Introduction
Ovarian cancer is one of the most common gynecologic
malignancies worldwide. According to recent global statistics,
>230,000 new patients are diagnosed with this disease every
year, and ovarian cancer accounts for ~140,000 mortalities annually
(1). Although detection techniques
and chemotherapy regimens for ovarian cancer have improved in
recent years, the 5-year survival rate of patients with advanced
stage ovarian cancer is ~30% (2).
This low survival rate is primarily due to the number of patients
diagnosed with ovarian cancer at late stages (3,4).
Notably, cancer cells become more resistant to conventional
chemotherapeutic agents at later stages (5). Therefore, the identification of novel
therapeutic methods is critical to improve the prognosis of
patients with ovarian cancer.
Minichromosome maintenance complex component 2
(MCM2) is one of the six proteins comprising the MCM complex, a
stable heterohexamer crucial for the regulation of DNA replication
(6,7). In eukaryotic cells, DNA synthesis is
initiated from defined sites, called replication origins. During
the G1 phase, replication origins interact with the origin
recognition complex (ORC), which induces the sequential recruitment
of cell division cycle 6, chromatin licensing and DNA replication
factor 1 and MCM2-7 to form a prereplication complex (8–10).
During the S phase, the activation of the MCM complex by cell cycle
kinases triggers the initiation of the DNA replication (11). Additionally, the MCM complex
restricts chromosome replication to only once per cell cycle
(11,12). Previous studies have demonstrated
that in yeast MCM mutations lead to chromosome loss, DNA damage and
increased recombination (13,14).
Consistent with previous studies on yeast, reduced expression
levels of MCM2 in mice result in lymphomas (15,16).
A large number of studies have confirmed that MCM2 is a reliable
proliferation and prognostic marker of oral, gastric, colon and
breast cancer, suggesting that it may represent a more reliable
marker than traditional ones, such as marker of proliferation Ki-67
(MKI67) and proliferating cell nuclear antigen (17–20).
A number of previous studies have reported that the expression
level of MCM2 is significantly higher in ovarian adenocarcinomas
compared with low malignant tumors (21,22)
and a high level of MCM2 expression in malignant tumors is
associated with higher grades, more advanced stages and poor
prognosis (23,24).
Since the regulation of the expression levels of
MCM2-7 is involved in ensuring proper genome replication preventing
genome instability, the present study aimed to investigate whether
agents that inhibit MCM2 gene expression may suppress cellular
proliferation, influencing the cell cycle and leading to cell
apoptosis. Moreover, the present study aimed to examine whether,
under replication stress conditions, a reduction in MCM2 expression
could sensitize cells to the chemotherapeutic drug carboplatin. In
the present study, the expression level of MCM2 was knocked down in
the human ovarian cancer cell line A2780. Collectively, the aim of
the present study was to develop a novel therapeutic strategy for
treating ovarian cancer.
Materials and methods
Cell culture
The human ovarian cancer cell line A2780 and the
293T cell line were obtained from The Chinese Academy of Sciences
Committee and were verified by STR profiling. A2780 cells were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin (Gibco; Thermo Fisher Scientific,
Inc.). The 293T cells were cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.). All cells were incubated at 37°C in a humidified
incubator with 5% CO2.
Construction of the MCM2 short hairpin
(sh)-RNA lentivirus vector and cell infection
RNA interference (RNAi) was used to downregulate the
expression level of MCM2 in A2780 cells. Two shRNAs were used to
target MCM2 and a scrambled shRNA was used as control (Table I; all from Sangon Biotech Co.,
Ltd). These shRNA oligos were annealed and ligated using
AgeI and EcoRI restriction sites into the linearized
pLKO.1-puro vector (Addgene) to generate pLKO.1-puro-MCM2 (shMCM2-1
and shMCM2-2) and pLKO.1-puro-control (shCON) recombinant vectors.
The constructed shRNA-expressing vectors were then confirmed by DNA
sequencing (Sangon Biotech Co., Ltd). The shRNA-expressing
recombinant plasmids (2 µg) were transfected together with two
helper plasmids, psPAX2 (1.5 µg) and pMD2.G (0.5 µg; both from
Addgene) into 293T cells with Lipofectamine 2000™ transfection
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Cell culture media containing
lentiviral particles were collected 48 h after transfection. For
cell infection, A2780 ovarian cancer cells were incubated in 6-well
plates at a density of 4×105 cells/well. Subsequently,
media containing 1×106 IFU/ml lentivirus in 8 µg/ml
polybrene (Sigma-Aldrich; Merck KGaA) was added to A2780 cells for
24 h. Stably infected cells were selected using 1 µg/ml puromycin
(Sigma-Aldrich; Merck KGaA) for 3–5 days and the RNAi knockdown
efficiency was detected by western blot analysis.
 | Table I.shRNA sequences. |
Table I.
shRNA sequences.
| shRNA | Sequence
(5′-3′) |
|---|
| shMCM2-1 |
CCGGGCTCTTCATACTGAAGCAGTTCTCGAGAACTGCTTCAGTATGAAGAGCTTTTT |
| shMCM2-2 |
CCGGCTATCAGAACTACCAGCGTATCTCGAGATACGCTGGTAGTTCTGATAGTTTTT |
| shCON |
CCTAAGGTTAAGTCGCCCTCG |
Western blot analysis
A2780 cells were treated with various concentrations
of carboplatin (0, 20, 30 and 40 µg/ml) for 48 h at 37°C.
Subsequently, the cells were lysed, and the total protein was
extracted. Alternatively, A2780 cells were treated with various UV
intensities (0, 20, 40 and 80 J). After 24 h, the cells were lysed,
and the total protein was extracted. Cells were lysed for total
protein extraction with a cell lysis buffer for Western and IP
(cat. no. P0013J; Beyotime Institute of Biotechnology) containing
protease inhibitor cocktail and PMSF (1%). The concentrations of
total protein were quantified using a bicinchoninic protein assay
kit (Beyotime Institute of Biotechnology). Extracted proteins were
mixed with SDS-PAGE Sample Loading Buffer (cat. no. P0015; Beyotime
Institute of Biotechnology) and boiled at 100°C for 10 min. The
protein samples (20 µg/well) were separated by SDS-PAGE on 10–12%
gels and transferred onto PVDF membranes (EMD Millipore). After
blocking with 5% skimmed milk for 1 h at room temperature, the
membrane was incubated at 4°C overnight with the following primary
antibodies: MCM2 (1:1,000; cat. no. 3619; Cell Signaling
Technology, Inc.), p53 (1:300; cat. no. 2527; Cell Signaling
Technology, Inc.), γ-H2A histone family member X (γ-H2AX; 1:400;
cat. no. 9718; Cell Signaling Technology, Inc.); H2AX (1:2,000;
cat. no. ab124781; Abcam) and β-actin (1:3,000; cat. no. ab8224;
Abcam). After washing with TBS-0.2% Tween-20 (TBST) three times,
the membranes were incubated with horseradish peroxidase-conjugated
goat anti-mouse (1:4,000; cat. no. ab205719; Abcam) or anti-rabbit
secondary antibodies (1:4,000; cat. no. ab205718; Abcam) for 1 h at
room temperature. Then, the membranes were washed with TBST three
times. Specific bands were visualized using an ECL detection kit
(Thermo Fisher Scientific, Inc.). The protein bands were visualized
by autoradiography. The band intensity was measured using the
ImageJ software (version 1.48; National Institutes of Health).
