Introduction
Hypertension is one of the most common chronic
diseases that is accompanied with a variety of severe
complications, including kidney failure, myocardial infarction and
atherosclerosis (1). Structural
alterations or vascular remodeling, increased stiffness and
endothelial dysfunction are key features of hypertension (2,3).
Vascular endothelial cells form a layer of flat squamous cells
located in the lining of the blood vessels and, serve roles in
maintaining the integrity and permeability of the vessel wall and
regulating vascular tone (4). Once
the vascular endothelial cells experience damage, the synthesis and
secretion angiotensin (Ang) II is promoted, leading to
vasoconstriction and increases in blood pressure (5,6). Ang
II was reported to be the most important element in the
renin-angiotensin system (RAS) (7). Several clinical applications revealed
that blocking the angiotensin II receptor could not only suppress
variations in and reduce blood pressure, but also decreased the
morbidity and mortality associated with cardiovascular diseases
(8,9).
Recently, the (pro) renin receptor (PRR) has
received widespread attention as a novel component of the RAS. It
was reported that both the knockdown and overexpression of PRR gene
in local tissues had notable effects on local-tissue RAS and blood
pressure regulation (10,11). Increased expression of PRR has been
detected in the nephritic collecting duct of Ang II-induced rat
models and the plasma of patients with hypertension (12,13).
PRR knockout could significantly suppress Ang II-induced
hypertension and the production of Ang II in the brain of human
renin-angiotensinogen double transgenic mice (14). Interestingly, high-expression PRR
also has a promoting effect on Ang II production, suggesting an
augmentation in positive feedback between PRR and Ang II (15,16).
MicroRNA (miRNA/miR) is a single-strand RNA molecule
of 21–24 nucleotides, which plays a vital role in the development
and homeostasis of tissues and organs by inhibiting protein
translation at the posttranscriptional level, or promoting mRNA
degradation (17). Previous
studies indicated that the marked difference in the miRNA
expression profile in the peripheral blood of patients with
hypertension contributed to the early diagnosis and the prognosis
of complications of this condition (18,19).
At present, investigations with different experimental models have
revealed that miR-133a protected against myocardial fibrosis and
modulated electrical repolarization (20,21).
In vivo experiments demonstrated that Ang II caused an
increase in systolic blood pressure and myocardial fibrosis in
under conditions of downregulated miR-133a and miR-29b (14). However, in the experimental model
of Ang II-dependent hypertension, the expression of miR-133a was
notably reduced under Ang II treatment, suggesting that high levels
of Ang II are negatively related to miR-133a expression (14,22).
To investigate the molecular mechanism of Ang
II-induced hypertension, in this study, we employed miR-133a mimic
and inhibitor to assess the inhibitory effect of Ang II on the
levels of miR-133a. We also measured the expression of PRR under
Ang II treatment and miR-133a mimic/inhibitor to explore the
possible role of PRR in Ang II-dependent hypertension.
Materials and methods
Cell culture and identification
Human umbilical vein endothelial cells (HUVECs) were
obtained from the Global Bioresource Center [American Type Culture
Collection (ATCC) PCS-100-010™]. The cells were maintained in
Dulbecco's modified Eagle's medium (DMEM; ATCC 30–2002™) containing
10% fetal bovine serum (FBS; ATCC 30-2020™) in a humidified
atmosphere of 5% CO2 at 37°C. The medium was replaced
every 2 days until the cells reached ~80-90% confluency. After
treatment with trypsin, the cells were transferred to multi-well
plates for further culture.
After 1 week of culture, the HUVECs were identified
using immunofluorescence staining. HUVECs were rinsed with 0.1 M
PBS. HUVECs were fixed in 100% methanol at 4°C for 30 min after
attaining 90% confluence. Cells were treated with PBS containing
0.25% Triton X-100 (T8200; Beijing Solarbio Science &
Technology, Co., Ltd.) for 30 min at room temperature to increase
cell permeability. Cells were blocked with 3% bovine serum albumin
(cat. no. 37525; Thermo Fisher Scientific, Inc.) in PBS for 60 min
at room temperature. The cells were then incubated with rabbit
anti-Fac VIII antibody (1:400, ab6994; Abcam) overnight at 4°C.
After removing the primary antibody by rinsing with PBS, the cells
were cultured with secondary Goat anti rabbit IgG (Alexa
Fluor® 488, 1:400, ab150077; Abcam) at room temperature
for 2 h in the dark. DAPI (Wuhan Boster Bio Engineering Co., Ltd.)
was used to stain the cell nuclei (blue) at room temperature for 10
min. After washing with PBS, the cells were analyzed under a
fluorescence microscope (Olympus Corporation, magnification, ×400)
and images were captured with a DP70 digital camera (Olympus
Corporation), 5 areas of per section were captured. PBS was used in
place of the antibodies in the control condition.
Survival of HUVECs with Ang II
treatment
The effects of Ang II (IA0380, Beijing Solarbio
Science & Technology, Co., Ltd.) on HUVECs were examined using
an MTT assay (cat. no. M2128, Sigma-Aldrich; Merck KGaA). HUVECs
were seeded in 96-well plate, and adjusted to 4×103
cells/well with DMEM containing 10% FBS. The cell suspension was
divided into several groups for Ang II pretreatment of different
concentrations (0, 10−7, 10−6,
10−5 and 10−4 M); the control group remained
untreated. As described previously (23), three time points were set (24, 48
and 72 h) to analyze the potential time-dependent effects of the
Ang II on cell viability. Subsequently, the plates were incubated
with MTT solution for 4 h. The supernatant was removed, and DMSO
(cat. no. D2650, Sigma-Aldrich; Merck KGaA) was added to dissolve
formazan crystals. The absorbance was measured at 450 nm in a
microplate reader.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
To investigate the effect of Ang II with different
concentrations on miR-133a expression, TRIzol reagent (Thermo
Fisher Scientific, Inc.) was used to extract total miRNAs and mRNAs
from Ang II-treated HUVECs. cDNA was synthesized from RNA using the
PrimeScript™ 1st Strand cDNA Synthesis kit (Takara Bio, Inc.) with
miRNA-specific primers (Table I).
