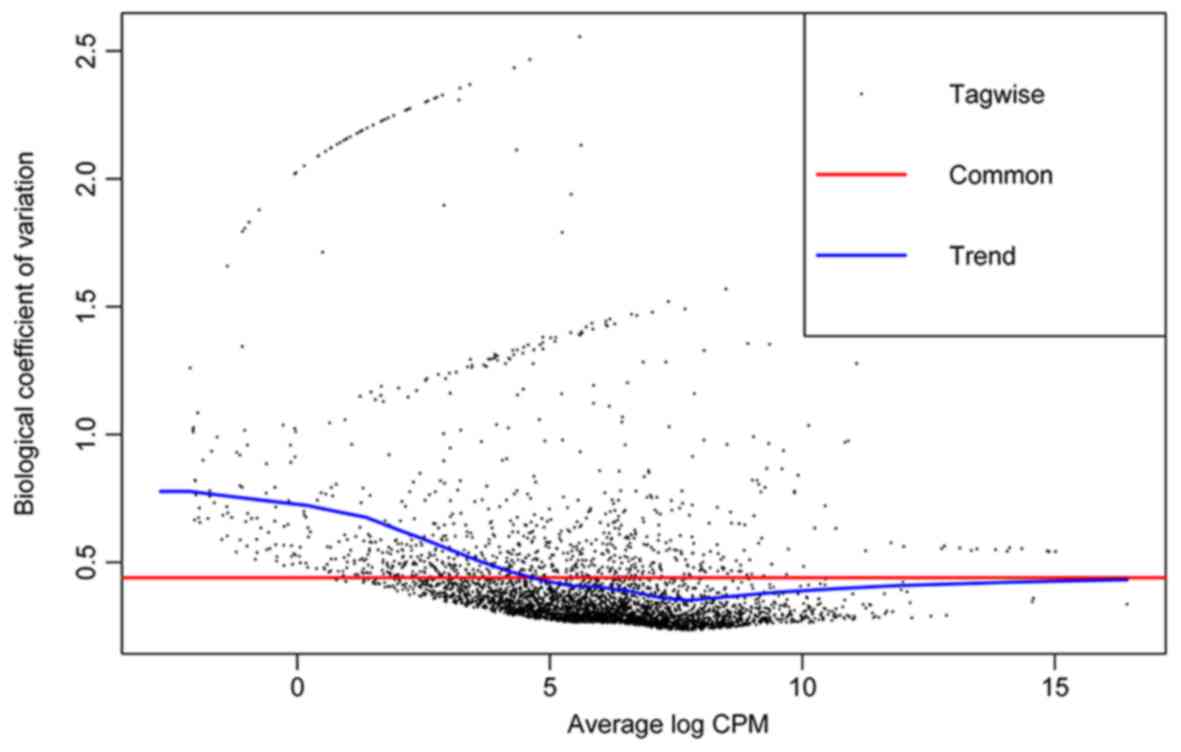
edgeR calculates the quadratic mean-variance
(dispersion) relationship to moderate the degree of dispersion
across features (genes) (Fig. 2).
With the threshold of |log2 FC|>1 and a P-value
<0.05, a total of 225 DEGs were identified. The most significant
DEG was SMDB11_RS09300 (GTP cyclohydrolase FolE2) with a
log2 FC of 6.4. The heat map is presented in Fig. 1C.
