Introduction
Periodontal diseases are a group of chronic
inflammatory diseases induced by Porphyromonus gingivalis
(P. gingivalis) that affect the tooth-supporting tissues.
P. gingivalis secrete a variety of toxic substances such as
lipopolysaccharide (LPS), gingival hormone and protease (1). LPS (also referred to as endotoxin) is
one of the important virulence factors that is known to mediate
inflammation and apoptosis (2).
Several cell types are involved in the development of
periodontitis. Studies have shown that periodontitis is associated
with altered expression of inflammatory mediators in gingival
fibroblasts and periodontal membrane cells (3,4).
However, few studies have investigated LPS-induced apoptosis of
vascular endothelial cells. Furthermore, periodontitis has been
shown to be associated with systemic cardiovascular disease
(5–7), which suggests its close association
with vascular endothelial cells.
Apoptosis refers to the self-regulated and orderly
death of cells controlled by genes in order to maintain the
stability of internal environment (8). The apoptosis of endothelial cells is
a key event that may impair the integrity of the vessel wall and
lead to the formation of atherosclerotic plaques (9). There are two typical apoptotic
pathways: The mitochondrial pathway and the death receptor pathway
(10,11). The death receptor pathway plays an
important role in cell apoptosis (12). A series of caspases proteins have
been shown to be activated by the death induced signal complex [a
form of death ligand that binds to the corresponding death receptor
on the cell surface (13)], which
in turn induces apoptosis. Throughout this process, caspase-3 acts
as an apoptotic executor (14).
The mitochondrial pathway is also referred to as the endogenous
apoptosis pathway (15). A recent
study revealed that apoptosis can be induced by multiple
pro-apoptotic stimuli affecting the mitochondria; these induce the
release of cytochrome c and activation of caspase enzymes,
which ultimately leads to cell apoptosis (16). Moreover, migration of Bax (a
pro-apoptotic member of the Bcl-2 family) from cytoplasm to
mitochondria induces activation of the caspase cascade system,
leading to apoptosis (15). In
contrast, the anti-apoptotic member Bcl-2 binds to the mitochondria
and inhibits death signals (17).
Collectively, these findings indicate an integral role of
caspase-3, Bcl-2-associated X (Bax) and Bcl-2 in modulating
apoptosis.
Mitogen-activated protein kinases (MAPKs; including
p38, JNK, ERKs) are serine/threonine protein kinases that serve a
critical role in response to extracellular stimulation (18). The degree of MAPKs phosphorylation
is a key determinant of cell fate: Death or survival (19,20).
A review of previous studies identified that activation of p38 MAPK
pathway is also related to apoptosis (21).
N-acetyl cysteine (NAC), a precursor for glutathione
biosynthesis, can scavenge oxygen free radicals, control
inflammatory reactions and inhibit apoptosis (22). NAC has been used as an antioxidant
in neurodegenerative diseases, chronic lung diseases,
cardiovascular diseases and other oxidative diseases (23–25).
Apart from the antioxidant properties, NAC has been shown to induce
hemodynamic improvements in a rabbit model of acute pulmonary
thromboembolism; the effect was mediated via the p38MAPK pathway
(26). The role of NAC in
preventing endothelial apoptosis and its beneficial effect on cell
survival by acting through the p38MAPK signaling pathway has evoked
considerable interest in recent years. Moreover, in the vascular
endothelial cells, nitic oxide (NO) is involved in modulating the
vascular tone, vascular proliferation, vascular apoptosis and
platelet aggregation (27).
Endothelial NO synthase (eNOS) is a critical enzyme for the
synthesis of vasoprotective molecule NO in the endothelial cells
(27). The main features of
endothelial dysfunction are loss of NO production and impaired eNOS
activity (28). Therefore, it was
hypothesized that NAC may also affect the function of vascular
endothelial cells by regulating the production of NO derived from
endothelial NO synthase.
No study, to the best of our knowledge, has
systematically investigated the effect of NAC on LPS-mediated
apoptosis in vitro by using the human umbilical vein
endothelial (HUVECs) culture system. The purpose of the present
study was to establish cell apoptosis model by LPS stimulation of
HUVECs and to evaluate the effect of NAC in LPS-induced apoptosis
and NO production in HUVECs. In addition, we investigated the
underlying molecular mechanisms.
Materials and methods
Materials
HUVECs were purchased from Otwo Biotech (Shenzhen)
Inc. LPS and NAC were purchased from Sigma-Aldrich (Merck KGaA).
SB203580 was purchased from Selleckchem. Fetal bovine serum (FBS)
was purchased from Gibco (Thermo Fisher Scientific, Inc.).
Dulbecco's Modified Eagle's Medium (DMEM) was purchased from
HyClone (GE Healthcare Life Sciences). Penicillin (100 U/ml) and
streptomycin (100 µg/ml) were purchased from Beyotime Institute of
Biotechnology. The Cell Counting Kit-8 (CCK-8) was purchased from
Dojindo Molecular Technologies, Inc. An NO assay kit (cat. no.
A013-1) was purchased from Nanjing Jiancheng Bioengineering
Institute. Antibodies against caspase-3, Bcl-2, Bax, phosphorylated
(p-)-p38, total (t)-p38, p-eNOS and t-eNOS were obtained from Cell
Signaling Technology, Inc. A fluorescein isothiocyanate (FITC)
Annexin V apoptosis detection kit was purchased from BD Pharmingen
(BD Biosciences). All other reagents were of ultrapure grade.
Cell culture
HUVECs were cultured in high-glucose DMEM containing
10% (v/v) FBS and 1% (v/v) penicillin-streptomycin solution at 37°C
in a 5% CO2 atmosphere in accordance with the
manufacturer's recommendation. To select the optimal concentration
of NAC for cell viability, HUVECs were incubated with high-glucose
DMEM containing 10% FBS at a density of
1×105/cm2 at 37°C in 5% CO2
humidified air. After the cells had adhered to the wall for 24 h at
37°C, different concentrations of NAC (0.25, 0.5, 1, 2.5 or 5 mM)
were used to stimulate the cells. For the induction of apoptosis
using LPS, cells were stimulated with LPS (1 µg/ml) for 24 h at
37°C.
Six experimental groups were examined, as presented
in Table I: i) Control group; ii)
LPS (1 µg/ml) group; iii) NAC (1 mM) group; iv) SB203580 (10 µM)
group; v) NAC (1 mM) + LPS (1 µg/ml) group; and vi) SB203580 (10
µM) + LPS (1 µg/ml) group. In the control group, cells were
cultured for 24 h in DMEM containing 10% FBS and 1%
penicillin-streptomycin solution. The LPS, NAC and SB203580 groups
were treated for 24 h with LPS, NAC and SB203580, respectively. The
NAC + LPS and SB203580 + LPS groups were treated with NAC and
SB203580, respectively, for 1 h prior to LPS exposure. Cells were
then incubated with LPS for 24 h. All treatments were preformed at
37°C with 5% CO2.
 | Table I.Experimental groups employed in the
present study. |
Table I.