The 20 µl reaction mixture was incubated under the following
parameters: Firstly, 65°C for 5 min, secondly, 30°C for 6 min and
50°C for 50 min. The mRNA levels were amplified using the SYBR
Premix Ex Taq™ II in the 7500 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The 20 µl qPCR
reaction solution contained 1 µl each of the forward and reverse
primers (10 µM), 10 µl SYBR fluorescent dye, 2 µl cDNA and RNase
Free dH2O. qPCR reactions were performed as follows:
Initial denaturation at 95°C for 10 min, repeated amplification by
40 cycles of 95°C for 13 sec, 60°C for 30 sec and 72°C for 2 min
and finally 72°C for 10 min. GAPDH was employed as an internal
control. The relative levels of miR-133a were calculated with the
2−ΔΔCq method (24).
 | Table I.Primers employed for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers employed for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Primer
sequences |
|---|
| MicroRNA-133a | Forward:
5′-ATAAGAATGCGGCCGCATTCCAAACTAGCAGCACTA-3′ |
|
| Reverse:
5′-AGCTTTGTTTAAACTTAACCATTCTAGCTTTTCC-3′ |
| Prorenin
receptor | Forward:
5′-CAGACGTGGCTGCATTGTCC-3′ |
|
| Reverse:
5′-CTGGGGGTAGAGCCAGTTTGTT-3 |
| GAPDH | Forward:
5′-ATGGCACCGTCAAGGCTGAG-3′ |
|
| Reverse:
5′-TGTCAGGTACGGTAGTGACG-3 |
Transfection of miR-133a mimics and
inhibitor
The miR-133a mimics/inhibitor (cat. nos. 4464066 and
4464084) were obtained from Thermo Fisher Scientific, Inc. HUVECs
were incubated in 6-well plates at 50% confluence. miR-133a mimics,
miR-133a inhibitor and negative miRNA control (cat. no. 4464061)
were diluted into 250 µl DMEM to a concentration of 50 nM and 5 µl
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) at room temperature. The mixture of diluted miRNA
and transfection reagents were dispensed into plates and incubated
at 37°C for 6 h in a humidified atmosphere of 5% CO2;
the media was then replaced complete medium (DMEM with 10% FBS).
The next day, cells were harvested and the efficacy of transfection
with the miR-133a mimics and inhibitor were tested by RT-qPCR. An
MTT assay was used to perform the cell viability of all groups'
cells at 24, 48 and 72 h.
Effects of miR-133a mimics/inhibitor
on the viability of Ang II-induced HUVECs
HUVECs transfected with miR-133a mimics/inhibitor
and empty vector were incubated with Ang II and FBS-free DMEM to
assess the effects of miR-133a mimics/inhibitor on Ang II
(10−5 M)-induced HUVECs. The viabilities of treated
cells were compared with the control group (untreated cells), and
cells transfected with miR-133a mimics/inhibitor and empty vector
(without Ang II treatment) groups were determined via an MTT assay
every 24 h as aforementioned.
Flow cytometry for the detection of
apoptotic Ang II-induced HUVECs
Cells in all groups were diluted to 4×103
cells/well with complete medium in 96-well plates. After 48 h,
HUVECs were trypsinized and harvested by centrifugation at 1,000 ×
g for 10 min at 4°C. Cells were resuspended in binding buffer with
propidium iodide (PI) and Annexin V-fluorescein isothiocyanate. The
stained cells were incubated at dark place for 15 min at room
temperature. Then, the apoptosis of various treated HUVECs was
assessed immediately with a FACScalibur flow cytometer (BD
Biosciences, San Jose, CA, USA) equipped with CellQuest software
(version 3.3; BD Biosciences).
miR-133a target prediction and a dual
luciferase assay
TargetScan (version 7.1, http://www.targetscan.org/vert_71/) was used to
predict potential target genes of miR-133a and identified PRR
(ATPase H+ transporting accessory protein 2) as a potential target.
Firstly, we cloned miR-133a into the GV272 vector (GV268-miR-133a;
Shanghai GeneChem Co., Ltd.). In addition, we inserted the target
sequences of miR-133a (accession no. NM_005765) present in the
3′untranslated region of PRR into the GV268 plasmid vector
(GV272-PRR 3′UTR). Secondly, after mutation using the QuikChange
Multi Site-Directed Mutagenesis kit (Agilent Technologies, Inc.)
the hsa-miR-133a-3p.1 (miRbase, accession no. MIMAT0000427)
sequences used were changed from 5′-UUGGUCCCCUUCAACCAGCUG-3′ to
5′-UUCCACCCCUUCAACCAGCUG-3′, which was predicted to abolish
binding; the mutated fragment was inserted into GV268 plasmid
vector (GV268-miR-133a-mut).
Luciferase reporter experiments were performed in a
human kidney epithelial cell line (293T, ATCC CRL-3216™; ATCC)
using Dual-Luciferase® Reporter Assay System (Promega
Corporation). 293T cells were cultured in an incubator with 5%
CO2 at 37°C and harvested at 80% confluence, and then
seeded into a 24-well plate with complete medium. Cells were
transfected with 50 ng of GV272-PRR 3′UTR plasmid, 40 nM of control
or GV268-miR-133a or GV268-miR-133a-mut were transfected into cells
using Lipofectamine 2000 as aforementioned. After 48 h
post-transfection, 293T cells were lysed and separated by
centrifugation at 15,000 × g for 3 min at 4°C. The prepared dual
luciferase reporter mixture was added into the supernatant of lysed
cells and the relative luciferase activity was immediately measured
by Sinergy 2 luminometer (Biotek Instruments, Inc.), Renilla
luciferase activity was used for normalization.
PRR expression in the miR-133a
mimics/inhibitor transfected HUVECs
The expression levels of PRR in HUVECs transfected
with miR-133a mimics, miR-133a inhibitor, or empty vector were
measured by RT-qPCR as described above and western blotting. The
primers of PRR were presented in Table
I. A BCA protein assay was used to extract the total proteins
from all cell groups. 10% SDS-PAGE was used to separate proteins
(30 µg), and the membrane was transferred to a polyvinylidene
difluoride membrane (EMD Millipore) and blocked with 5% nonfat milk
in TBST at room temperature for 60 min. The membrane was incubated
with rabbit anti-PRR (1:1,000, HPA003156; Sigma-Aldrich; Merck
KGaA) primary antibodies at 4°C overnight. Subsequently, the
membrane was washed three times in TBST for 10 min at room
temperature, and then was incubated with the goat anti-rabbit IgG,
HRP-linked antibody (1:10,000, cat. no. 31460; Thermo Fisher
Scientific, Inc.) for 2 h at room temperature. Proteins were
detected using ECL™ western blot detection reagents (cat. no.
32106; Thermo Fisher Scientific, Inc.). Optical band density was
quantified by ImageJ software (version 1.46; National Institutes of
Health). GAPDH was considered as an internal control.