β-Actin was used to normalize the protein expression levels and the
relative expression levels were subsequently calculated.
Cell proliferation assay
Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc.) was used to assess cell proliferation. The
cells were seeded in 96-well plates in triplicate at a density of
~1×103 cells/well and cultured in 100 µl RPMI-1640 for
24, 48, 72, 96 and 120 h. After incubation, the culture medium was
replaced with 110 µl RPMI-1640 supplemented with CCK-8 reagent (10
µl CCK-8 in 100 µl RPMI-1640), and the cells were incubated at 37°C
for 2 h. A microplate reader (BioTek Instruments, Inc.) was used to
measure the optical density value at a wavelength of 450 nm. All
experiments were performed in triplicate.
Cell cycle assay
Exponentially growing cells were seeded into 6-well
plates at a density of 4×105 cells/well and synchronized
in serum-free RPMI-1640 for 16–24 h. Then, the medium was replaced
with RPMI-1640 containing 10% FBS and antibiotics. Following
incubation for 24 h, cells were harvested, washed with ice-cold
PBS, and fixed with precooled 70% ethanol overnight at 4°C. The
next day, the fixed cells were washed with ice-cold PBS and stained
with 500 µl propidium iodide (PI)/RNase Staining Buffer (BD
Pharmingen; BD Biosciences) for 30 min at room temperature in the
dark. Cell cycle analyses were performed with a flow cytometer (BD
Biosciences). Cell cycle was analyzed using the ModFit software
(version 4.1; Verity Software House, Inc.). The experiments were
repeated three times.
Cell apoptosis assay
To examine apoptosis, the cells were harvested
(4×105 cells/well) and washed twice with ice-cold PBS.
Then, the cells were resuspended with 1X Annexin V binding buffer
(BD Biosciences) and stained with PI and Annexin V-FITC according
to the manufacturer's protocol (BD Biosciences). Cell apoptosis was
detected by flow cytometry (BD Biosciences) and the assays were
repeated three times. Cells negative for both PI and Annexin V were
considered viable cells. PI-negative and Annexin V-positive cells
were considered early apoptotic cells. PI-positive and Annexin
V-positive cells were considered late-stage apoptotic cells. Cell
apoptosis data were analyzed using BD CellQuest™ Pro software
(version 6.1; BD Biosciences).
Colony formation assay
The control and knockdown group cells were seeded in
6-well plates at a density of 1,000 cells/well. Cells were then
exposed to 0, 2.5, 3.5, 5 and 7.5 µg/ml carboplatin (Qilu
Pharmaceutical Co., Ltd.) for 48 h at 37°C. Then, the medium was
replaced with complete culture medium. Following incubation at 37°C
for 10–14 days, the cells were fixed with 4% paraformaldehyde at
room temperature for 15 min and then stained with 0.5% crystal
violet dye at room temperature for 10 min. Colonies containing
>50 cells were counted under a light microscope (magnification,
×100).
Statistical analysis
Statistical analyses were performed using SPSS 23.0
software (IBM Corp.). Data are presented as the mean ± standard
deviation from three experiments. Two-tailed Student's t-test was
used to evaluate the differences between two groups. One-way ANOVA
was used to evaluate the differences between multiple groups, and
Dunnett's test was used as the post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Lentivirus-mediated MCM2 knockdown in
human ovarian cancer A2780 cells
To investigate the biological function of MCM2 in
human ovarian cancer cells, shRNA targeting human MCM2 (shMCM2-1
and shMCM2-2) and a negative control (shCON) were infected into
A2780 ovarian cancer cells to knockdown the expression of MCM2. The
protein expression levels of MCM were investigated in stably
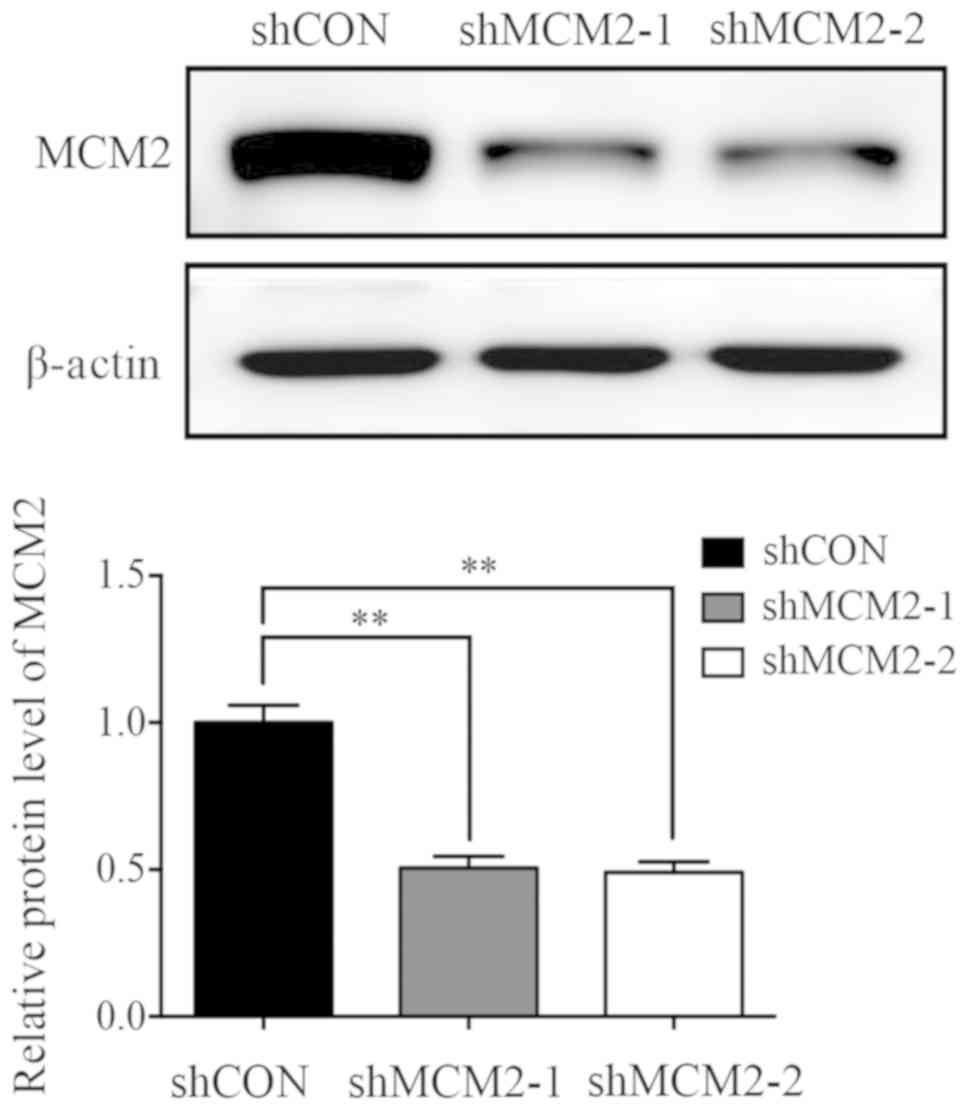
infected A2780 cells by western blot analysis (Fig. 1), and the protein expression level
of MCM2 was decreased by ~50% in shMCM2-1 (50.59±4%) and shMCM2-2
(49.1±3.6%) cells compared with cells infected with shCON (Fig. 1).