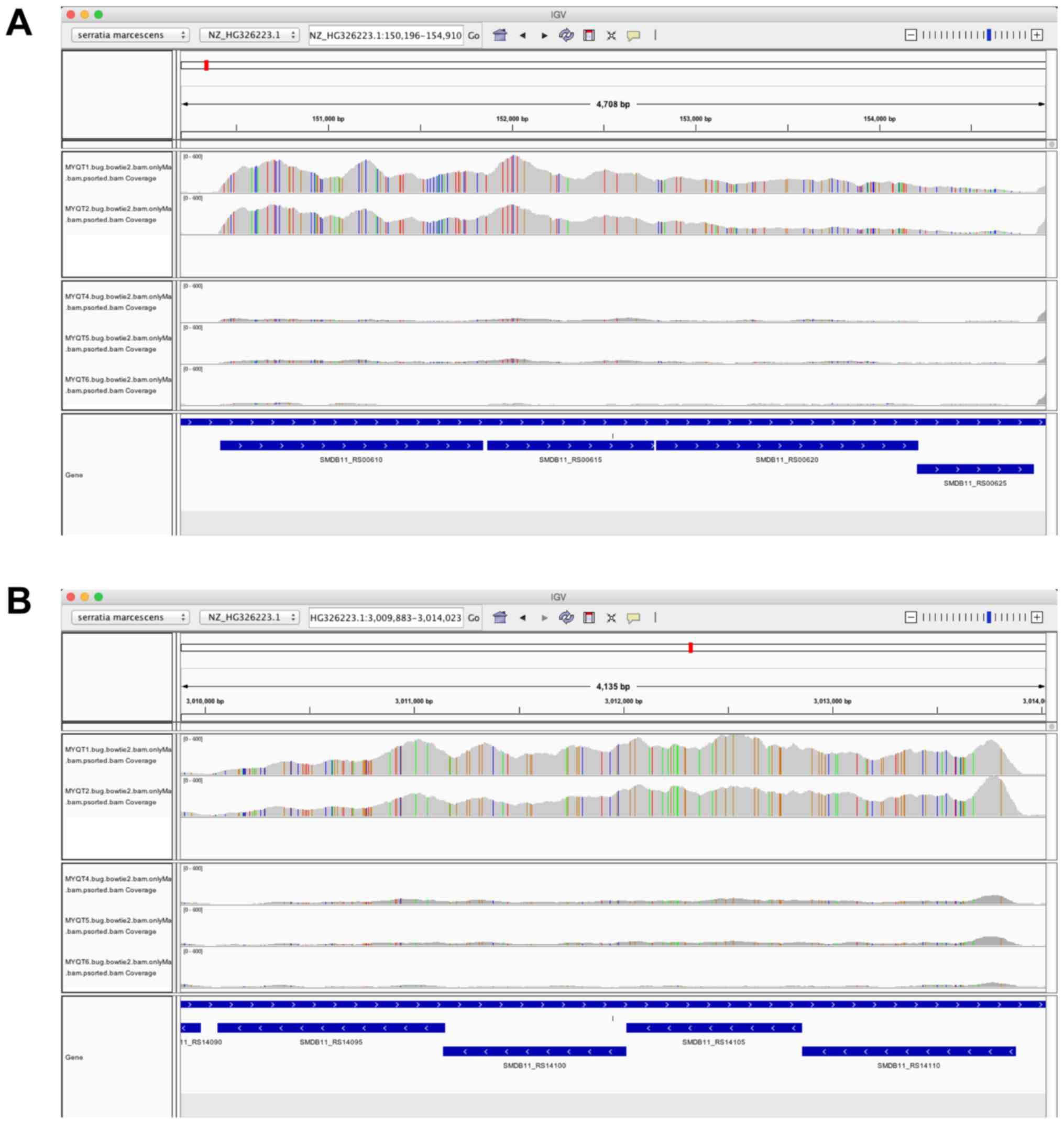
Using IGV, we identified the genomic region where
the most significant DE gene was located, which was the region from
locus tag SMDB11_RS00610 to SMDB11_RS00625 (siroheme synthase,
sulfate adenylyltransferase subunit 2, sulfate adenylyltransferase,
and adenylyl-sulfate kinase) (Fig.
3A). The four genes were all identified as DEGs by edgeR, and
the four genes were highly expressed in the MYQT1 and MYQT2 groups
compared to that in the other groups (MYQT4, MYQT5, and MYQT6). The
IGV plot of this region strongly supported the conclusion that the
four genes belonged to the same operon. Additionally, the genomic
region from locus tag SMDB11_RS14095 to SMDB11_RS14110
(sulfate/thiosulfate transporter subunit, sulfate/thiosulfate
transporter permease subunit, sulfate/thiosulfate transporter
subunit, and thiosulfate transporter subunit) (Fig. 3B) was also assessed. The four genes
were all identified as DEGs by edgeR, and the four genes were
highly expressed in the MYQT1 and MYQT2 groups compared to that in
the other groups (MYQT4, MYQT5, and MYQT6). Once again, the IGV
plot of this region strongly supported the conclusion that the four
genes belonged to the same operon.