Experimental groups employed in the
present study.
| Groups | Control | LPS | NAC | SB203580 | NAC+LPS | SB203580 + LPS |
|---|
| NAC | − | − | + | − | + | − |
| LPS | − | + | − | − | + | + |
| SB203580 | − | − | − | + | − | + |
Cell viability assay
A CCK-8 assay was performed to assess cell
viability. The experiments were divided into three groups: Blank,
control and experimental groups (NAC and NAC + LPS groups). First,
different concentrations of NAC (0.25, 0.5, 1, 2.5, 5 mM) were
applied in the NAC group, respectively; after 1 h, 1 µg/ml LPS was
added to the NAC + LPS group. HUVECs were seeded in a 96-well plate
at a density of 1×105/cm2; the cells adhered
to the plate for 24 h in 5% CO2 at 37°C. Subsequently,
the DMEM medium was removed and CCK-8 solution (10 µl CCK-8 + 100
µl DMEM) added to each well. Then, incubation was continued for 2 h
in a 5% CO2 atmosphere at 37°C. The absorbance at 450 nm
was detected by microplate reader (Synergy H4, BioTek Instruments,
Inc.). All experiments were conducted in triplicate.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
mRNA expressions of target genes (caspase-3, Bax and
Bcl-2) were quantified using RT-qPCR. For RNA extraction, cells
were plated in a six-well plate at a density of
1×105/cm2 in DMEM containing 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and 1% penicillin-streptomycin
solution (Beyotime Institute of Biotechnology) for 24 h at 37°C.
Subsequently, cells were treated according to the six experimental
groups. The total RNA was extracted from cultured HUVECs with
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Electrophoresis was performed to examine the integrity of
the RNA extracted. First, cDNA was synthesized from total RNA in
each sample using the PrimeScript RT reagent kit (Takara Bio, Inc.)
according to the manufacturer's instructions incubating the samples
for 1 h at 42°C, for 5 min at 70°C and for 1 min at 4°C. Total RNA
(500 ng) was reverse transcribed to cDNA with 5X Reaction Buffer (4
µl), Oligo DT18 primer (1 µl), dNTP Mix (2 µl), RiboLock RNase
Inhibitor (1 µl), RevertAid M-MuL V RT (1 µl) and ddH2O
to supplement to a total volume of 20 µl. SYBR Green Mix (Toyobo
Life Science) was used for RT-qPCR of cDNA samples following the
manufacturer's manual. The amplification assay was performed using
an ABI Prism 7300 sequence detection PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) The reaction conditions
included 95°C for 5 min, followed by 40 cycles of two-step PCR
(95°C for 10 sec and 60°C for 30 sec) and a final extension at 75°C
for 10 min. The primer sequences employed for qPCR are listed in
Table II. RT-qPCR for each sample
was performed in triplicate. Glyceraldehyde-phosphate dehydrogenase
(GAPDH) was used as an internal reference control and the relative
expressions of mRNA were quantified using the 2−∆∆Cq
method (29).
| Table II.Primer Sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table II.
Primer Sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Control | Primer sequence
(5′-3′) |
|---|
| Caspase-3 | Reverse |
CTGAATGTTTCCCTGAGGTTTG |
|
| Forward |
CCAAAGATCATACATGGAAGCG |
| Bax | Reverse |
CAGTTTGCTGGCAAAGTAGAAA |
|
| Forward |
CGAACTGGACAGTAACATGGAG |
| Bcl-2 | Reverse |
GAACTCAAAGAAGGCCACAATC |
|
| Forward |
GACTTCGCCGAGATGTCCAG |
| GAPDH | Reverse |
AGGCGCCCAATACGACCAA |
|
| Forward |
CCACTAGGCGCTCACTGTTC |
Protein isolation and western blot
analysis
The expression levels of target proteins (caspase-3,
Bax, Bcl-2, p-p38 MAPK/t-p38MAPK and p-eNOS/t-eNOS) were determined
by western blotting. The cells were harvested and the total
proteins were extracted using the radioactive immunoprecipitation
assay buffer (Thermo Scientific, Inc.). Total protein was boiled
for 5 min and then cooled for another 5 min. A bicinchoninic acid
protein concentration assay kit (Pierce; Thermo Fisher Scientific,
Inc.) was used for protein quantification and the same amount of
protein (20 µg) was separated from each sample by 12% SDS-PAGE
(Beyotime Institute of Biotechnology). Following electrophoresis,
the proteins were transferred to polyvinylidene fluoride membranes.
These membranes were blocked with 5% non-fat dried milk in PBST
(0.1% Tween 20 in PBS) for 2 h at room temperature and then
incubated overnight at 4°C with primary antibodies against
caspase-3 (1:1,000; rabbit, cat. no. PAB33236; Bioswamp; Wuhan
Beinglay Biological Technology Co.), Bax (1:1,000; rabbit, cat. no.
PAB30861; Bioswamp; Wuhan Beinglay Biological Technology Co.),
Bcl-2 (1:1,000; rabbit, cat. no. PAB33482; Bioswamp; Wuhan Beinglay
Biological Technology Co.), p38MAPK (1:2,000; rabbit, cat. no.
14064-1-AP; ProteinTech Group, Inc.)/p-p38 MAPK (1:1,000; rabbit,
cat. no. ab178867; Abcam), eNOS (1:1,000; rabbit, cat. no.
PAB32306; Bioswamp; Wuhan Beinglay Biological Technology
Co.)/p-eNOS (1:1,000; rabbit, cat. no. PAB36348-P; Bioswamp; Wuhan
Beinglay Biological Technology Co.) and β-actin (1:1,000; rabbit,
cat. no. PAB36265; Bioswamp; Wuhan Beinglay Biological Technology
Co.), followed by washing with PBST three times (5 min each time).
Subsequently, the membrane was incubated for 1 h at room
temperature with horseradish peroxidase-conjugated goat anti-rabbit
IgG secondary antibody (1:20,000; goat. no. PAB160011; Bioswamp;
Wuhan Beinglay Biological Technology Co.). After washing with TBST
for three times (5 min each time), the target bands were detected
using the Clarity Western Enhanced Chemiluminescence kit (Analytik
Jena AG) and the band intensities were quantified using ImageJ
software. (version 1.44; National Institutes of Health). The
expression levels of target proteins were determined relative to
that of β-actin.
Cell apoptosis assay
The apoptosis rate of cells was evaluated using the
Annexin V-FITC/PI apoptosis detection kit. The cells were collected
and centrifuged for 5 min at 1,000 × g at 37°C and rinsed twice
with cold PBS. Subsequently, the cells at a density of
1.0×107 cells/ml were resuspended in 1X Binding Buffer.