Effects of MCM2 knockdown on
proliferation, cell cycle distribution and cell apoptosis in A2780
cells
Since MCM2 was shown to serve an important role in
DNA replication in eukaryotes, and as a previous study reported
that knockdown of MCM2 causes cell death (25), the present study investigated
whether knockdown of MCM2 influenced cell proliferation, cell cycle
and cell apoptosis. Therefore, CCK-8 assay was performed at
different time points to measure cell viability. Notably, the
proliferation of A2780 cells was significantly inhibited following
knockdown of MCM2 compared with the control group at 4 and 5 days
(Fig. 2A). To investigate the role
of MCM2 in the regulation of the cell cycle, flow cytometry was
performed. The present results suggested that inhibition of MCM2 in
A2780 cells caused an increase in the G1 phase, whereas the number
of cells in the G2 phase decreased compared with the control
(Fig. 2B). The cell apoptosis
assay suggested that cells infected with shMCM2 exhibited similar
apoptotic rates compared with control cells (Fig. 2C). Therefore, MCM2 knockdown did
not induce the apoptosis of A2780 cells. Notably, the present cell
proliferation and apoptosis results were not in line with previous
studies on hepatocellular carcinoma, lung carcinoma and esophageal
cancer cells (25).
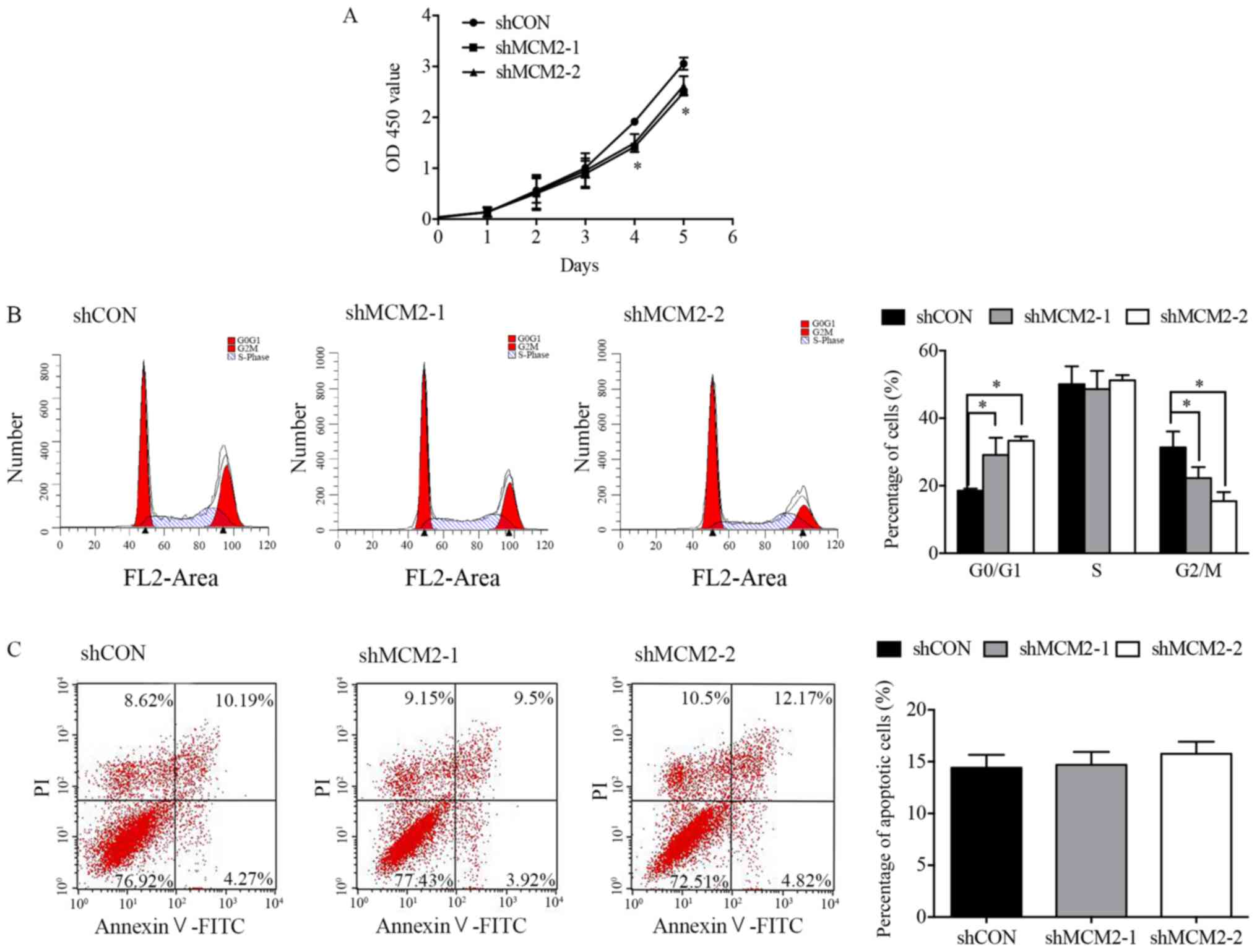 | Figure 2.Effect of MCM2 knockdown on A2780
cell proliferation, cell cycle and apoptosis. (A) Cell
proliferation was assessed using the Cell Counting Kit-8 assay at
0, 1, 2, 3, 4 and 5 days after infection. Cell proliferation was
inhibited by MCM2 knockdown at 4 and 5 days. (B) Cell cycle was
analyzed by flow cytometry. Representative cell cycle histograms of
A2780 cells are presented. Cells treated with MCM2 shRNA exhibited
a higher number of cells in the G0/G1-phase. (C) Cell apoptosis was
determined by flow cytometry. Infection with shMCM2 did not induce
apoptosis in A2780 cells. A representative flow cytometry analysis
is presented. Data are presented as the mean ± standard deviation
from three experiments. *P<0.05 vs. shCON. MCM2, minichromosome
maintenance complex component 2; shRNA, short hairpin RNA; shCON,
control shRNA; shMCM2, shRNA targeting MCM2; OD, optical
density. |
Knockdown of MCM2 enhances carboplatin
sensitivity in A2780 cells
Since the knockdown of MCM2 did not significantly
influence cell proliferation at 1–3 days, and the downregulation of
MCM2 did not affect cell apoptosis under normal circumstances, the
present study investigated whether the effects of MCM2 knockdown
would increase if replication forks were put under stress by