Functional enrichment analysis of
DEGs
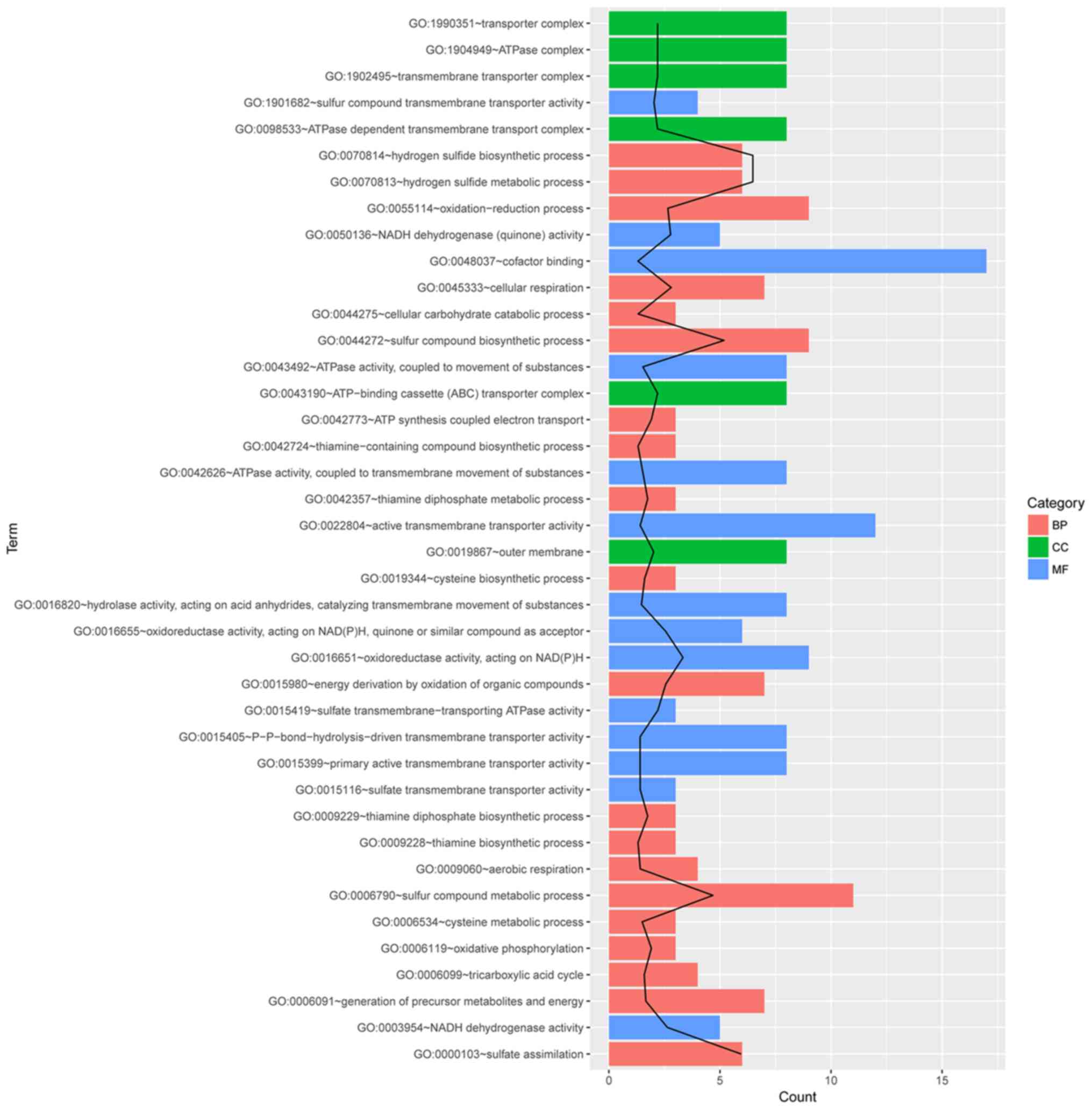
The 225 DEGs were used to perform functional
enrichment analysis, and several GO terms related to
antibiotic-resistant mechanisms were identified. The initial
assessment of the two sets of significant DE genes and their gene
names (in the two IGV plots) indicated that hydrogen sulfide
metabolic process and sulfate transmembrane-transporting ATPase
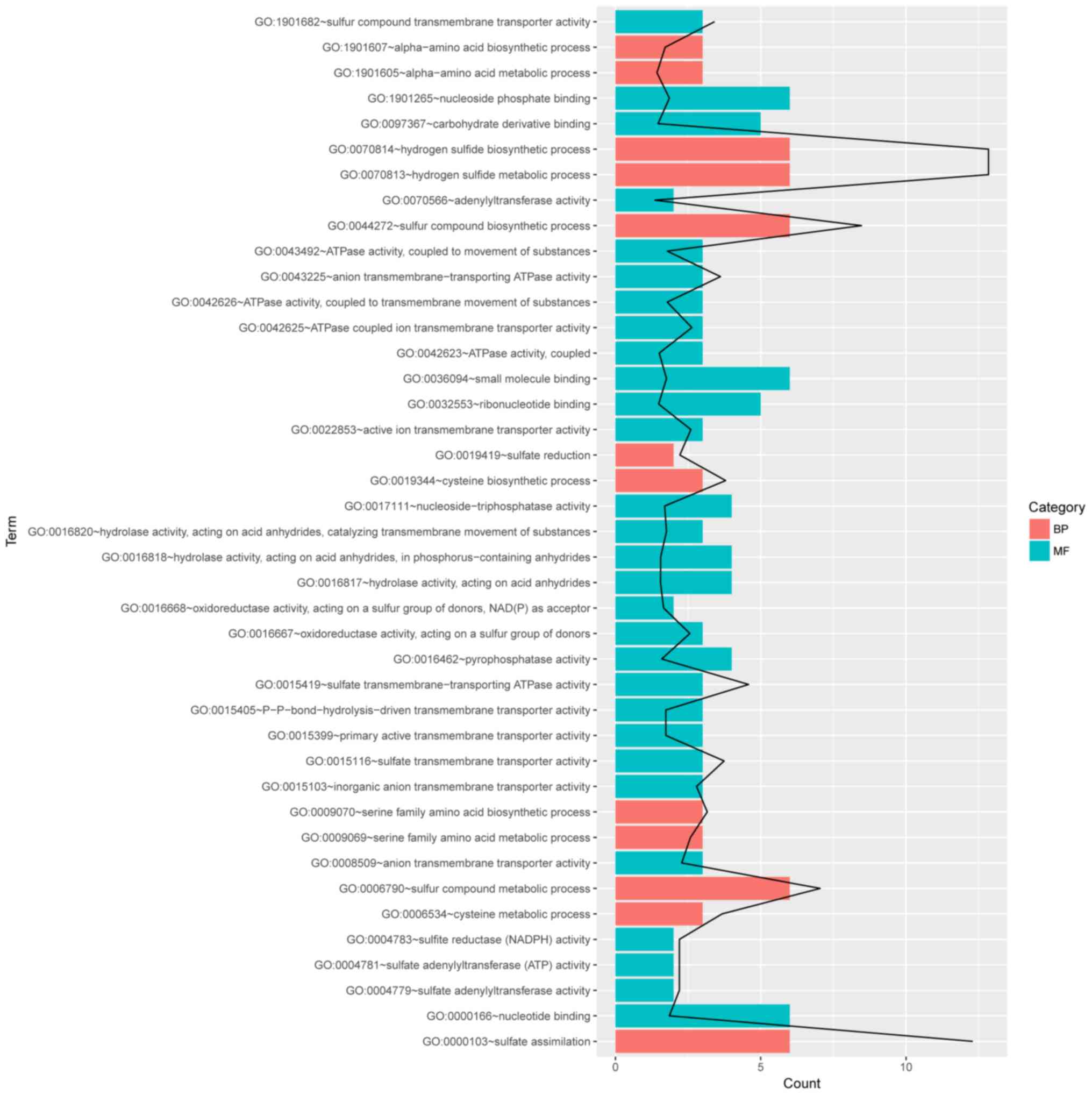
activity may be enriched. Specifically, as revealed in Fig. 4, the first two enriched functional
groups were GO:0070814~hydrogen sulfide biosynthetic process and
GO:1901682~sulfur compound transmembrane transporter activity. The
first two GO terms mostly reflected the two sets of DEGs (operons)
we identified in the DEG analysis. The other GO term related to
antibiotics-resistant mechanisms was GO:0043190~ATP-binding
cassette (ABC) transporter complex.
Construction and analysis of the PPI
network and extraction of significant modules
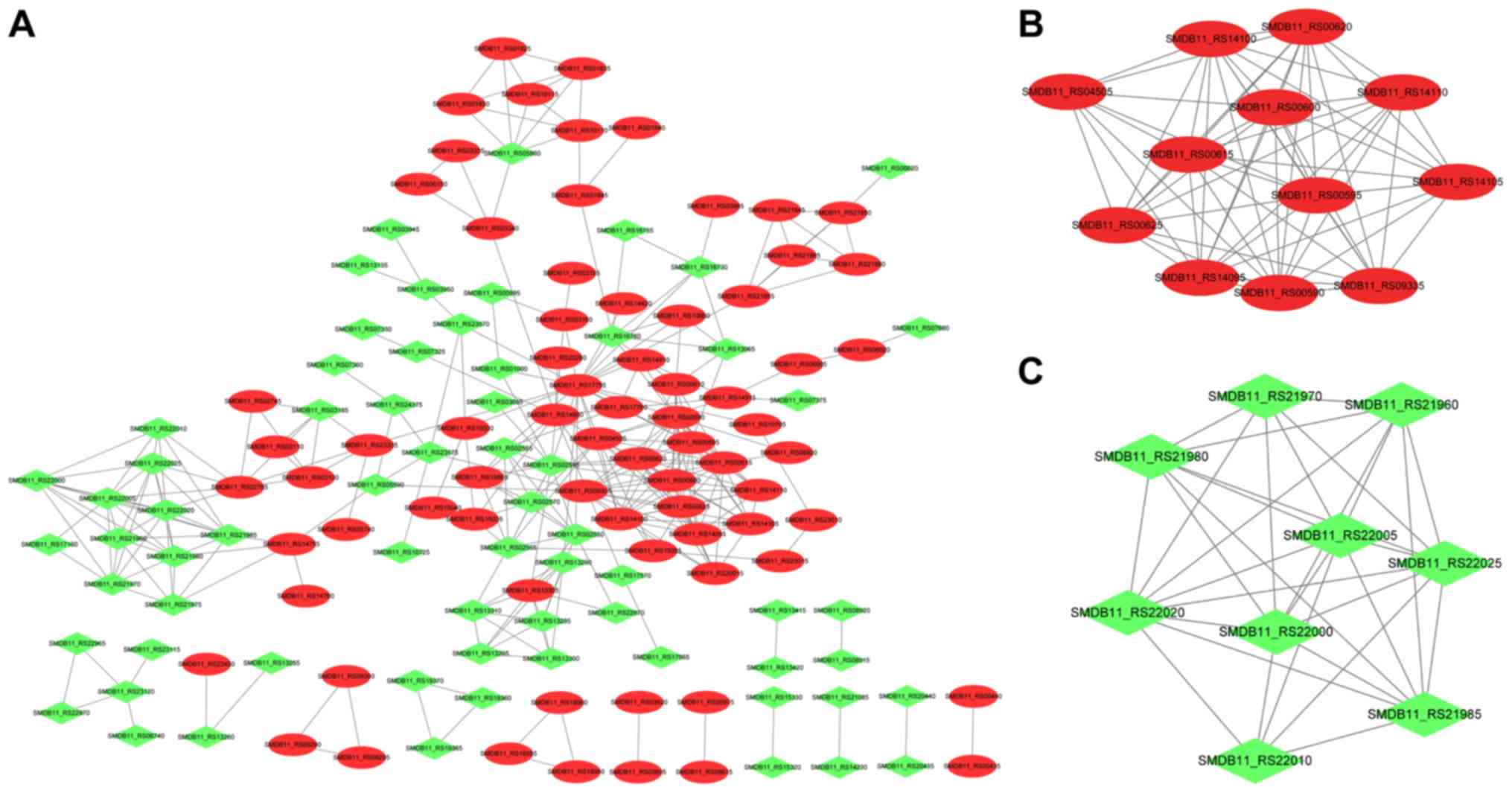
The PPI network was constructed (Fig. 5A) and consisted of 140 nodes
(proteins) and 318 edges (interactions). After analyzing three