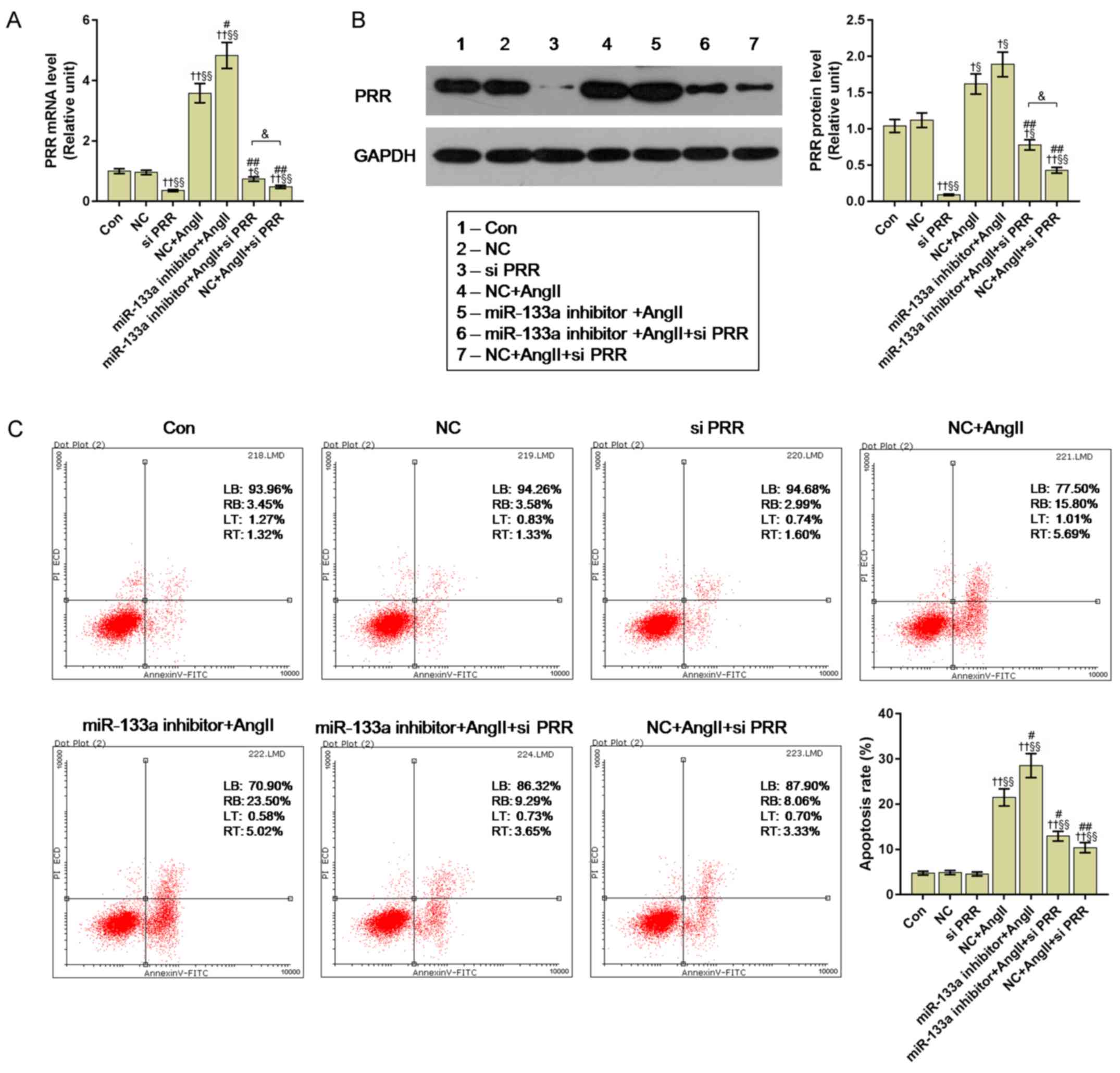
Effects of PRR silencing on miR-133a
inhibitor-transfected HUVECs under Ang II treatment
Small interfering RNA specifically targeting PRR
(siPPR; cat. no. 138075) was obtained from Invitrogen (Thermo
Fisher Scientific, Inc.). Lipofectamine 2000, the restriction
enzymes BamHI (cat. no. ER0051) and EcoRI (cat. no.
ER0271) (both from Thermo Fisher Scientific, Inc.) and the pcDNA3.1
(+) vector (Shanghai GenePharma Co., Ltd.) was used to transfect
siPRR (50 nM) into untreated cells and miR-133a inhibitor-treated
cells (miR-133a inhibitor + siPRR), according to the manufacturer's
protocols. Following transfection for 48 h, cells were harvested
and used for subsequently experiments. To explore the effects of
siPRR, miR-133a inhibitor and Ang II treatment on PRR expression,
seven groups were generated, including control, empty vector (NC),
siPRR, NC + Ang II, miR-133a inhibitor + Ang II, NC + Ang II +
siPRR and miR-133a inhibitor + Ang II + siPRR. The relative levels
of PRR were determined by RT-qPCR and western blotting as described
above. We also measured the rate of apoptosis of these cell groups
by flow cytometry as described above, the advanced apoptotic cells
in the upper right quadrant and early apoptotic cells in the lower
right quadrant, the apoptotic rate was the sum of the advanced
apoptotic rate and the early apoptotic.
Statistical analysis
Statistical significance was analyzed by using
one-way analysis of variance between groups, followed by a
Bonferroni's post-hoc test using SPSS version 20.0 (IBM Corp.). All
data analysis were performed three times. All data were presented
as the mean ± standard error of the mean. P<0.05 was considered
to indicate a statistically significant difference.