supplementation with DNA-damaging drugs. To examine whether
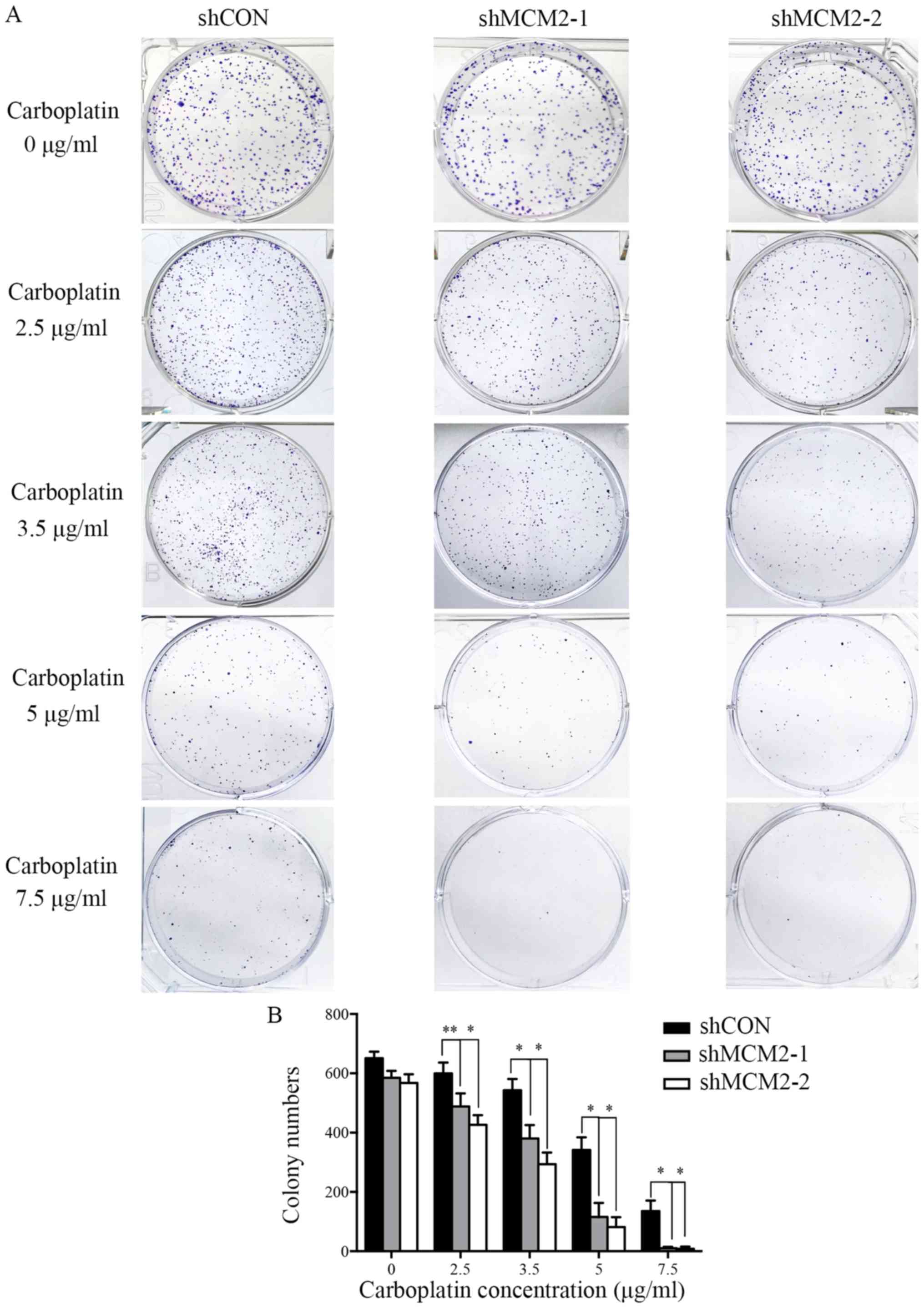
knockdown of MCM2 could sensitize A2780 cells to the common ovarian
cancer chemotherapeutic drug carboplatin (5,26), a
colony formation assay was performed. Control cells and MCM2
knockdown cells were treated with different concentrations of
carboplatin. In the absence of carboplatin, all cells formed
colonies (Fig. 3). Compared with
the control group, the knockdown of MCM2 significantly decreased
the colony formation of A2780 cells in response to various
concentrations of carboplatin (Fig.
3). The present results suggested that the combined treatment
of MCM2-shRNA and carboplatin significantly reduced the colony
formation of A2780 cells compared with carboplatin treatment alone.
Therefore, the present results support the hypothesis that the
downregulation of MCM2 level can enhance the sensitivity of A2780
cells to carboplatin.
Knockdown of MCM2 increases
carboplatin and UV irradiation-induced double-strand breaks (DSBs)
in A2780 cells
Next, the present study sought to identify the
mechanism underlying carboplatin sensitivity in MCM2-knockdown
cells. DNA damage and repair were investigated following treatment
with two genotoxic agents, carboplatin and UV irradiation.
Carboplatin treatment reduced the protein expression level of MCM2
in a dose-dependent manner (Fig. 4A
and B). The present results suggested that MCM2 may serve a
role in DNA damage and repair. However, the protein expression
level of MCM2 was not affected in UV-irradiated cells (Fig. 5A and B). γ-H2AX is a sensitive
marker of DNA DSBs (27).
Therefore, to investigate the effects of MCM2 knockdown on DNA
damage and repair, γ-H2AX expression following DNA damage was
analyzed by western blotting, and γ-H2AX expression was normalized
to the total protein expression level of H2AX. The present results
suggested that the γ-H2AX/total H2AX ratio increased in
MCM2-deficient cells compared with control cells after treatment
with carboplatin and UV (Figs. 4A
and 5A). The present results
suggested that the ratio of γ-H2AX/total H2AX in cells treated with
carboplatin and infected with shMCM2 was higher compared with
control cells treated with carboplatin (Fig. 4A and D). Similarly, the UV
irradiation experiment suggested that MCM2 knockdown significantly
increased the ratio of γ-H2AX/total H2AX in A2780 cells exposed to
UV irradiation at 20, 40 and 80 J (Fig. 5A and D). Therefore, the present
results suggested that the enhanced sensitivity of MCM2-knockdown
A2780 cells to carboplatin may have been caused by the accumulation
of damaged DNA.