types of network topology properties of nodes in the PPI network,
15 important nodes were identified, as revealed in Table I. The results indicated that four
DEGs (SMDB11_RS17755, SMDB11_RS00590, SMDB11_RS04505, and
SMDB11_RS02545) belonged to the top 15 genes regardless of the
calculation method used.
 | Table I.Top 15 DEGs identified using three
different calculation methods. |
Table I.
Top 15 DEGs identified using three
different calculation methods.
|
Gene_symbol | Degree | Gene_symbol | Betweenness | Gene_symbol |
Closeness |
|---|
|
SMDB11_RS17755 | 21 | SMDB11_RS17755 | 5978.15 | SMDB11_RS17755 |
0.028501127 |
|
SMDB11_RS00590 | 16 | SMDB11_RS23335 | 2504.5398 | SMDB11_RS04505 |
0.028292285 |
|
SMDB11_RS00615 | 15 | SMDB11_RS04505 | 2078.2375 | SMDB11_RS00590 |
0.028280772 |
|
SMDB11_RS00595 | 15 | SMDB11_RS14800 | 2021.148 | SMDB11_RS00595 |
0.028234817 |
|
SMDB11_RS04505 | 14 | SMDB11_RS02765 | 1661.6104 | SMDB11_RS14100 |
0.02822335 |
|
SMDB11_RS00620 | 14 | SMDB11_RS16760 | 1243.2932 | SMDB11_RS02545 |
0.028194726 |
|
SMDB11_RS00600 | 14 | SMDB11_RS09335 | 1066.5082 | SMDB11_RS16760 |
0.028114887 |
|
SMDB11_RS09335 | 13 | SMDB11_RS23570 | 1013.2664 | SMDB11_RS23335 |
0.028103517 |
|