Then, 100 µl of cell suspension was transferred into a 5 ml test
tube and 5 µl Annexin V/FITC and 5 µl PI solution were added. The
test tube was gently vortexed to mix the cells with the reagent and
incubated at room temperature for 15 min in the dark. Subsequently,
400 µl of 1X Binding Buffer was added to each test tube prior to
analysis using a flow cytometer (BD FACSCanto II; BD Biosciences)
and FACSDiva software (version 6.1; BD Biosciences) to detect the
apoptotic rate. Flow cytometry results suggested that the advanced
apoptotic cells were in the upper right quadrant, and the early
apoptotic cells were in the lower right quadrant. The apoptotic
rate was calculated as the sum of early and advanced apoptotic
cells.
Measurement of NO
The cell supernatants were collected and NO
concentration was detected with the NO detection kit following the
manufacturer's recommendation. The absorbance was measured at 540
nm with microplate reader (ELx800, General Electric Company). The
concentration of NO in each sample was calculated by comparing the
absorption with a standard curve line.
Statistical analysis
All data are presented as mean ± standard deviation
values from three experimental replicates. One-way analysis of
variance followed by a Student-Newman-Keuls post hoc test, or a
Student's-test was used to analyze the experimental data using SPSS
17.0 software (SPSS, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of NAC on viability of
HUVECs
The results of NAC cell toxicity assay are shown in
Fig. 1A. The results showed no
significant changes in the viability of HUVECs after incubation
with NAC alone at various concentrations (0.25, 0.5, 1, 2.5, 5 mM)
for 24 h. The effects of NAC on HUVEC viability under LPS (1 µg/ml)
treatment are shown in Fig. 1B.
The data revealed that LPS treatment (1 µg/ml) significantly
decreased the cell viability compared with the control group.
Additionally, it was found that NAC pretreatment (0.25, 0.50, 1,
2.5, 5 mM) for 1 h significantly prevented the LPS-induced decline
in cell viability compared with the LPS group. The cell viability
remained essentially unchanged over a specific concentration range
of NAC (1–5 mM). Therefore, 1 mM NAC was used in the subsequent
experiments.
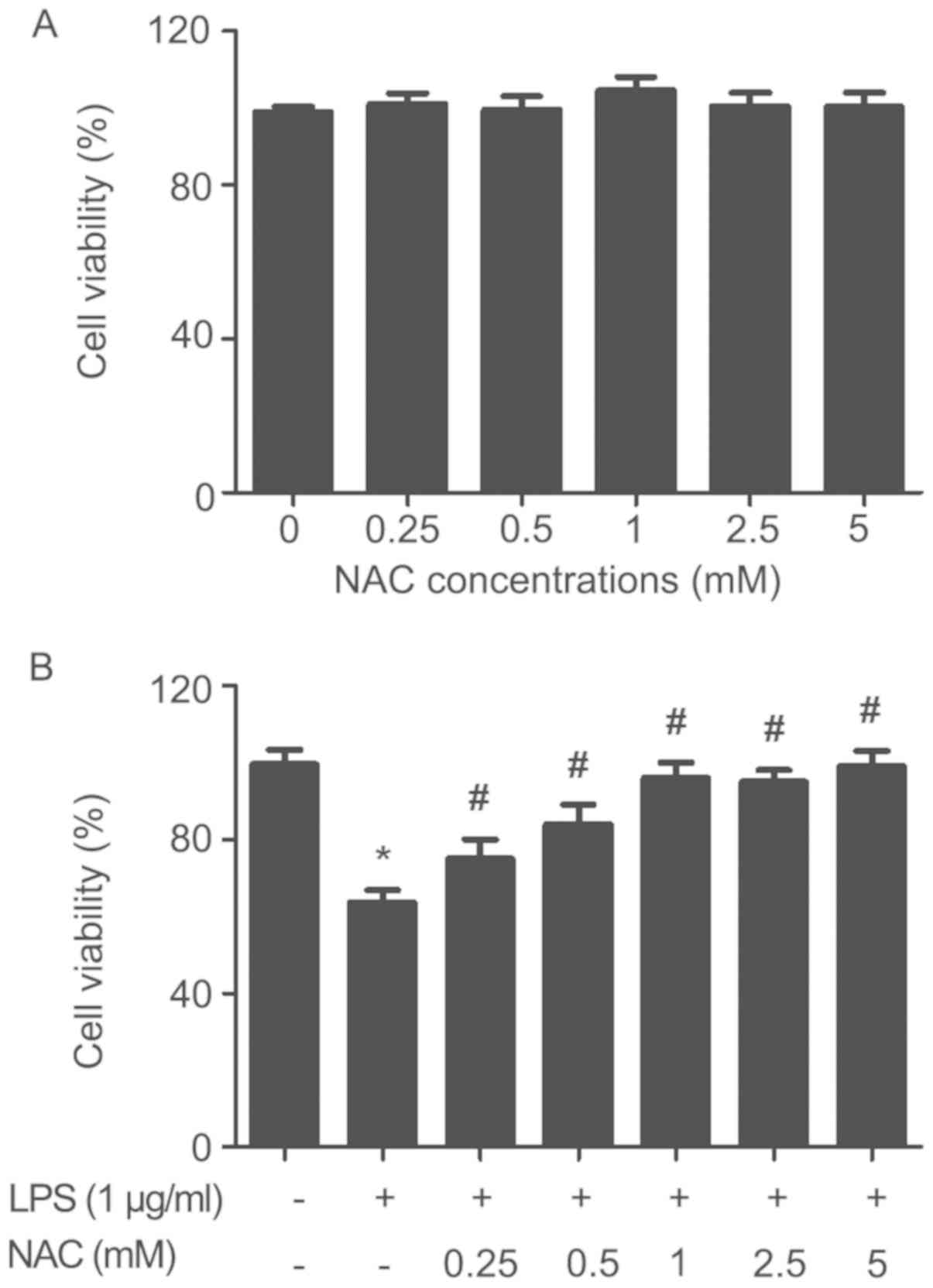 | Figure 1.Effect of NAC on viability of HUVECs.