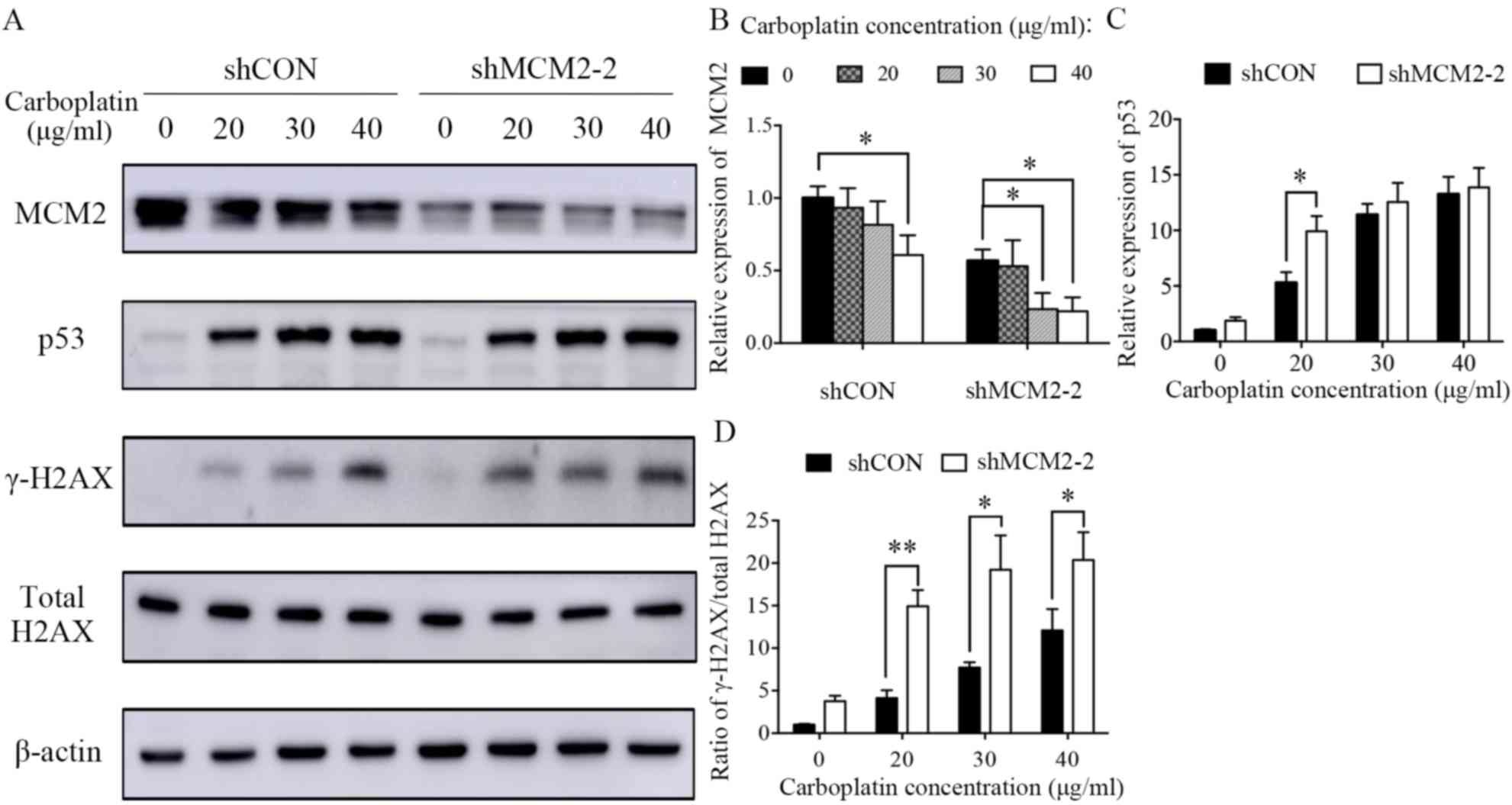 | Figure 4.Knockdown of MCM2 increases the
protein expression levels of γ-H2AX and p53 after treatment with
carboplatin. (A) A2780 cells were treated with various
concentrations of carboplatin (0, 20, 30 and 40 µg/ml) for 48 h.
Subsequently, the cells were lysed and the total protein was
extracted. Protein expression levels of MCM2, p53, γ-H2AX and total
H2AX were detected by western blotting. MCM2 knockdown increased
DNA damage-associated markers in response to carboplatin. (B)
Quantification of the protein expression levels of MCM2, (C) p53
and (D) the γ-H2AX/H2AX ratio. γ-H2AX was used as an indicator of
double-strand breaks. Data are presented as the mean ± standard
deviation from three experiments. *P<0.05 and **P<0.01 vs.
the respective control. MCM2, minichromosome maintenance complex
component 2; shRNA, short hairpin RNA; shCON, control shRNA;
shMCM2, shRNA targeting MCM2; H2AX, H2A histone family member
X. |
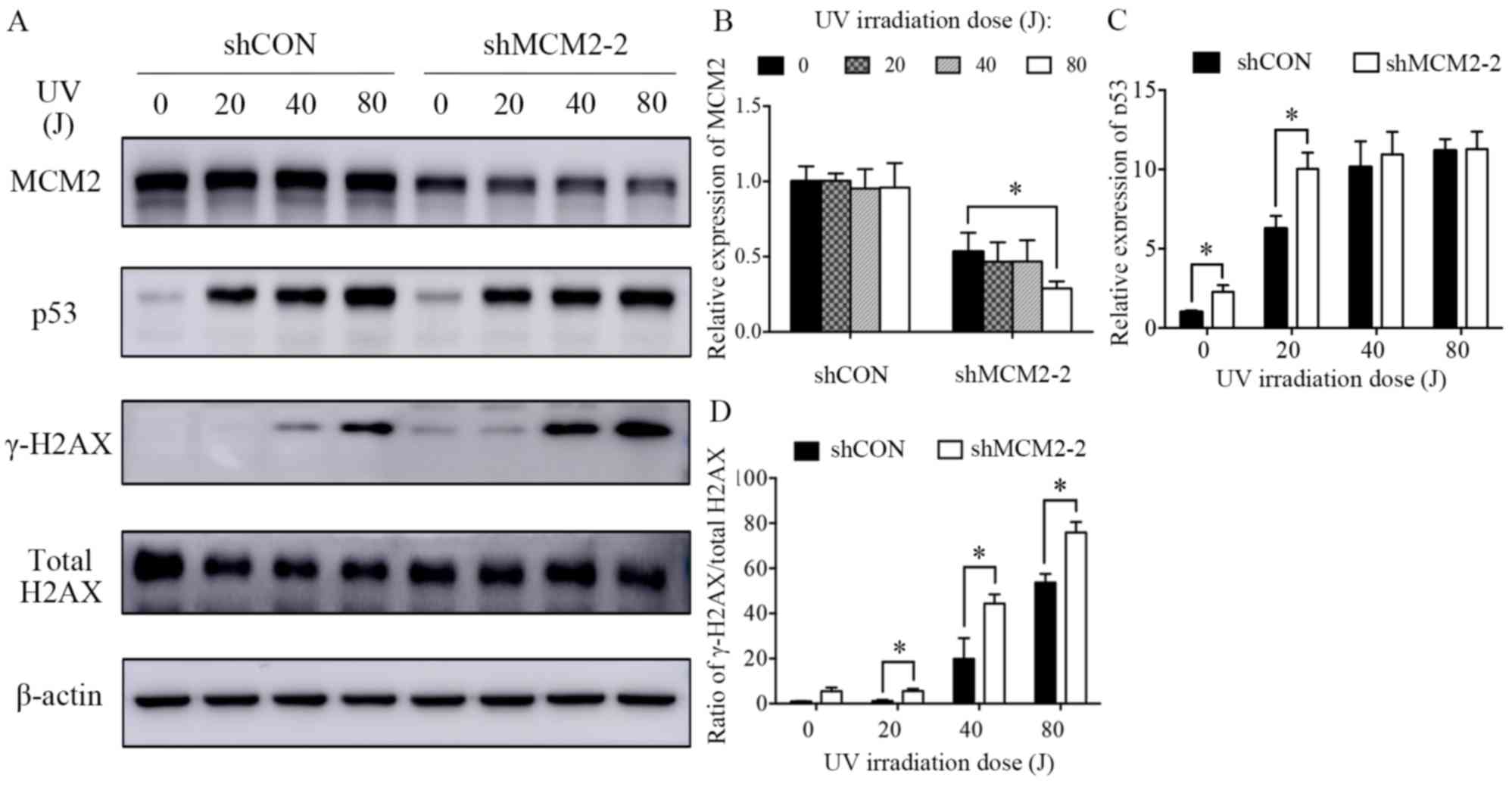 | Figure 5.Knockdown of MCM2 increases the
protein expression levels of γ-H2AX and p53 after treatment with
UV. (A) A2780 cells were treated with various UV intensities (0,
20, 40 and 80 J) for 24 h. Subsequently, the cells were lysed and
the total protein was extracted. Protein expression levels of MCM2,
p53, γ-H2AX and total H2AX were detected by western blotting. MCM2
knockdown increased DNA damage-associated markers in response to UV
irradiation. Quantification of the protein expression levels of (B)
MCM2, (C) p53 and (D) the γ-H2AX/H2AX ratio. γ-H2AX was used as an
indicator of double-strand breaks. Data are presented as the mean ±
standard deviation from three experiments. *P<0.05 vs. the
respective control. MCM2, minichromosome maintenance complex
component 2; shRNA, short hairpin RNA; shCON, control shRNA;
shMCM2, shRNA targeting MCM2; H2AX, H2A histone family member
X. |
DNA damage response in MCM2-deficient
A2780 cells in vitro
The increased DNA damage in A2780 MCM2-knockdown
cells after treatment with carboplatin and UV irradiation suggested
that MCM2 may serve a role in mediating the DNA damage response. In
a previous study, MCM2 was identified as a mediator of the DNA
damage response when the replisome interacts with a genetic lesion
(28). Therefore, the present
study investigated the influence of MCM2 knockdown on the
expression levels of proteins associated with the DNA damage repair
pathway. The present results suggested that the protein expression
levels of the tumor suppressor gene p53 were increased in MCM2
deficient cells compared with control cells after carboplatin
treatment (Fig. 4A and C) and UV
irradiation (Fig. 5A and C). An
important DNA damage repair pathway involves p53 (29–31),
which in turn serves an important role in cell cycle arrest and
apoptotic response following cellular damage. The increased protein
expression level of p53 suggested the activation of the
p53-dependent apoptotic response.