Results
Cultured cells are identified as
HUVECs by immunofluorescence analysis
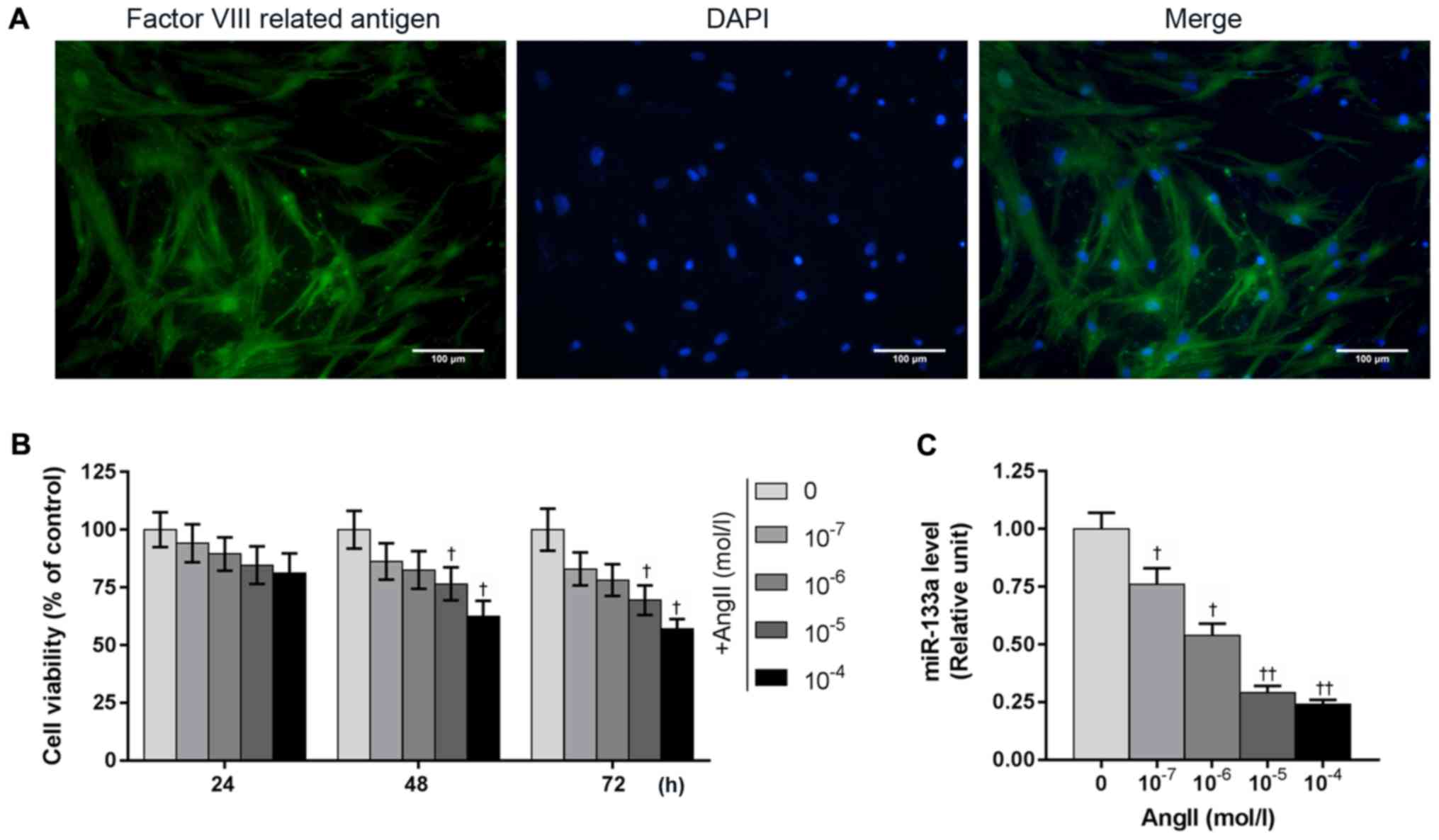
As shown in Fig. 1,
the notable green and blue fluorescence indicated positive
identification of factor VIII-related antigen. Cell nuclei were
stained with DAPI, in which the blue fluorescence was evenly
distributed in the nucleus. Our results demonstrated the obtained
cells were highly purified HUVECs.
Ang II suppresses cell viability and
miR-133a expression in HUVECs
Our results showed that Ang II inhibited the
viability of HUVECs in a time- and concentration-dependent manner,
while the expression of miR-133a was reduced with increasing
concentrations of Ang II. In addition, Ang II of 10−5 M
significantly suppressed the viability of HUVECs at 48 h compared
with the control; a significant decrease in miR-133a expression was
also noted (Fig. 1B and C).
Therefore, Ang II of 10−5 M was selected for subsequent
experiments.
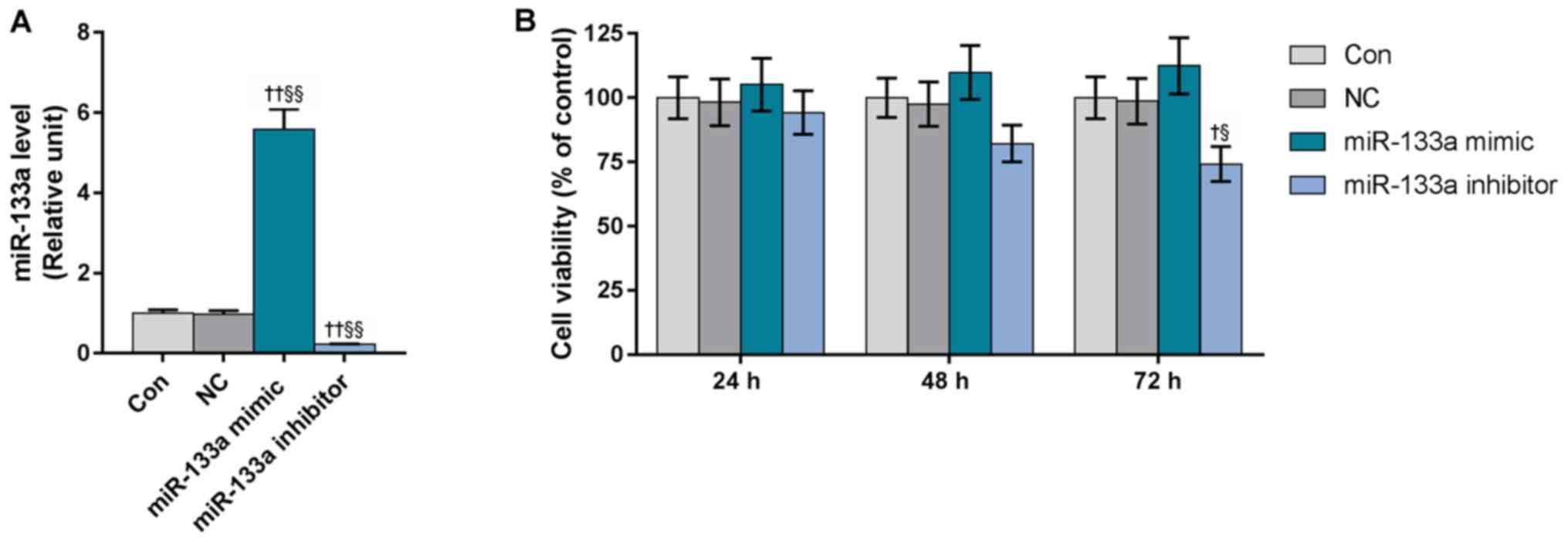
Downregulated miR-133a suppresses the
viability of HUVECs
To investigate the effects of miR-133a on the
viability of HUVECs, cells were transfected with miR-133a mimics or
inhibitors (Fig. 2A). Our results
revealed that overexpression of miR-133a markedly enhanced the
viability of HUVECs, whereas miR-133a inhibitors exerted opposing
effects on HUVECs and significantly reduced the cell viability at
72 h, compared with the control and miR-133a mimics groups
(Fig. 2B).