(A) Effects of NAC (0.25, 0.5, 1, 2.5, 5 mM) on the viability of
HUVECs at 24 h. (B) HUVECs were pretreated with NAC (0.25, 0.5, 1,
2.5, 5 mM) for 1 h, followed by stimulation with LPS (1 µg/ml) for
24 h. Mean ± standard deviation values from three independent
experiments are presented. *P<0.05 vs. untreated,
#P<0.05 vs. LPS. NAC, N-acetyl cysteine; HUVECs,
human umbilical vein endothelial cells; LPS,
lipopolysaccharide. |
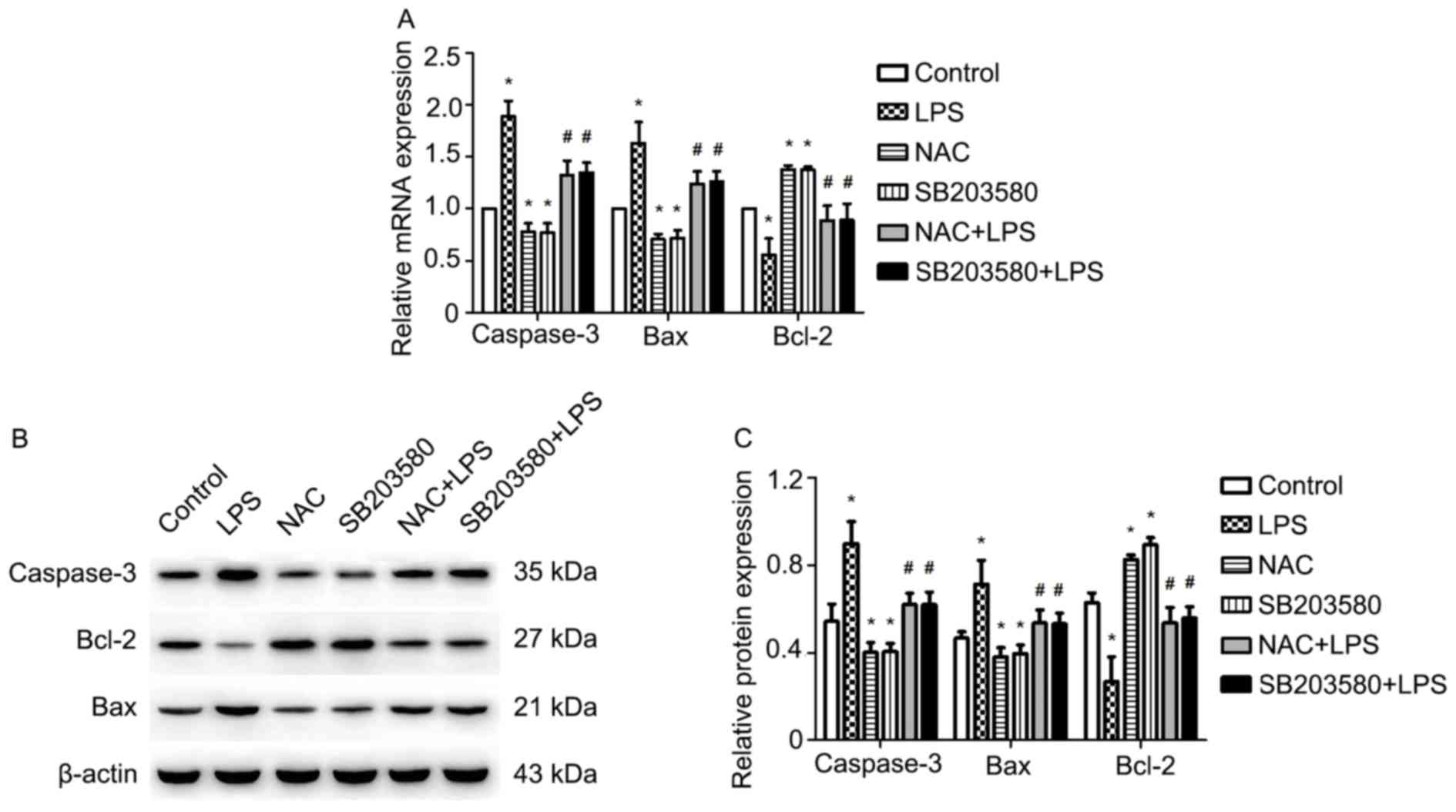
NAC attenuates LPS-induced increases
in caspase-3 and Bax, and decreases in Bcl-2 expression in
HUVECs
The relative expression levels of caspase-3, Bax and
Bcl-2 are shown in Fig. 2A and C.
The RT-qPCR results indicated that LPS significantly induced the
expression of caspase-3 and Bax, but downregulated that of Bcl-2
compared with the control group; however, the effects of LPS were
significantly attenuated by pretreatment with NAC or SB203580. In
addition, pretreatment of HUVECs with NAC or SB203580 significantly
inhibited the LPS-induced upregulation of caspase-3 and Bax, and
the downregulation of Bcl-2 compared with LPS treatment alone
(Fig. 2A). Western blotting
analysis of protein expression levels were consistent with the mRNA
results (Fig. 2B and C).
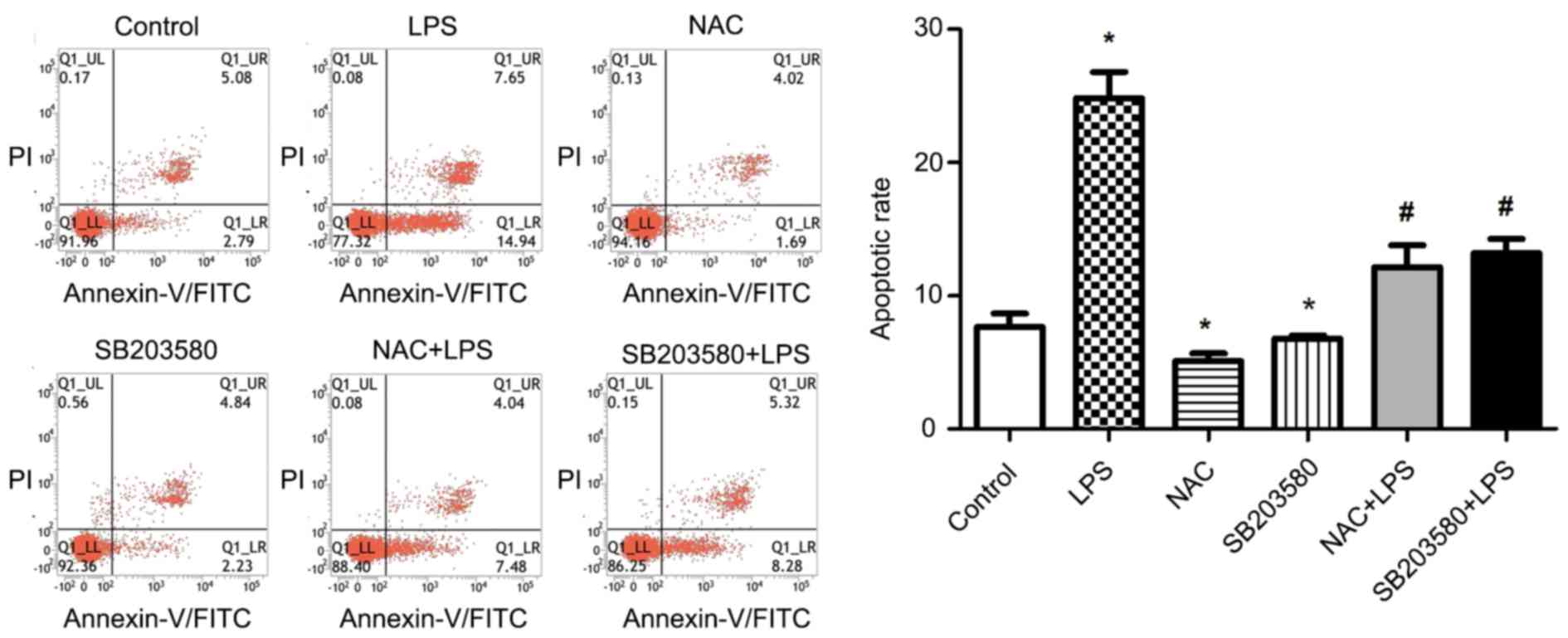
NAC inhibits LPS-induced apoptosis of
HUVECs
It was further examined whether NAC reduces the rate
of apoptosis using flow cytometry. The results showed that
treatment of HUVECs with LPS alone significantly increased the rate
of apoptosis compared with control group. However, pretreatment of
cells with NAC for 1 h significantly decreased the apoptosis rate
compared with LPS treatment alone. Similar results were observed
after pretreatment with SB203580. The results suggest that NAC
plays an important anti-apoptotic role (Fig. 3).