Discussion
Carboplatin-based chemotherapy is the standard
first-line treatment for patients with advanced stage ovarian
cancer, tumor relapse or tumor metastasis. However,
carboplatin-based chemotherapy resistance may occur and, in many
cases, can result in treatment failure (26). For this reason, reversing
carboplatin resistance in ovarian cancer is the major focus of
multiple research studies. A previous study demonstrated that in
the cisplatin-resistant ovarian carcinoma cell line
PE01CDDP, the MCM2 expression level was increased by
>2 folds compared with normal PE01 cells (32). Therefore, the present study
hypothesized that MCM2 may be associated with chemotherapy
resistance in patients with ovarian cancer.
MCM2-7 is a key factor during replication initiation
and elongation (33). MCM2-7 was
shown to interact with DNA during the G1 phase and is activated
during the S phase, triggering DNA replication origin licensing and
firing (33). It has been reported
that the MCM2/5 active site functions as an ATP-dependent ‘gate’ of
the MCM2-7 ring, and that the status of this gate regulates the
function of MCM2-7 (34). Mutated
MCM4 in mice causes a reduction in the overall levels of MCMs,
leading to a high incidence of mammary adenocarcinomas (35). Additionally, when the expression
levels of MCM2 are decreased to 33%, the average mouse lifespan is
significantly reduced due to the occurrence of various types of
cancer, in particular T- and B-cell lymphoma (16). Therefore, loss of MCM function in
mice induces genome instability and cancer predisposition (16). However, increased expression levels
of MCM2 have been detected in various types of cancer, and were
identified to be associated with advanced stages and poor prognosis
(21,36,37).
Additionally, increased expression levels of MCM2 were identified
to be associated with the upregulation of MKI67, an important
marker of cell proliferation (21,36,37).
These previous studies indicated that the precise control of MCM2
expression is important to maintain the stability of the genome.
Therefore, in the present study, the expression level of MCM2 was
knocked down in A2780 ovarian cancer cells. RNAi was used to
downregulate the expression level of MCM2, and the biological
effects of MCM2 were investigated in A2780 cells. The present
results demonstrated that MCM2 knockdown exhibited a limited effect
on A2780 cell proliferation and cell cycle, and did not affect cell
apoptosis. The present findings suggested that targeted inhibition
of MCM2 may not be an adequate therapy without additional
treatments.
Therefore, the present study hypothesized that the
acute downregulation of MCM2 may allow the formation of a limited,
but sufficient, amount of active MCM2-7 complexes. The present
results are consistent with various previous studies that have
reported that normal DNA replication rates and cell proliferation
in vitro are maintained also when the protein expression
levels of MCM are significantly decreased (38–41).
The present findings can be explained by the fact that the MCM2-7
complex is abundant in proliferating cells and is recruited to the
chromatin at levels that are 3–20 folds higher than the levels
required to unwind the DNA at the replication fork (42–45).
Furthermore, MCM hexamers are homogenously distributed on DNA, and
are not accumulated at the levels of the replication origins and
ORCs (46,47). Previous studies have reported that
MCM2-7 complexes can be found on DNA at significant distances from
the ORCs (46,47). Moreover, each genomic site
containing MCM2-7 bound to the DNA is a candidate replication
origin that can potentially initiate replication.
Next, the present study aimed to identify the
function of the excess levels of MCMs compared with the number of
replication origins. The present results suggested that a partial
reduction in MCM2 had limited effects on cell proliferation, cell
cycle and apoptosis. Therefore, the present study investigated
effects of MCM2 knockdown in the presence of replication stress.
Thus, various concentrations of carboplatin were added to A2780
control cells and MCM2-knockdown cells. After exposure to
carboplatin, the survival rate of MCM2-knockdown cells decreased
significantly compared with the control cells.
Collectively, the results of the present and
previous studies have suggested that under normal conditions,
sufficient MCM complexes are recruited to the chromatin at various
potential origins of replication (42,43).
A previous study reported that following the initiation of
replication at one replication origin, a signal is sent to inhibit
the activation of additional replication origins (45). However, when cells experience
replication stress and the replication forks are stalled, the
dormant origins of replication may be activated to rescue stalled
or damaged primary replication forks (45). In conclusion, cells maintain an
excessive number of MCM2-7 complexes bound to dormant replication
origins that can be activated during replication stress (41,45).
The present data revealed that after decreasing the expression
level of MCM2, A2780 cells exhibited significantly reduced
proliferation following treatment with carboplatin, a replication
inhibitor. Under limited DNA replication licensing conditions, a
reduced number of potential replication origins are available, and
cells may require the additional dormant replication origins used
to rescue collapsed replication forks. This hypothesis could
explain the hypersensitivity of MCM2-knockdown cells to replication
inhibitors. An excessive number of potential DNA replication
origins not only serves as a backup system in case of collapsed
replication forks, but are also important in the maintenance of
genome stability, since mice with reduced MCM levels or function
exhibit high rates of cancer incidence (16). Additionally, a previous study
reported that the increased number of potential DNA replication
origins serves a role in suppressing genetic damage (48).