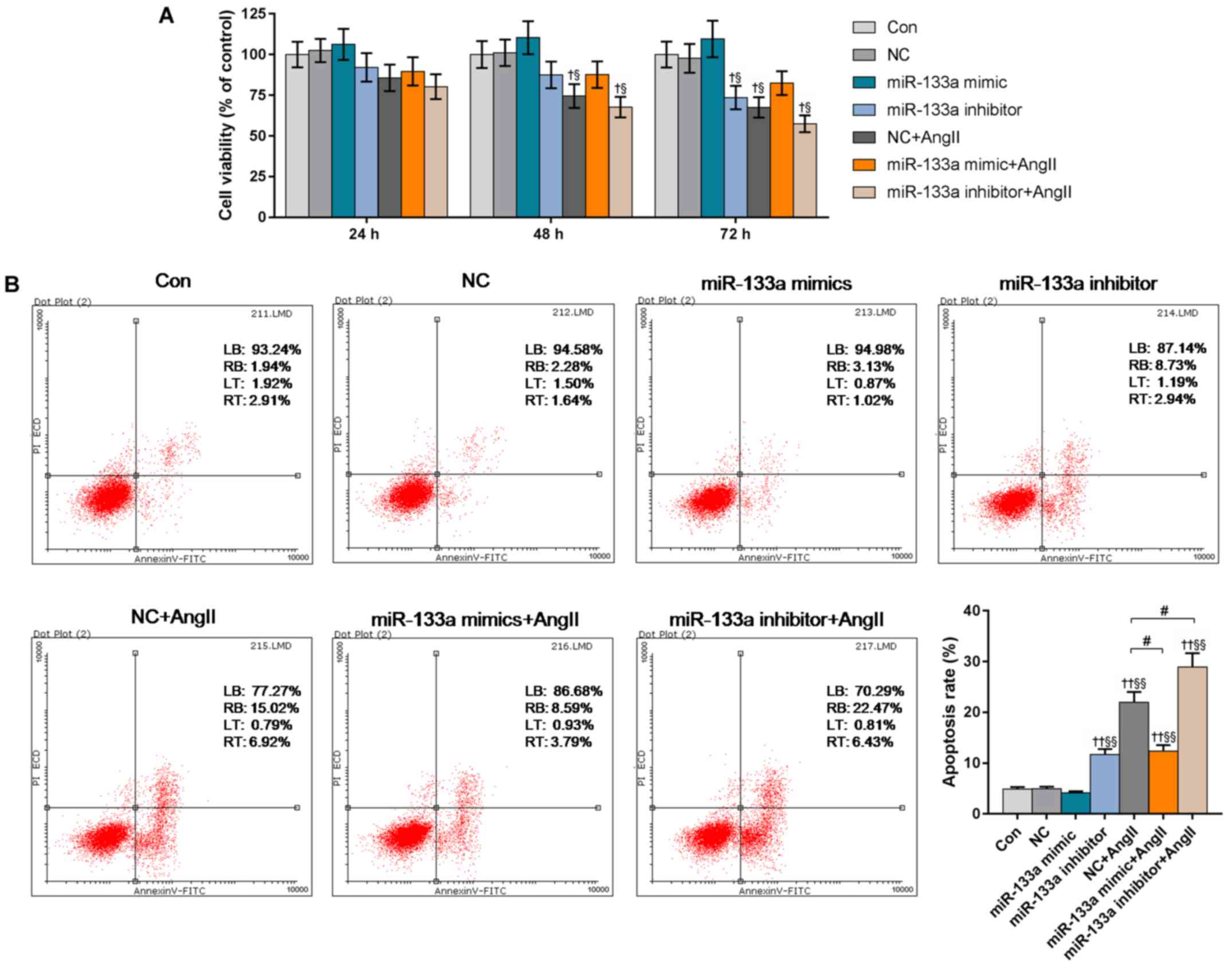
Upregulated miR-133a enhances the
viability of Ang II-induced HUVECs
Our results showed the cell viabilities in the
miR-133a inhibitor + Ang II and NC + Ang II groups to be
significantly decreased at 48 h, compared with the control and NC
groups. miR-133a mimic had a notably positive effect on cell
viability. The cell viability in miR-133a inhibitor group was
significantly reduced at 72 h compared with the control; the cell
viability in miR-133a inhibitor + Ang II group was the lowest
(Fig. 3A).
Flow cytometry was used to determine the apoptotic
rates of the various treated HUVECs (Fig. 3B). The apoptotic rate of miR-133a
inhibitor-transfected cells significantly increased from 4.85% in
the control to 11.67% (P<0.01). miR-133a mimic markedly reduced
the Ang II-induced apoptotic rate from 21.94% in the NC + Ang II
group to 12.38% (P<0.05). The miR-133a inhibitor + Ang II group
had the highest apoptotic rate (28.9%).