NAC inhibits LPS-induced p38MAPK
phosphorylation in HUVECs
To further explore the mechanism of the
anti-apoptotic effect of NAC, the protein expression levels of
p-p38MAPK/t-p38MAPK were investigated (Fig. 4A and B). LPS treatment
significantly increased the phosphorylation of p38MAPK, compared
with the control, without altering t-p38MAPK expression in HUVECs.
However, this effect of LPS was abolished by pretreatment with NAC
or the p38MAPK inhibitor (SB203580). In brief, pretreatment with
NAC reduced the expression of p-p38MAPK compared with cells exposed
to LPS alone.
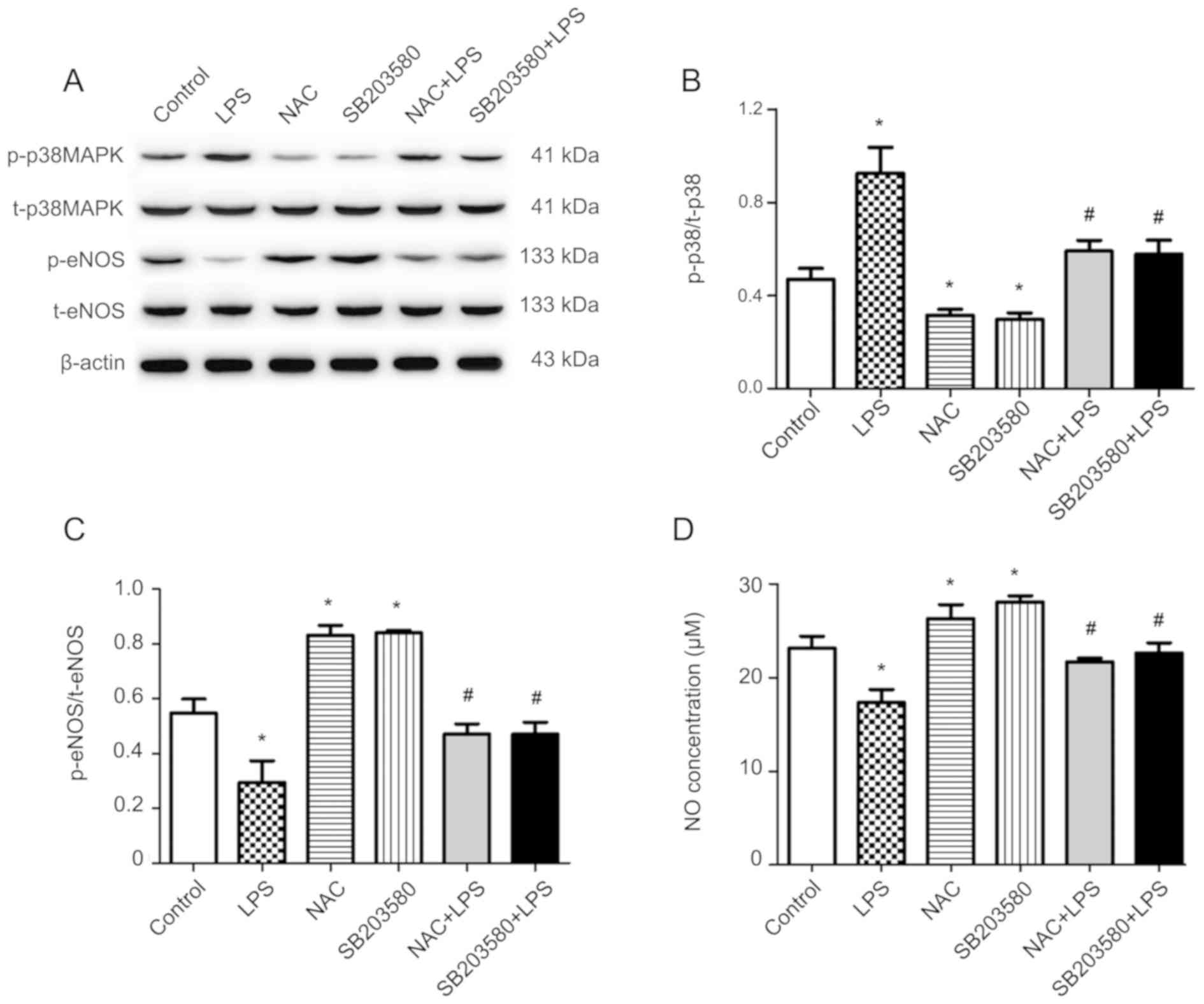 | Figure 4.Effects of NAC pretreatment on the
expression of p38MAPK, eNOS and NO production in HUVECs stimulated
with LPS. Cells were pretreated with 1 mM NAC or 10 µM SB203580 for
1 h, then co-treated with 1 µg/ml LPS for 24 h. (A) Levels of
p-p38MAPK, t-p38MAPK, p-eNOS and t-eNOS protein were determined by
western blot analysis. (B) Densitometry analysis of band intensity
was expressed as the ratio of p-p38/t-p38. (C) Densitometry
analysis of band intensity was expressed as the ratio of
p-eNOS/t-eNOS. (D) NO concentration was determined via an NO assay
kit. Mean ± standard deviation values from three independent
experiments are presented. *P<0.05 vs. untreated;
#P<0.05 vs. LPS. NAC, N-acetyl cysteine; MAPK,
mitogen-activated protein kinase; HUVECs, human umbilical vein
endothelial cells; LPS, lipopolysaccharide; eNOS, endothelial
nitric oxide synthase; NO, nitric oxide; p-, phosphorylated; t-,
total. |
NAC alleviates LPS-induced inhibition
of eNOS phosphorylation and NO production in HUVECs
The protein expression levels of p-eNOS/t-eNOS are
shown in Fig. 4A and C. LPS
treatment significantly inhibited the phosphorylation of eNOS
without altering t-eNOS expression, compared with the control.