Additionally, in the present study, carboplatin
treatment decreased the protein expression level of MCM2 and
increased the level of p53 in a dose-dependent manner. According to
a previous study, DNA DSB signals elicit ATM serine/threonine
kinase-dependent phosphorylation of checkpoint kinase 2 (Chk2)
(49). Activated Chk2 can
transduce the signal via p53, which activates the transcription of
the cell cycle inhibitor p21, leading to the downregulation of the
protein expression level of MCM (49). However, the decrease in the protein
expression level of MCM2 was not significant in UV-irradiated
cells, suggesting that MCM2 may serve various roles in response to
different forms of DNA damage. Moreover, both the
carboplatin-treated and UV-irradiated cells showed higher protein
expression levels of γ-H2AX and p53 in MCM2-knockdown cells
compared with control cells. γ-H2AX is a sensitive marker of DNA
DSB (27), and thus, the increased
levels of γ-H2AX suggested that a higher frequency of DNA damage
was present in MCM2-knockdown cells compared with control cells.
The mechanism underlying MCM2 knockdown-mediated chemosensitivity
in A2780 cells is unclear, but the present results suggested that
the regulation of p53 was involved in this mechanism. The tumor
suppressor gene p53 serves a pivotal role in the DNA damage
response pathway, causing G1 cell cycle arrest, promoting DNA
damage repair (29) or inducing
cell apoptosis to protect genome stability (30,31).
A previous study reported that defective apoptosis may contribute
to resistance to platinum cytotoxicity, (50) and cells with p53 deletions
(51) or mutations (52) are more resistant to platinum,
possibly due to the inactivation of the p53-dependent apoptotic
response. Additionally, a previous study suggested that p53
signaling may sensitize cells to cisplatin (52). Therefore, the sensitivity of
MCM2-knockdown cells to carboplatin may be promoted by the
activation of the p53-dependent apoptotic pathway. However, the
specific mechanisms underlying MCM2 knockdown-mediated sensitivity
of A2780 cells to carboplatin require further investigation.
In conclusion, the present data suggested that
combined treatment with the chemotherapeutic agent carboplatin and
MCM2 shRNA significantly enhanced the chemosensitivity of A2780
cells via upregulation of p53. Notably, the combined treatment was
more effective than single treatment with either carboplatin or
MCM2 shRNA alone. To the best of our knowledge, the present study
is the first to suggest the inhibition of MCM2 as a strategy to
potentiate the efficacy of carboplatin in ovarian cancer. However,
only one cell type was used in the present study, and the reduction
in the protein expression level of MCM2 following MCM2 was not
optimal. Therefore, further experiments investigating various cell
lines and overexpression models are required in order to identify
the effects of MCM2 overexpression on cell proliferation, cell
cycle and carboplatin resistance. The present findings suggested
that the combination of MCM2 knockdown and carboplatin treatment
may represent a novel therapeutic strategy to treat ovarian cancer.
Additionally, MCM2 could represent a potential target for designing
new drugs and for the development of novel therapeutic methods for
ovarian cancer treatment.
Acknowledgements
The authors would like to thank Miss Qian Ma
(Department of Oncology, Shanghai Medical College, Fudan
University) for her assistance with the western blotting assay.
Funding
The present study was supported by a grant from The
National Natural Science Foundation of China (grant nos.
NSF-81772808, 81572552 and 81772774).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
MD performed the majority of the experiments, and
wrote and revised the manuscript. AZ and SX constructed the
recombinant lentiviral vector. JS performed the Cell Counting Kit-8
assay. HuZ and HoZ performed the cell cycle and apoptosis
experiments. YW and JS performed the statistical analysis. RL and
LG designed the study and wrote the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dahiya N and Morin PJ: MicroRNAs in
ovarian carcinomas. Endocr Relat Cancer. 17:F77–F89. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Greenlee RT, Hill-Harmon MB, Murray T and
Thun M: Cancer statistics, 2001. CA Cancer J Clin. 51:15–36. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wright JD, Shah M, Mathew L, Burke WM,
Culhane J, Goldman N, Schiff PB and Herzog TJ: Fertility
preservation in young women with epithelial ovarian cancer. Cancer.
115:4118–4126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iorio MV, Visone R, Di Leva G, Donati V,
Petrocca F, Casalini P, Taccioli C, Volinia S, Liu CG, Alder H, et
al: MicroRNA signatures in human ovarian cancer. Cancer Res.
67:8699–8707. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fung-Kee-Fung M, Oliver T, Elit L, Oza A,
Hirte HW and Bryson P: Optimal chemotherapy treatment for women
with recurrent ovarian cancer. Curr Oncol. 14:195–208. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Forsburg SL: Eukaryotic MCM proteins:
Beyond replication initiation. Microbiol Mol Biol Rev. 68:109–131.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lei M: The MCM complex: Its role in DNA
replication and implications for cancer therapy. Curr Cancer Drug
Targets. 5:365–380. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bell SP and Dutta A: DNA replication in
eukaryotic cells. Annu Rev Biochem. 71:333–374. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dimitrova DS, Todorov IT, Melendy T and
Gilbert DM: Mcm2, but not RPA, is a component of the mammalian
early G1-phase prereplication complex. J Cell Biol. 146:709–722.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mendez J and Stillman B: Perpetuating the
double helix: Molecular machines at eukaryotic DNA replication
origins. Bioessays. 25:1158–1167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Parker MW, Botchan MR and Berger JM:
Mechanisms and regulation of DNA replication initiation in
eukaryotes. Crit Rev Biochem Mol Biol. 52:107–144. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Romanowski P and Madine MA: Mechanisms
restricting DNA replication to once per cell cycle: The role of
Cdc6p and ORC. Trends Cell Biol. 7:9–10. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hennessy KM, Lee A, Chen E and Botstein D:
A group of interacting yeast DNA replication genes. Genes Dev.
5:958–969. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gibson SI, Surosky RT and Tye BK: The
phenotype of the minichromosome maintenance mutant mcm3 is
characteristic of mutants defective in DNA replication. Mol Cell
Biol. 10:5707–5720. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kunnev D, Rusiniak ME, Kudla A, Freeland
A, Cady GK and Pruitt SC: DNA damage response and tumorigenesis in
Mcm2-deficient mice. Oncogene. 29:3630–3638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pruitt SC, Bailey KJ and Freeland A:
Reduced Mcm2 expression results in severe stem/progenitor cell
deficiency and cancer. Stem Cells. 25:3121–3132. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yousef EM, Furrer D, Laperriere DL, Tahir
MR, Mader S, Diorio C and Gaboury LA: MCM2: An alternative to Ki-67
for measuring breast cancer cell proliferation. Mod Pathol.
30:682–697. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Andrade BA, León JE, Carlos R,
Delgado-Azañero W, Mosqueda-Taylor A and de Almeida OP: Expression
of minichromosome maintenance 2, Ki-67, and geminin in oral nevi
and melanoma. Ann Diagn Pathol. 17:32–36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Czyzewska J, Guzińska-Ustymowicz K,
Pryczynicz A, Kemona A and Bandurski R: Immunohistochemical
evaluation of Ki-67, PCNA and MCM2 proteins proliferation index
(PI) in advanced gastric cancer. Folia Histochem Cytobiol.