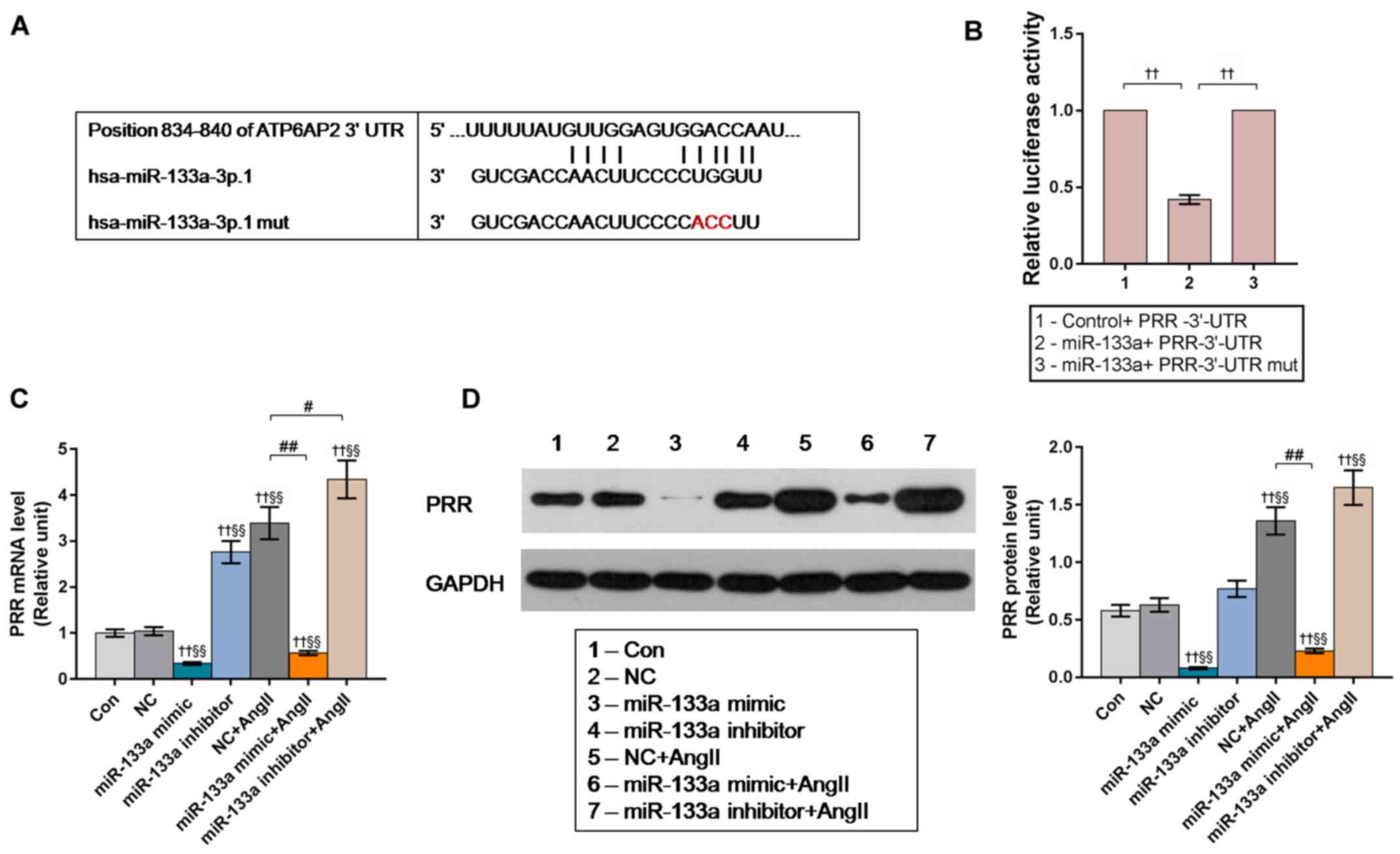
miR-133a targets the 3′UTR of PRR
mRNA
In the first step of target prediction, we used
TargetScan and identified two potential binding sites for miR-133a
in the position 834–840 of human PRR 3′UTR. Bioinformatic analysis
for the target site of miR-133a in the 3′-UTR of PRR was shown in
Fig. 4A. We also performed a
dual-luciferase reporter assay to further verify the putative
direct binding of miR-133a to the 3′UTR of PRR mRNA.
293T cells were co-transfected with plasmids
containing the 3′-UTR of PRR (GV272-PRR 3′UT), and miR-133a
(GV268-miR-133a) or miR-133a-mut plasmid (GV268-miR-133a-mut). The
results of the luciferase assay revealed that the activity of
firefly luciferase in the miR-133a-transfected group was
significantly reduced compared with the control, while no
significant changes were observed in miR-133a-mut transfected group
(Fig. 4B).
We further examined the expression levels of PRR in
the miR-133a mimic/inhibitor-transfected HUVECs. The results of
RT-qPCR and western blotting demonstrated that the levels of PRR in
miR-133a mimic-transfected HUVECs were significantly lower than the
control groups, while the mRNA level of PRR in miR-133a inhibitor
group and NC + Ang II group were significantly higher compared with
the control groups; and miR-133a inhibitor significantly enhanced
the promotive effect of Ang II on PRR expression (Fig. 4C and D). These results indicated
that Ang II promotes PRR expression by inhibiting miR-133a.
siPRR weakens the proapoptotic effects
of Ang II and miR-133a inhibitor on HUVECs
We employed siPRR to investigate the molecular
mechanism of Ang II acting on PRR expression. Our results showed
that Ang II induced significantly increased the expression of PRR,
compared with the control and NC groups (P<0.01); however, the
miR-133a inhibitor + Ang II group had a significantly higher
expression of PRR than the NC + Ang II group (P<0.05). After
being transfected with siPRR, the PRR levels were significantly
decreased compared with the control; the relative expression levels
of PRR in the miR-133a inhibitor + Ang II + siPRR group was
significantly higher than in the NC + Ang II + siPRR group
(P<0.05) (Fig. 5A and B).
We further measured the apoptosis of all cell groups
by flow cytometry. The apoptotic rate of the siPRR group exhibited
no significant difference compared with the control and NC groups,
but Ang II treatment significantly enhanced apoptosis from 4.77% in
the control to 21.49%. miR-133a inhibitor significantly enhanced
the apoptosis-promoting effects of Ang II (P<0.05). Importantly,
siPRR significantly reduced the apoptotic rate from 21.49% in the
NC + Ang II group and 28.52% in the miR-133a inhibitor + Ang II
group to 11.39 and 12.94%, respectively (P<0.01) (Fig. 5C).
Discussion
Endothelial cells play a crucial role in regulating
the production of Ang and vasopermeability (25,26).
Endothelial dysfunction was considered as an initiation factor in
the pathophysiology of essential hypertension and subsequently
induced various complications (27). Previous studies have reported that
miRNAs were extensively involved in the occurrence of hypertension,
and served an important role in the regulation of blood pressure
(28,29). In present study, we observed that
miR-133a had the ability to directly modulate the expression of PRR
at the transcriptional level; Ang II was proposed to significantly
upregulate PRR expression to enhance the apoptosis of HUVECs
through the dysregulation of miR-133a levels.
Numerous reports have suggested that Ang II, as a
major effector peptide of the RAS, was closely related to the
occurrence and development of hypertension (30,31).
Our results showed that Ang II treatment could significantly
suppress the viability of HUVECs with increasing concentrations of
Ang II, which may promote the process of endothelial dysfunction
and hypertension. The effects of Ang II on HUVEC viability also
occurred in a time-dependent manner. Several previous studies
showed that the endogenous miRNAs produced by endothelial cells
regulated the expression of hypertension-associated genes (18,32,33).
In 2012, Castoldi et al (14) revealed that downregulated miR-133a
was detected in cardiac hypertrophy in transgenic mice with cardiac
overexpression of an active mutant protein kinase B; Kontaraki
et al (20) demonstrated
that miR-133a was negatively associated with left ventricular
hypertrophy. These findings were consistent with our results, in
which the expression of miR-133a was significantly inhibited in Ang
II-induced HUVECs at the concentration of 10−5 M. To
explore the relationship between miR-133a expression and the
suppressed viability of HUVECs induced by Ang II, miR-133a mimics
and inhibitor were employed. We reported that miR-133a mimics
counteracted the inhibitory effects of Ang II on miR-133a
expression, but miR-133a inhibitor could promote the apoptosis
induced by Ang II treatment. These results suggested that Ang II
enhanced the apoptosis of HUVECs through the dysregulation of
miR-133a expression, while increased miR-133a levels may suppress
apoptosis.