However, NAC pretreatment significantly antagonized the decrease in
LPS-induced eNOS phosphorylation; similar results were obtained
after pretreatment with SB203580. The NO concentration in the cell
supernatant in various groups is presented in Fig. 4D. HUVECs treated with LPS exhibited
significant decreases in the synthesis of NO compared with the
untreated group. Correspondingly, pretreatment of HUVECs with NAC
significantly attenuated the LPS-induced reductions in NO
synthesis, which was consistent with the results observed after
pretreatment with SB203580.
Discussion
The current study examined the effect of NAC on
LPS-induced apoptosis of HUVECs. Additionally, we investigated the
potential molecular mechanisms that mediate the effects of NAC. LPS
significantly promoted the synthesis of caspase-3 and Bax, while
suppressing that of Bcl-2 in HUVECs. However, NAC pretreatment
preempted these effects, which suggests that NAC has an
anti-apoptotic effect on endothelial cells. This result is
consistent with the results of flow cytometry. Moreover, it was
also observed that NAC inhibited LPS-induced upregulation of
p-p38MAPK signaling protein in HUVECs, an effect that was similar
to that of SB203580. In addition, it was also found that NAC
pretreatment significantly restored the phosphorylation of eNOS and
production of NO, which suggests that the anti-apoptotic effect of
NAC on HUVECs may be mediated via its effect on NO synthesis.
Periodontitis is closely related to cardiovascular
diseases (30). Endothelial
dysfunction and apoptosis play a vital role in the progression of
atherosclerosis (31). Moreover,
Bcl-2 and Caspase protein families serve an important role in the
induction of apoptosis (32).
Caspase-3 is considered as the main caspase-executor in apoptosis
(14) and the most reliable
determinant of apoptosis (33).
The Bcl-2 protein family comprises of both anti-apoptotic (such as
Bcl-2) and pro-apoptotic (such as Bax) members. Decreased
expression of Bcl-2 and increased expression of Bax has been shown
to stimulate the release of cytochrome c from mitochondria,
which can activate caspase-9 (34). Caspase-9 catalyzes the activation
of caspase-3, which eventually induces apoptosis (35,36).
Previous studies have shown that overexpression of tumor necrosis
factor α can increase the expression of Bax and promote
cardiomyocyte apoptosis, eventually contributing to an imbalance
between Bcl-2 and Bax (37). It
has been shown that the imbalance between Bcl-2 and Bax determines
whether the cells undergo apoptosis (38). In the present study, western blot
analysis and RT-qPCR demonstrated that LPS negatively regulated the
expression of Bcl-2 at the protein and the mRNA levels. In
contrast, LPS appeared to have regulated the expression of
caspase-3 and Bax at the protein and mRNA levels. However, these
effects were suppressed by pretreatment with NAC or SB203580.
Periodontal inflammation was shown to be associated with the
increased concentration of important atherosclerosis biomarkers in
the blood; furthermore, the concentration of these markers (such as
CRP) was shown to decrease after periodontal treatment (39). The results of the present study
suggested that NAC may have an anti-apoptotic effect, which might
be mediated via the p38MAPK signal pathway. This conclusion is
consistent with our results of flow cytometric analysis.
MAPKs (including ERK1/2, JNK and p38) play a key
role in determining the cell fate, such as proliferation,
differentiation, survival and apoptosis (40). Activation of p38 and JNK is
considered as a pro-apoptotic signal, while activation of ERK1/2 is
considered a pro-survival signal (17). The p38MAPK signaling pathway is
involved in the functional response of macrophages and neutrophils,
including adhesion and apoptosis (41,42),
which is implicated in a variety of cardiovascular diseases
(43). It has also been shown to
regulate a variety of cellular biological events including cell
migration, proliferation and differentiation (44–46).
Phosphorylation of p38MAPK in endothelial cells can be activated by
physiological stress, LPS, osmotic stress and ischemia reperfusion,
which can affect the function of endothelial cells and eventually
promote cell apoptosis (47).
p38MAPK not only downregulates anti-apoptotic proteins, but also
upregulates pro-apoptotic proteins (48,49).
The present study assessed whether the anti-apoptotic effect of NAC
is partly mediated via the p38MAPK signal pathway; HUVECs were
treated with LPS, NAC and SB203580 and the changes in
p-p38MAPK/t-p38MAPK examined. As expected, the expression of
p-p38MAPK in HUVECs was increased after treatment with LPS.
However, the expression of p-p38 was downregulated by pretreatment
with NAC or SB203580. The results suggested that NAC can inhibit
the phosphorylation of p38MAPK signal protein. This result is
consistent with a previous study in which NAC was shown to
attenuate hepatocyte apoptosis by inhibiting the expression of
p-p38MAPK (50); the role of
p38MAPK in vascular inflammation and endothelial cell apoptosis has
been reported (51,52). These results suggest that NAC can
attenuate LPS-induced apoptosis by inhibiting the activation of
p-p38MAPK.
NOS consists of neuronal NOS (nNOS), eNOS and
inducible NOS (53). eNOS is the
main subtype of NOS expressed in vascular endothelial cells and
accounts for most of NO production. The critical role of NO in
regulating the growth, function, apoptosis and survival of
endothelial cells is well documented (54,55).
Atherosclerosis stimulates endothelial cell injury, which impairs
eNOS bioactivity and the synthesis of NO (56). Decreased bioavailability of NO is
an important feature of vascular dysfunction (57). Decreased vascular NO
bioavailability has long been considered as a common pathogenetic
mechanism of endothelial dysfunction, leading to the occurrence of
cardiovascular risk factors such as hypertension, atherosclerosis
and diabetes (58). The regulation
of NO production by activating eNOS phosphorylation is a key
strategy for treating cardiovascular diseases (59). Oxidative free radical-mediated
apoptosis or cellular dysfunction, which is the main cause of
decreased endothelial-derived NO production and bioavailability,
can lead to impaired endothelial-dependent vascular reactivity
(60). NAC has an antioxidant
effect, can scavenge oxygen radicals and reduce apoptosis (18). However, to the best of our
knowledge, no studies have directly investigated whether the
anti-apoptotic effect of NAC in endothelial cells is mediated via
activation of eNOS phosphorylation and NO production. To determine
whether the vasculoprotective effect of NAC is mediated via
endothelium-derived NO synthesis, the phosphorylation of eNOS and
was examined and NO concentration in the cell supernatant was
assessed. The results showed that LPS treatment significantly
reduces the expression of p-eNOS protein. Pretreatment of HUVECs
with NAC or SB203580 enhanced the expression of p-eNOS protein, the
result of NO concentration analysis was consistent with the changes
in the expression levels of p-eNOS. A previous study showed that
endothelium-derived NO plays an important physiological role in
regulating vascular tension as well as endothelial cell survival
and migration (61). The current
study indicated that the protective effects of NAC on vascular
endothelial cells may be mediated via modulation of
endothelium-derived NO synthesis.