47:289–296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guzińska-Ustymowicz K, Pryczynicz A,
Kemona A and Czyzewska J: Correlation between proliferation
markers: PCNA, Ki-67, MCM-2 and antiapoptotic protein Bcl-2 in
colorectal cancer. Anticancer Res. 29:3049–3052. 2009.PubMed/NCBI
|
|
21
|
Gakiopoulou H, Korkolopoulou P, Levidou G,
Thymara I, Saetta A, Piperi C, Givalos N, Vassilopoulos I, Ventouri
K, Tsenga A, et al: Minichromosome maintenance proteins 2 and 5 in
non-benign epithelial ovarian tumours: Relationship with cell cycle
regulators and prognostic implications. Br J Cancer. 97:1124–1134.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scott IS, Heath TM, Morris LS, Rushbrook
SM, Bird K, Vowler SL, Arends MJ and Coleman N: A novel
immunohistochemical method for estimating cell cycle phase
distribution in ovarian serous neoplasms: Implications for the
histopathological assessment of paraffin-embedded specimens. Br J
Cancer. 90:1583–1590. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dudderidge TJ, Stoeber K, Loddo M,
Atkinson G, Fanshawe T, Griffiths DF and Williams GH: Mcm2,
Geminin, and KI67 define proliferative state and are prognostic
markers in renal cell carcinoma. Clin Cancer Res. 11:2510–2517.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kodani I, Osaki M, Shomori K, Araki K,
Goto E, Ryoke K and Ito H: Minichromosome maintenance 2 expression
is correlated with mode of invasion and prognosis in oral squamous
cell carcinomas. J Oral Pathol Med. 32:468–474. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feng D, Tu Z, Wu W and Liang C: Inhibiting
the expression of DNA replication-initiation proteins induces
apoptosis in human cancer cells. Cancer Res. 63:7356–7364.
2003.PubMed/NCBI
|
|
26
|
McGuire WP III and Markman M: Primary
ovarian cancer chemotherapy: Current standards of care. Br J
Cancer. 89 (Suppl 3):S3–S8. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mah LJ, El-Osta A and Karagiannis TC:
GammaH2AX: A sensitive molecular marker of DNA damage and repair.
Leukemia. 24:679–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cortez D, Glick G and Elledge SJ:
Minichromosome maintenance proteins are direct targets of the ATM
and ATR checkpoint kinases. Proc Natl Acad Sci USA.
101:10078–10083. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bates S and Vousden KH: p53 in signaling
checkpoint arrest or apoptosis. Curr Opin Genet Dev. 6:12–18. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gottlieb TM and Oren M: p53 and apoptosis.
Semin Cancer Biol. 8:359–368. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Janus F, Albrechtsen N, Dornreiter I,
Wiesmüller L, Grosse F and Deppert W: The dual role model for p53
in maintaining genomic integrity. Cell Mol Life Sci. 55:12–27.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Macleod K, Mullen P, Sewell J, Rabiasz G,
Lawrie S, Miller E, Smyth JF and Langdon SP: Altered ErbB receptor
signaling and gene expression in cisplatin-resistant ovarian
cancer. Cancer Res. 65:6789–6800. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Labib K: How do Cdc7 and cyclin-dependent
kinases trigger the initiation of chromosome replication in
eukaryotic cells? Genes Dev. 24:1208–1219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bochman ML and Schwacha A: The Mcm2-7
complex has in vitro helicase activity. Mol Cell. 31:287–293. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shima N, Alcaraz A, Liachko I, Buske TR,
Andrews CA, Munroe RJ, Hartford SA, Tye BK and Schimenti JC: A
viable allele of Mcm4 causes chromosome instability and mammary
adenocarcinomas in mice. Nat Genet. 39:93–98. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang C, Wen Y, Li H, Zhang D, Zhang N, Shi
X, Jiang B, Ma X, Yang P, Tang H, et al: Overexpression of
minichromosome maintenance 2 predicts poor prognosis in patients
with gastric cancer. Oncol Rep. 27:135–142. 2012.PubMed/NCBI
|
|
37
|
Obermann EC, Went P, Zimpfer A, Tzankov A,
Wild PJ, Stoehr R, Pileri SA and Dirnhofer S: Expression of
minichromosome maintenance protein 2 as a marker for proliferation
and prognosis in diffuse large B-cell lymphoma: A tissue microarray
and clinico-pathological analysis. BMC Cancer. 5:1622005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Crevel G, Hashimoto R, Vass S, Sherkow J,
Yamaguchi M, Heck MM and Cotterill S: Differential requirements for
MCM proteins in DNA replication in Drosophila S2 cells. PLoS One.
2:e8332007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tsao CC, Geisen C and Abraham RT:
Interaction between human MCM7 and Rad17 proteins is required for
replication checkpoint signaling. EMBO J. 23:4660–4669. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Oehlmann M, Score AJ and Blow JJ: The role
of Cdc6 in ensuring complete genome licensing and S phase
checkpoint activation. J Cell Biol. 165:181–190. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ibarra A, Schwob E and Méndez J: Excess
MCM proteins protect human cells from replicative stress by
licensing backup origins of replication. Proc Natl Acad Sci USA.
105:8956–8961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lei M, Kawasaki Y and Tye BK: Physical
interactions among Mcm proteins and effects of Mcm dosage on DNA
replication in Saccharomyces cerevisiae. Mol Cell Biol.
16:5081–5090. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rowles A, Chong JP, Brown L, Howell M,
Evan GI and Blow JJ: Interaction between the origin recognition
complex and the replication licensing system in Xenopus. Cell.
87:287–296. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stoeber K, Tlsty TD, Happerfield L, Thomas
GA, Romanov S, Bobrow L, Williams ED and Williams GH: DNA
replication licensing and human cell proliferation. J Cell Sci.
114:2027–2041. 2001.PubMed/NCBI
|
|
45
|
Woodward AM, Göhler T, Luciani MG,
Oehlmann M, Ge X, Gartner A, Jackson DA and Blow JJ: Excess Mcm2-7
license dormant origins of replication that can be used under
conditions of replicative stress. J Cell Biol. 173:673–683. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hyrien O, Marheineke K and Goldar A:
Paradoxes of eukaryotic DNA replication: MCM proteins and the
random completion problem. Bioessays. 25:116–125. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Masata M, Malínský J, Fidlerová H, Smirnov
E and Raska I: Dynamics of replication foci in early S phase as
visualized by cross-correlation function. J Struct Biol. 151:61–68.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kunnev D, Freeland A, Qin M, Leach RW,
Wang J, Shenoy RM and Pruitt SC: Effect of minichromosome
maintenance protein 2 deficiency on the locations of DNA
replication origins. Genome Res. 25:558–569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kwon HJ, Hong YK, Park C, Choi YH, Yun HJ,
Lee EW and Kim BW: Widdrol induces cell cycle arrest, associated
with MCM down-regulation, in human colon adenocarcinoma cells.
Cancer Lett. 290:96–103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Minagawa Y, Kigawa J, Itamochi H, Kanamori
Y, Shimada M, Takahashi M and Terakawa N: Cisplatin-resistant HeLa
cells are resistant to apoptosis via p53-dependent and -independent
pathways. Jpn J Cancer Res. 90:1373–1379. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Asada N, Tsuchiya H and Tomita K: De novo
deletions of p53 gene and wild-type p53 correlate with acquired
cisplatin-resistance in human osteosarcoma OST cell line.
Anticancer Res. 19:5131–5137. 1999.PubMed/NCBI
|
|
52
|
Kigawa J, Sato S, Shimada M, Takahashi M,
Itamochi H, Kanamori Y and Terakawa N: p53 gene status and
chemosensitivity in ovarian cancer. Hum Cell. 14:165–171.
2001.PubMed/NCBI
|