Recently, PRR was reported in the molecular
mechanism of hypertension as a member of RAS (34,35).
Several researches detected upregulated PRR in Ang II-dependent
hypertension (11,36), which was consistent with our
experiments whereby the expression levels of PRR were significantly
increased following Ang II treatment. In 2015, Li et al
(10) designed and developed a
novel PRR inhibitory peptide (PRO20), which could efficiently
inhibit PRR binding to prorenin and Ang II-dependent hypertension.
Of note, miR-133a inhibitor was observed to induce the expression
of PRR, while, miR-133a mimics suppressed PRR expression with or
without Ang II treatment. It has been known that miRNAs typically
bind to the mRNA 3′-UTR of its target gene or lead to the
degradation the mRNA at the post-transcriptional level, thereby
inhibiting the expression of the target gene (37–39).
A previous study found that miR-152 could modulate the RAS through
directly targeting PRR under hyperglycemic conditions (40). In 2018, Wang et al (36) reported that 30 differentially
expressed miRNAs were predicted to target RAS components, and
transfection of miR-181a-5p and miR-663 into HTR-8/SVneo
trophoblast cells suppressed the mRNA expression of genes encoding
prorenin, and prorenin protein production (41). However, no sufficient role of
miR-133a in the PRR-protein-angiotensin system was determined.
Therefore, we evaluated the targeting ability of miR-133a to PRR
mRNA by using TargetScan and conducting a dual-luciferase assay,
and confirmed that PRR was a target of miR-133a. The PRR 3′ UTR was
determined to contain two predicted conserved target sites for
miR-133a.
In present study, siPRR was successfully transfected
into HUVECs to investigate the role of PRR in cell apoptosis
mediated by Ang II. Our results demonstrated that siPRR had no
notable effect on cell apoptosis compared with the control and NC
groups, yet the apoptosis of the NC + Ang II and miR-133a inhibitor
+ Ang II groups were significantly decreased by PRR silencing.
Taken together, our results suggested that siPRR could
significantly suppress the proapoptotic ability of Ang II and
miR-133a inhibitor.
There are some limitations to our study. The
observation that miR-133a downregulation promoted HUVECs injury was
only supported by in vitro experiments only. The effects of
miR 133 on myocardial fibrosis in vivo and in vitro
were also not evaluated, which poses as a limitation. Additionally,
the mechanism we proposed requires further investigation.
In conclusion, we investigated the roles of Ang II,
miR-133a and PRR in the molecular mechanism of hypertension. We
reported that Ang II may serve in a negative feedback mechanism in
association with miR-133a in the context of HUVEC viability. Ang II
had the ability to gradually reduce cell viability of HUVECs with
increasing concentrations, but this could be counteracted with
increased miR-133a expression. miR-133a was demonstrated to be able
to directly bind the PRR 3′UTR and inhibit PRR expression at the
post-transcriptional level. Most importantly, our results suggested
a possible mechanism in which Ang II inhibited the expression of
miR-133a, while increased PRR promoted the apoptosis of HUVECs in
the absence of miR-133a downregulation. These findings may improve
understanding of the mechanism underlying Ang II-dependent
hypertension and could aid developments into treatments for this
condition.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
BL and ML made substantial contributions to
conception and design of the present study. HW, DZ, JL and JT
performed data acquisition, data analysis and interpretation. BL
and ML drafted the article/critically revised it for important
intellectual content. All authors approved the final version to be
published. All authors agree to be accountable for all aspects of
the work in ensuring that questions related to the accuracy or
integrity of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hodgson TA and Cai L: Medical care
expenditures for hypertension, its complications, and its
comorbidities. Medical Care. 39:599–615. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ponticos M and Smith BD: Extracellular
matrix synthesis in vascular disease: Hypertension, and
atherosclerosis. J Biomed Res. 28:25–39. 2014.PubMed/NCBI
|
|
3
|
Rizzoni D and Agabiti-Rosei E: Structural
abnormalities of small resistance arteries in essential
hypertension. Intern Emerg Med. 7:205–212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang NP, Li H, Qiu YL, Zhou GM, Wang Y, Ma
J and Mei QB: The effects of microgravity on blood vessels and
vascular endothelial cells. Sheng Li Ke Xue Jin Zhan. 45:385–390.
2014.(In Chinese). PubMed/NCBI
|
|
5
|
Bali A and Jaggi AS: Angiotensin
II-triggered kinase signaling cascade in the central nervous
system. Rev Neurosci. 27:301–315. 2016.PubMed/NCBI
|
|
6
|
Zimmerman MC, Lazartigues E, Lang JA,
Sinnayah P, Ahmad IM, Spitz DR and Davisson RL: Superoxide mediates
the actions of angiotensin II in the central nervous system. Circ
Res. 91:1038–1045. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Becari C, Oliveira EB and Salgado MC:
Alternative pathways for angiotensin II generation in the
cardiovascular system. Braz J Med Biol Res. 44:914–919. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Naik P, Murumkar P, Giridhar R and Yadav
MR: Angiotensin II receptor type 1 (AT1) selective nonpeptidic
antagonists-a perspective. Bioorg Med Chem. 18:8418–8456. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Messerli FH and Bangalore S: Angiotensin
receptor blockers reduce cardiovascular events, including the risk
of myocardial infarction. Circulation. 135:2085–2087. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li W, Sullivan MN, Zhang S, Worker CJ,
Xiong Z, Speth RC and Feng Y: Intracerebroventricular infusion of
the (Pro)renin receptor antagonist PRO20 attenuates
deoxycorticosterone acetate-salt-induced hypertension.
Hypertension. 65:352–361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li XC and Zhuo JL: Recent updates on the
proximal tubule Renin-angiotensin system in angiotensin
II-Dependent hypertension. Curr Hypertens Rep. 18:632016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gonzalez AA, Lara LS, Luffman C, Seth DM
and Prieto MC: Soluble form of the (pro)renin receptor is augmented
in the collecting duct and urine of chronic angiotensin
II-dependent hypertensive rats. Hypertension. 57:859–864. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morimoto S, Ando T, Niiyama M, Seki Y,
Yoshida N, Watanabe D, Kawakami-Mori F, Kobori H, Nishiyama A and
Ichihara A: Serum soluble (pro)renin receptor levels in patients
with essential hypertension. Hypertens Res. 37:642–648. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Castoldi G, Di Gioia CR, Bombardi C,
Catalucci D, Corradi B, Gualazzi MG, Leopizzi M, Mancini M, Zerbini
G, Condorelli G, et al: MiR-133a regulates collagen 1A1: Potential
role of miR-133a in myocardial fibrosis in angiotensin II-dependent
hypertension. J Cell Physiol. 227:850–856. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li W, Peng H, Mehaffey EP, Kimball CD,
Grobe JL, van Gool JM, Sullivan MN, Earley S, Danser AH, Ichihara A
and Feng Y: Neuron-specific (pro)renin receptor knockout prevents
the development of salt-sensitive hypertension. Hypertension.