In conclusion, the findings of present study
indicated that NAC attenuated LPS-induced vein endothelial cells
apoptosis via the p38MAPK signaling pathway. The findings also
suggested that NAC may be valuable for the prevention and treatment
of periodontitis. However, other relevant mechanisms underlying the
effects of NAC require further investigation.
Acknowledgements
Not applicable.
Funding
This project was supported by a grant from the
Office of Science & Technology and Intellectual Property of
Luzhou [grant no. 2010-s-16(1/3)], the Luzhou-school Union (grant
no. 2016LZXNYD-J21) and the Southwest Medical University, Luzhou,
China (grant no. 23).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TX and LG made substantial contributions to the
design of the experiments. TX, ZZ, RZ and JH performed the
experiments and collected the data. TX, ZZ and JH analyzed and
interpreted the data. TX produced the manuscript. TX, ZZ, RZ, JH
and LG revised the manuscript. All authors read and approved the
final manuscript to be published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hajishengallis G: Periodontitis: From
microbial immune subversion to systemic inflammation. Nat Rev
Immunol. 15:30–44. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bi C, Jiang Y, Fu T, Hao Y, Zhu X and Lu
Y: Naringin inhibits lipopolysaccharide-induced damage in human
umbilical vein endothelial cells via attenuation of inflammation,
apoptosis and MAPK pathways. Cytotechnology. 68:1473–1487. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang F, Guan M, Wei L and Yan H: IL-18
promotes the secretion of matrix metalloproteinases in human
periodontal ligament fibroblasts by activating NF-κB signaling. Mol
Med Rep. 19:703–710. 2019.PubMed/NCBI
|
|
4
|
Zhan D, Guo L and Zheng L: Inhibition of
the receptor for advanced glycation promotes proliferation and
repair of human periodontal ligament fibroblasts in response to
high glucose via the NF-κB signaling pathway. Arch Oral Biol.
87:86–93. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dietrich T, Jimenez M, Krall Kaye EA,
Vokonas PS and Garcia RI: Age-dependent associations between
chronic periodontitis/edentulism and risk of coronary heart
disease. Circulation. 117:1668–1674. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dietrich T, Sharma P, Walter C, Weston P
and Beck J: The epidemiological evidence behind the association
between periodontitis and incident atherosclerotic cardiovascular
disease. J Periodontol 40 (4 Suppl). S70–S84. 2013.
|
|
7
|
Tonetti MS and Van Dyke TE; Working group
1 of the joint EFP/AAP workshop, : Periodontitis and
atherosclerotic cardiovascular disease: Consensus report of the
Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J
Clin Periodontol. 40 (Suppl 14):S24–S29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2010. View Article : Google Scholar
|
|
9
|
Rotllan N, Wanschel AC, Fernández-Hernando
A, Salerno AG, Offermanns S, Sessa WC and Fernández-Hernando C:
Genetic evidence supports a major role for Akt1 in VSMCs during
atherogenesis. Circ Res. 116:1744–1752. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xue M, Ge Y, Yu C, Zheng Z, He X and Zhao
J: Apoptosis is induced by docosahexaenoic acid in breast cancer
cells via death receptor and mitochondria-mediated pathways. Mol
Med Rep. 16:9782017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mughal MJ, Xi P, Yi Z and Jing F:
Aflatoxin B1 invokes apoptosis via death receptor pathway in
hepatocytes. Oncotarget. 8:8239–8249. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wnęk A, Andrzejewska E, Kobos J, Taran K
and Przewratil P: Molecular and immunohistochemical expression of
apoptotic proteins Bax, Bcl-2 and Caspase 3 in infantile hemangioma
tissues as an effect of propranolol treatment. Immunol Lett.
185:27–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang X, Chen Y, Cai G, Li X and Wang D:
Carnosic acid induces apoptosis of hepatocellular carcinoma cells
via ROS-mediated mitochondrial pathway. Chem Biol Interact.
277:91–100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu Y, Zhang P, Yang H, Ge Y and Xin Y:
Effects of demethoxycurcumin on the viability and apoptosis of skin
cancer cells. Mol Med Rep. 16:539–546. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang D and Bi Z: Bufalin inhibited the
growth of human osteosarcoma MG-63 cells via down-regulation of
Bcl-2/Bax and triggering of the mitochondrial pathway. Tumor Biol.
35:4885–4890. 2014. View Article : Google Scholar
|
|
18
|
Ono K and Han J: The p38 signal
transduction pathway: Activation and function. Cell Signal.
12:1–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dickinson RJ and Keyse SM: Diverse
physiological functions for dual-specificity MAP kinase
phosphatases. J Cell Sci. 119:4607–4615. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rose BA, Force T and Wang Y:
Mitogen-activated protein kinase signaling in the heart: Angels
versus demons in a heart-breaking tale. Physiol Rev. 90:1507–1546.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo C, Wang SL, Xu ST, Wang JG and Song
GH: SP600125 reduces lipopolysaccharide-induced apoptosis and
restores the early-stage differentiation of osteoblasts inhibited
by LPS through the MAPK pathway in MC3T3-E1 cells. Int J Mol Med.
35:1427–1434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Millea PJ: N-acetylcysteine: Multiple
clinical applications. Am Fam Physician. 80:265–269.
2009.PubMed/NCBI
|
|
24
|
Nigwekar SU and Kandula P:
N-acetylcysteine in cardiovascular-surgery-associated renal
failure: A meta-analysis. Ann Thorac Surg. 87:139–47. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zafarullah M, Li WQ, Sylvester J and Ahmad
M: Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life
Sci. 60:6–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang R, Wang Y, Pan L and Tian H:
N-Acetylcysteine potentiates the haemodynamic-improving effect of
sildenafil in a rabbit model of acute pulmonary thromboembolism via
the p38 MAPK pathway. Clin Exp Pharmacol Physiol. 46:163–172. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bian K, Doursout MF and Murad F: Vascular
system: Role of nitric oxide in cardiovascular diseases. J Clin
Hypertens (Greenwich). 10:304–310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Taguchi K, Hida M, Hasegawa M, Matsumoto T
and Kobayashi T: Dietary polyphenol morin rescues endothelial
dysfunction in a diabetic mouse model by activating the Akt/eNOS
pathway. Mol Nutr Food Res. 60:580–588. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta DeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chistiakov DA, Orekhov AN and Bobryshev
YV: Links between atherosclerotic and periodontal disease. Exp Mol
Pathol. 100:220–235. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang K, Zhang H, Luo Y, Zhang J, Wang M,
Liao P, Cao L, Guo P, Sun G and Sun X: Gypenoside XVII prevents
atherosclerosis by attenuating endothelial apoptosis and oxidative
stress: Insight into the ERα-Mediated PI3K/Akt pathway. Int J Mol
Sci. 18(pii): E772017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kaufmann SH, Lee SH, Meng XW, Loegering
DA, Kottke TJ, Henzing AJ, Ruchaud S, Samejima K and Earnshaw WC:
Apoptosis-associated caspase activation assays. Methods.