SMDB11_RS00625 | 13 | SMDB11_RS21855 | 1010 | SMDB11_RS14910 |
0.028103517 |
|
SMDB11_RS02545 | 13 | SMDB11_RS01845 | 915.4805 | SMDB11_RS02550 |
0.028097836 |
|
SMDB11_RS14100 | 13 | SMDB11_RS05590 | 890.28687 | SMDB11_RS00615 |
0.028080808 |
|
SMDB11_RS14095 | 12 | SMDB11_RS03340 | 874.0195 | SMDB11_RS00620 |
0.028075136 |
|
SMDB11_RS22005 | 10 | SMDB11_RS14915 | 855.25476 | SMDB11_RS00600 |
0.028075136 |
|
SMDB11_RS22020 | 10 | SMDB11_RS02545 | 767.4091 | SMDB11_RS02555 |
0.028075136 |
|
SMDB11_RS14105 | 10 | SMDB11_RS00590 | 710.21094 | SMDB11_RS15030 |
0.028058134 |
With the use of the MCODE plug-in, the two modules
with the highest score were obtained (Fig. 5B and C). As revealed in Fig. 5B, cluster 1 (score=11.27) had 12
nodes and 62 interactions. Cluster 2 (score=8), as revealed in
Fig. 4C, consisted of 9 nodes and
32 interactions. In special, SMDB11_RS00590 and SMDB11_RS04505
belonged to cluster 1. Moreover, enrichment analysis of the genes
in the enriched clusters (Fig. 6)
was performed. No GO terms were identified for genes in cluster 2,
and there were 41 GO terms enriched for cluster 1, such as hydrogen
sulfide metabolic process, sulfate assimilation, and sulfur
compound biosynthetic process.