63:316–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gonzalez AA and Prieto MC: Roles of
collecting duct renin and (pro)renin receptor in hypertension: Mini
review. Ther Adv Cardiovasc Dis. 9:191–200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bartel DP: MicroRNA target recognition and
regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kriegel AJ, Baker MA, Liu Y, Liu P, Cowley
AW Jr and Liang M: Endogenous microRNAs in human microvascular
endothelial cells regulate mRNAs encoded by hypertension-related
genes. Hypertension. 66:793–799. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dorn GW II: MicroRNAs in cardiac disease.
Transl Res. 157:226–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kontaraki JE, Marketou ME, Parthenakis FI,
Maragkoudakis S, Zacharis EA, Petousis S, Kochiadakis GE and Vardas
PE: Hypertrophic and antihypertrophic microRNA levels in peripheral
blood mononuclear cells and their relationship to left ventricular
hypertrophy in patients with essential hypertension. J Am Soc
Hypertens. 9:802–810. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matkovich SJ, Wang W, Tu Y, Eschenbacher
WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM and Dorn GW II:
MicroRNA-133a protects against myocardial fibrosis and modulates
electrical repolarization without affecting hypertrophy in
pressure-overloaded adult hearts. Circ Res. 106:166–175. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu Y, Huang A, Li T, Su X, Ding H, Li H,
Qin X, Hou L, Zhao Q, Ge X, et al: MiR-152 reduces human umbilical
vein endothelial cell proliferation and migration by targeting
ADAM17. FEBS Lett. 588:2063–2069. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han G, Wei Z, Cui H, Zhang W, Wei X, Lu Z
and Bai X: NUSAP1 gene silencing inhibits cell proliferation,
migration and invasion through inhibiting DNMT1 gene expression in
human colorectal cancer. Exp Cell Res. 367:216–221. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shan H, Zhang S, Wei X, Li X, Qi H, He Y,
Liu A, Luo D and Yu X: Protection of endothelial cells against Ang
II-induced impairment: Involvement of both PPARa and PPARγ via
PI3K/Akt pathway. Clin Exp Hypertens. 38:571–577. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pourgholami MH, Khachigian LM, Fahmy RG,
Badar S, Wang L, Chu SW and Morris DL: Albendazole inhibits
endothelial cell migration, tube formation, vasopermeability, VEGF
receptor-2 expression and suppresses retinal neovascularization in
ROP model of angiogenesis. Biochem Biophys Res Commun. 397:729–734.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gkaliagkousi E, Gavriilaki E,
Triantafyllou A and Douma S: Clinical Significance of endothelial
dysfunction in essential hypertension. Curr Hypertens Rep.
17:852015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Poliseno L, Tuccoli A, Mariani L,
Evangelista M, Citti L, Woods K, Mercatanti A, Hammond S and
Rainaldi G: MicroRNAs modulate the angiogenic properties of HUVECs.
Blood. 108:3068–3071. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ooi JY, Bernardo BC and Mcmullen JR: The
therapeutic potential of miRNAs regulated in settings of
physiological cardiac hypertrophy. Future Med Chem. 6:205–222.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Biancardi VC, Bomfim GF, Reis WL,
Al-Gassimi S and Nunes KP: The interplay between Angiotensin II,
TLR4 and hypertension. Pharmacol Res. 120:88–96. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cha SA, Park BM and Kim SH:
Angiotensin-(1–9) ameliorates pulmonary arterial hypertension via
angiotensin type II receptor. Korean J Physiol Pharmacol.
22:447–456. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Condorelli G, Latronico MV and Cavarretta
E: microRNAs in cardiovascular diseases: Current knowledge and the
road ahead. J Am Coll Cardiol. 63:2177–2187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dluzen DF, Kim Y, Bastian P, Zhang Y,
Lehrmann E, Becker KG, Noren Hooten N and Evans MK: MicroRNAs
modulate oxidative stress in hypertension through PARP-1
regulation. Oxid Med Cell Longev. 2017:39842802017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang T: Crosstalk between (Pro)renin
receptor and COX-2 in the renal medulla during angiotensin
II-induced hypertension. Curr Opin Pharmacol. 21:89–94. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Williamson CR, Khurana S, Nguyen P, Byrne
CJ and Tai TC: Comparative analysis of renin-angiotensin system
(RAS)-related gene expression between hypertensive and normotensive
rats. Med Sci Monit Basic Res. 23:20–24. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang L, Zhu Q, Lu A, Liu X, Zhang L, Xu C,
Liu X, Li H and Yang T: Sodium butyrate suppresses angiotensin
II-induced hypertension by inhibition of renal (pro)renin receptor
and intrarenal renin-angiotensin system. J Hypertens. 35:1899–1908.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Christodoulatos GS and Dalamaga M:
Micro-RNAs as clinical biomarkers and therapeutic targets in breast
cancer: Quo vadis? World J Clin Oncol. 5:71–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lauressergues D, Couzigou JM, Clemente HS,
Martinez Y, Dunand C, Bécard G and Combier JP: Primary transcripts
of microRNAs encode regulatory peptides. Nature. 520:90–93. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sharma NM, Nandi SS, Zheng H, Mishra PK
and Patel KP: A novel role for miR-133a in centrally mediated
activation of the renin-angiotensin system in congestive heart
failure. Am J Physiol Heart Circ Physiol. 312:H968–H979. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Haque R, Hur EH, Farrell AN, Iuvone PM and
Howell JC: MicroRNA-152 represses VEGF and TGFβ1 expressions
through post-transcriptional inhibition of (Pro)renin receptor in
human retinal endothelial cells. Mol Vis. 21:224–235.
2015.PubMed/NCBI
|
|
41
|
Wang Y, Lumbers ER, Arthurs AL, Corbisier
de Meaultsart C, Mathe A, Avery-Kiejda KA, Roberts CT, Pipkin FB,
Marques FZ, et al: Regulation of the human placental (pro)renin
receptor-prorenin-angiotensin system by microRNAs. Mol Hum Reprod.
24:453–464. 2018.PubMed/NCBI
|