44:262–272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Végran F, Boidot R, Solary E and
Lizard-Nacol S: A short caspase-3 isoform inhibits
chemotherapy-induced apoptosis by blocking apoptosome assembly.
PLoS One. 6:e290582011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cain K, Bratton SB and Cohen GM: The
Apaf-1 apoptosome: A large caspase-activating complex. Biochimie.
84:203–214. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rodriguez J and Lazebnik Y: Caspase-9 and
APAF-1 form an active holoenzyme. Genes Dev. 13:3179–3184. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu L, Zhao Y, Xu S, Jin C, Wang M and Fu
G: Leptin confers protection against TNF-α-induced apoptosis in rat
cardiomyocytes. Biochem Biophys Res Commun. 455:126–132. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Alladi PA, Roy T, Singh N and Wadhwa S:
Prenatal auditory enrichment with species-specific calls and sitar
music modulates expression of Bcl-2 and Bax to alter programmed
cell death in developing chick auditory nuclei. Int J Dev Neurosci.
23:363–373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Paraskevas S, Huizinga JD and Loos BG: A
systematic review and meta-analyses on C-reactive protein in
relation to periodontitis. J Clin Periodontol. 35:277–290. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Osaki LH and Patrícia G: MAPKs and signal
transduction in the control of gastrointestinal epithelial cell
proliferation and differentiation. Int J Mol Sci. 14:10143–10161.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bruns B, Hönle T, Kellermann P, Ayala A
and Perl M: Divergent effects of neutrophils on fas-induced
pulmonary inflammation, apoptosis, and lung damage. Shock.
47:225–235. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Qin Y, Zhou ZW, Pan ST, He ZX, Zhang X,
Qiu JX, Duan W, Yang T and Zhou SF: Graphene quantum dots induce
apoptosis, autophagy, and inflammatory response via p38
mitogen-activated protein kinase and nuclear factor-κB mediated
signaling pathways in activated THP-1 macrophages. Toxicology.
327:62–76. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wan Q, Liu Z, Yang Y and Cui X:
Suppressive effects of berberine on atherosclerosis via
downregulating visfatin expression and attenuating visfatin-induced
endothelial dysfunction. Int J Mol Med. 41:1939–1948.
2018.PubMed/NCBI
|
|
44
|
Zhang WB, Liu YQ, Zhang X, Lin L and Yin
SL: The role of β-adrenergic receptors and p38MAPK signaling
pathways in physiological processes of cardiosphere-derived cells.
J Cell Biochem. 119:1204–1214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xu Y, Xiao H, Luo H, Chen Y, Zhang Y, Tao
L, Jiang Y, Chen Y and Shen X: Inhibitory effects of oxymatrine on
TGF-β1-induced proliferation and abnormal differentiation in rat
cardiac fibroblasts via the p38MAPK and ERK1/2 signaling pathways.
Mol Med Rep. 16:5354–5362. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li X, Cheng XW, Hu L, Wu H, Guo-Pin g, Hao
CN, Jiang H, Zhu E, Huang Z, Inoue A, et al: Cathepsin S activity
controls ischemia-induced neovascularization in mice. Int J
Cardiol. 183:198–208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Palmieri D, Aliakbarian B, Casazza AA,
Ferrari N, Spinella G, Pane B, Cafueri G, Perego P and Palombo D:
Effects of polyphenol extract from olive pomace on anoxia-induced
endothelial dysfunction. Microvasc Res. 83:281–289. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu T, Zhou Y, Liu YC, Wang JY, Su Q, Tang
ZL and Li L: Coronary microembolization induces cardiomyocyte
apoptosis through the LOX-1-dependent endoplasmic reticulum stress
pathway involving JNK/P38 MAPK. Can J Cardiol. 31:1272–1281. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yokota T and Wang Y: p38 MAP kinases in
the heart. Gene. 575:369–376. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Du X, Shi Z, Peng Z, Zhao C, Zhang Y, Wang
Z and Li X, Liu G and Li X: Acetoacetate induces hepatocytes
apoptosis by the ROS-mediated MAPKs pathway in ketotic cows. J Cell
Physiol. 232:3296–3308. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dan Y, Ping X, Shubin G and Huihua L:
Induction of MAPK phosphatase-1 by hypothermia inhibits
TNF-alpha-induced endothelial barrier dysfunction and apoptosis.
Cardiovasc Res. 85:520–529. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ward NC, Croft KD, Blacker D, Hankey GJ,
Barden A, Mori TA, Puddey IB and Beer CD: Cytochrome P450
metabolites of arachidonic acid are elevated in stroke patients
compared with healthy controls. Clin Sci (Lond). 121:501–507. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dudzinski DM, Igarashi J, Greif D and
Michel T: The regulation and pharmacology of endothelial nitric
oxide synthase. Annu Rev Pharmacol Toxicol. 46:235–276. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ahanchi SS, Tsihlis ND and Kibbe MR: The
role of nitric oxide in the pathophysiology of intimal hyperplasia.
J Vasc Surg. 45 (Suppl A):A64–A73. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Slomiany BL and Slomiany A: Role of
constitutive nitric oxide synthase S-nitrosylation in Helicobacter
pylori-induced gastric mucosal cell apoptosis: Effect of ghrelin.
Inflammopharmacology. 18:233–240. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Napoli C, de Nigris F, Williams-Ignarro S,
Pignalosa O, Sica V and Ignarro LJ: Nitric oxide and
atherosclerosis: An update. Nitric Oxide. 15:265–279. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Guzzoni V, Cunha TS, das Neves VJ, Briet
L, Costa R, Moura MJCS, Oliveira V, Franco MDCP, Novaes PD and
Marcondes FK: Nandrolone combined with strenuous resistance
training reduces vascular nitric oxide bioavailability and impairs
endothelium-dependent vasodilation. Steroids. 131:7–13. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shi GF, An LJ, Jiang B, Guan S and Bao YM:
Alpinia protocatechuic acid protects against oxidative damage in
vitro and reduces oxidative stress in vivo. Neurosci Lett.
403:206–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Oche B, Chen L, Ma YK, Yang Y, Li CX, Geng
X, Qiu LZ, Gao XM and Wang H: Cryptotanshinone and wogonin
up-regulate eNOS in vascular endothelial cells via ERα and
down-regulate iNOS in LPS stimulated vascular smooth muscle cells
via ERβ. Arch Pharm Res. 39:249–258. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kim YW, West XZ and Byzova TV:
Inflammation and oxidative stress in angiogenesis and vascular
disease. J Mol Med (Berl). 91:323–328. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Nagarajan S, Rajendran S, Saran U, Priya
MK, Swaminathan A, Siamwala JH, Sinha S, Veeriah V, Sonar P, Jadhav
V, et al: Nitric oxide protects endothelium from cadmium mediated
leakiness. Cell Biol Int. 37:495–506. 2013. View Article : Google Scholar : PubMed/NCBI
|