Discussion
Understanding the genetic mechanisms that underlie
the antibiotic resistance of bacteria is a critical issue. In this
study, we used RNA-seq to investigate the patterns of gene
expression that may be associated with the antibiotic resistance
mechanisms of S. marcescens. A total of 225 DEGs were
identified, of which upregulated SMDB11_RS09300 (GTP cyclohydrolase
FolE2) was the most significant with a log2 FC of 6.4,
and these DEGs were enriched in different GO terms, including
hydrogen sulfide biosynthetic process, sulfur compound
transmembrane transporter activity, and ABC transporter complex.
Additionally, several genes were identified to be important genes
in the PPI network, including SMDB11_RS17755 (upregulated;
glutamate synthase large subunit), SMDB11_RS00590 (upregulated;
sulfite reductase subunit A), and SMDB11_RS04505 (upregulated;
cystathionine β-synthase). Functional enrichment analysis of genes
in significant clusters revealed that genes were associated with
sulfur metabolism.
SMDB11_RS09300 encodes GTP cyclohydrolase FolE2
(27), an enzyme involved in the
biosynthesis of folic acid and pteridines (28,29).
There is evidence that the folic acid biosynthesis pathway may be a
potential target for the development of antibiotics, and this has
been validated by the clinical use of several drugs (30). Rengarajan et al demonstrated
that folate metabolism was a target for resistance to a type of
antibiotic (31). In the present
study, upregulated SMDB11_RS09300 was revealed to have the highest
expression change in the multidrug-resistant S. marcescens,
which was consistent with previous studies indicating that the
upregulation of SMDB11_RS09300 may play a significant role in the
multidrug-resistant mechanisms of S. marcescens by
participating in folate metabolism.
Glutamate synthase is important as it provides
glutamate for glutamine synthetase reaction (32). A study showed that the export of
glutamine synthetase was associated with the formation of the
poly-L-glutamate/glutamine cell wall structure of organisms
(33). A decrease in extracellular
glutamine synthetase activity can inhibit bacterial growth
(34). Evidence has indicated that
several cell-wall-related genes can be strongly expressed in
Staphylococcus aureus in response to antibiotics, such as
gltD, which encodes the small subunit of glutamate synthase
(35). In the present study, it
was revealed that SMDB11_RS17755 (glutamate synthase large subunit)
was upregulated and was a hub protein in the PPI network,
suggesting that the upregulation of this gene may contribute to
cell wall metabolism, which may defend S. marcescens against
the antibacterial activities of the agents.
Moreover, SMDB11_RS00590 (sulfite reductase subunit
α) and SMDB11_RS04505 (cystathionine β-synthase) were also
upregulated and played important roles in the PPI network in this
study. The sulfite reductases catalyze the reduction of sulfite to
sulfide (36), and cystathionine
β-synthase is also a sulfur metabolism enzyme (37). Synthetic antimicrobial agents such
as the ‘sulfa drugs’ (sulfonamides) have also been brought into
wider usage, and they can inhibit steps in folic acid metabolism
(38). Sulfur metabolic pathways
are essential for survival and virulence of many pathogenic
bacteria and represent a promising new area for therapy against
multidrug resistant microbes (39). In the present study, the identified
225 DEGs and genes in the significant cluster 1 were mainly
associated with sulfur metabolism. Thus, it was concluded that the
upregulation of SMDB11_RS00590 and SMDB11_RS04505, which were
involved in the sulfur metabolism in S. Marcescens, may be
critical in its antibiotic resistance mechanisms. However, further
studies are required to validate the roles of these genes.
However, the major limitation of this study was that
a small number of replicates was used, which did not provide
sufficient statistical power to assess the significance of the
findings. Thus, the results of this study require confirmation in
the future, i.e., when more replicates and confirmatory experiments
using additional techniques (e.g., qPCR) are available. Another
limitation of this study was that the associations used to
construct the PPI network were derived from another species due to
the lack of S. marcescens in the database. Thus, more
studies are required to verify the biological interpretation of
these results.
In conclusion, this study profiled genes in
multidrug-resistant S. marcescens using RNA sequencing.
SMDB11_RS09300 may play a significant role in the
multidrug-resistant mechanisms of S. marcescens by
participating in folate metabolism. SMDB11_RS17755 may contribute
to the integrity of cell membranes, which is involved in the
multi-drug resistance of S. marcescens. The upregulation of
SMDB11_RS00590 and SMDB11_RS04505 in S. marcescens may be
critical in its antibiotic resistance mechanisms. Further studies
with a large panel of isolates are required to validate these
findings. However, these findings are important and will aid in
better understanding bacterial resistance to these drugs.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Medical and
Health Science and Technology Plan of Zhejiang Province (Program
no. 2019KY134) and The Hangzhou Health and Family Planning
Technology Plan (Program no. 2018Z07).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL and MD conceived the present study. ZL drafted
the manuscript. MX and HW collected and analyzed the data. LW
interpreted the data. All authors read and approved the final
manuscript, and agreed to be accountable for all aspects of the
work in ensuring that questions related to the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
KPC-2
|
Klebsiella pneumoniae
carbapenemase-2
|
|
LPS
|
lipopolysaccharide
|
|
DEGs
|
differentially expressed genes
|
|
PPI
|
protein-protein interaction
|
References
|
1
|
Courtney CM, Goodman SM, McDaniel JA,
Madinger NE, Chatterjee A and Nagpal P: Photoexcited quantum dots
for killing multidrug-resistant bacteria. Nat Mater. 15:529–534.
2016. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iguchi A, Nagaya Y, Pradel E, Ooka T,
Ogura Y, Katsura K, Kurokawa K, Oshima K, Hattori M, Parkhill J, et
al: Genome evolution and plasticity of Serratia marcescens,
an important multidrug-resistant nosocomial pathogen. Genome Biol
Evol. 6:2096–2110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Blair JM, Webber MA, Baylay AJ, Ogbolu DO
and Piddock LJ: Molecular mechanisms of antibiotic resistance. Nat
Rev Microbiol. 13:42–51. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alekshun MN and Levy SB: Molecular
mechanisms of antibacterial multidrug resistance. Cell.
128:1037–1050. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma XJ, Yang HF, Liu YY, Mei Q, Ye Y, Li
HR, Cheng J and Li JB: The emergence of the 16S rRNA
methyltransferase RmtB in a multidrug-resistant Serratia
marcescens isolate in China. Ann Lab Med. 35:172–174. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Su W, Zhu Y, Deng N and Li L:
Imipenem-resistance in Serratia marcescens is mediated by
plasmid expression of KPC-2. Eur Rev Med Pharmacol Sci.
21:1690–1694. 2017.PubMed/NCBI
|
|
7
|
Olaitan AO, Morand S and Rolain JM:
Mechanisms of polymyxin resistance: Acquired and intrinsic
resistance in bacteria. Front Microbiol. 5:6432014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rabbani B, Nakaoka H, Akhondzadeh S, Tekin
M and Mahdieh N: Next generation sequencing: Implications in
personalized medicine and pharmacogenomics. Mol Biosyst.
12:1818–1830. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Crofts TS, Gasparrini AJ and Dantas G:
Next-generation approaches to understand and combat the antibiotic
resistome. Nat Rev Microbiol. 15:422–434. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Crowley E, Bird P, Fisher K, Goetz K,
Boyle M, Benzinger MJ Jr, Juenger M, Agin J, Goins D and Johnson R:
Evaluation of the VITEK 2 Gram-negative (GN) microbial
identification test card: Collaborative study. J AOAC Int.
95:778–785. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McClure R, Balasubramanian D, Sun Y,
Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK and Tjaden B:
Computational analysis of bacterial RNA-Seq data. Nucleic Acids
Res. 41:e1402013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Langmead B and Salzberg SL: Fast
gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Robinson MD and Oshlack A: A scaling
normalization method for differential expression analysis of
RNA-seq data. Genome Biol. 11:R252010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gene Ontology Consortium: Gene ontology
consortium: Going forward. Nucleic Acids Res 43 (Database Issue).
D1049–D1056. 2015. View Article : Google Scholar
|
|
19
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res 43
(Database Issue). D447–D452. 2015. View Article : Google Scholar
|
|
20
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Opsahl T, Agneessens F and Skvoretz J:
Node centrality in weighted networks: Generalizing degree and
shortest paths. Soc Netw. 32:245–251. 2010. View Article : Google Scholar
|
|
22
|
Cukierski WJ and Foran DJ: Using
betweenness centrality to identify manifold shortcuts. Proc IEEE
Int Conf Data Min. 2008:949–958. 2008.PubMed/NCBI
|
|
23
|
Du Y, Gao C, Chen X, Hu Y, Sadiq R and
Deng Y: A new closeness centrality measure via effective distance
in complex networks. Chaos. 25:0331122015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang Y, Li M, Wang J, Pan Y and Wu FX:
CytoNCA: A cytoscape plugin for centrality analysis and evaluation
of protein interaction networks. Biosystems. 127:67–72. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Safari-Alighiarloo N, Taghizadeh M,
Rezaei-Tavirani M, Goliaei B and Peyvandi AA: Protein-protein
interaction networks (PPI) and complex diseases. Gastroenterol
Hepatol Bed Bench. 7:17–31. 2014.PubMed/NCBI
|
|
26
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sankaran B, Bonnett SA, Shah K, Gabriel S,
Reddy R, Schimmel P, Rodionov DA, de Crécy-Lagard V, Helmann JD,
Iwata-Reuyl D and Swairjo MA: Zinc-independent folate biosynthesis:
Genetic, biochemical, and structural investigations reveal new
metal dependence for GTP cyclohydrolase IB. J Bacteriol.
191:6936–6949. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rebelo J, Auerbach G, Bader G, Bracher A,
Nar H, Hösl C, Schramek N, Kaiser J, Bacher A, Huber R and Fischer
M: Biosynthesis of pteridines. Reaction mechanism of GTP
cyclohydrolase I. J Mol Biol. 326:503–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Babitzke P, Gollnick P and Yanofsky C: The
mtrAB operon of Bacillus subtilis encodes GTP cyclohydrolase I
(MtrA), an enzyme involved in folic acid biosynthesis, and MtrB, a
regulator of tryptophan biosynthesis. J Bacteriol. 174:2059–2064.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bermingham A and Derrick JP: The folic
acid biosynthesis pathway in bacteria: Evaluation of potential for
antibacterial drug discovery. Bioessays. 24:637–648. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rengarajan J, Sassetti CM, Naroditskaya V,
Sloutsky A, Bloom BR and Rubin EJ: The folate pathway is a target
for resistance to the drug para-aminosalicylic acid (PAS) in
mycobacteria. Mol Microbiol. 53:275–282. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pan FL and Coote JG: Glutamine synthetase
and glutamate synthase activities during growth and sporulation in
Bacillus subtilis. J Gen Microbiol. 112:373–377. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harth G, Zamecnik PC, Tang JY, Tabatadze D
and Horwitz MA: Treatment of Mycobacterium tuberculosis with
antisense oligonucleotides to glutamine synthetase mRNA inhibits
glutamine synthetase activity, formation of the
poly-L-glutamate/glutamine cell wall structure, and bacterial
replication. Proc Natl Acad Sci USA. 97:418–423. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harth G and Horwitz MA: An inhibitor of
exported Mycobacterium tuberculosis glutamine synthetase
selectively blocks the growth of pathogenic mycobacteria in axenic
culture and in human monocytes: Extracellular proteins as potential
novel drug targets. J Exp Med. 189:1425–1436. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Utaida S, Dunman PM, Macapagal D, Murphy
E, Projan SJ, Singh VK, Jayaswal RK and Wilkinson BJ: Genome-wide
transcriptional profiling of the response of Staphylococcus aureus
to cell-wall-active antibiotics reveals a cell-wall-stress
stimulon. Microbiology. 149:2719–2732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schnell R, Sandalova T, Hellman U,
Lindqvist Y and Schneider G: Siroheme- and [Fe4-S4]-dependent NirA
from Mycobacterium tuberculosis is a sulfite reductase with a
covalent Cys-Tyr bond in the active site. J Biol Chem.
280:27319–27328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bhattacharyya S, Saha S, Giri K, Lanza IR,
Nair KS, Jennings NB, Rodriguez-Aguayo C, Lopez-Berestein G, Basal
E, Weaver AL, et al: Cystathionine beta-synthase (CBS) contributes
to advanced ovarian cancer progression and drug resistance. PLoS
One. 8:e791672013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bisht R, Katiyar A, Singh R and Mittal P:
Antibiotic resistance-A global issue of concern. Asian J Pharm Clin
Res. 2:34–39. 2009.
|
|
39
|
Bhave DP, Muse WB III and Carroll KS: Drug
targets in mycobacterial sulfur metabolism. Infect Disord Drug
Targets. 7:140–158. 2007. View Article : Google Scholar : PubMed/NCBI
|