Introduction
Sepsis is an organ dysfunction caused by an
imbalance altered homeostasis in the host due to infection. Its
high morbidity and mortality, resulting in the increased use of
medical resources, is the main cause of death in pediatric
intensive care units (PICUs) (1).
Sepsis-associated encephalopathy (SAE) is a diffuse brain
dysfunction caused by sepsis, that can occur during its early
stages, even before the onset of clinical symptoms. The
pathogenesis of SAE involves multiple factors and is complex
(2,3). Many studies of both septic patients
and animal models have concluded that mitochondrial disruption
appears to be related to the progression and outcome of sepsis
(4,5). A previous study (6) demonstrated that elements of
mitochondrial disorder are conducive to SAE, including increases in
reactive oxygen species (ROS), and mitochondrial biogenesis,
generation of cellular ATP and reduction in the mitochondrial
membrane potential (MMP or ΔΨm). Astrocytes account for 90% of the
total brain cells and play an important role in the pathophysiology
of the central nervous system (CNS) (7). Therefore, we explored the changes in
mitochondrial function in astrocytes under septic conditions.
According to published data, uncoupling protein 2
(UCP2) plays a vital role under physiological and pathological
conditions (8). This main role of
UCP2 is to regulate the mitochondrial inner and outer membrane
proton gradients and establish a bridge between ATP and ROS
generation (4). Increased UCP2
expression has been observed during pathological states such as
atherosclerotic plaques (9)
cerebral ischemia (10),
experimental autoimmune encephalomyelitis (EAE) (11), and lipopolysaccharide (LPS)-induced
cardiomyocyte injury (12). Some
pathophysiological conditions can cause irritation that can lead to
increased UCP2 expression, resulting in a neuroprotective process.
UCP2 alleviates ROS generation to protect cells from oxidative
stress-induced damage. UCP2 has been implicated in intracellular
calcium regulation, neuronal plasticity, ATP production, apoptosis
and synaptic transmission (13).
Inflammatory signaling activation is an important
mechanism leading to increased ROS production and mitochondrial
dysfunction (14). In experimental
sepsis, after co-stimulation with LPS and interferon (IFN)-γ, a
potent inflammatory state and significant mitochondrial damage can
be observed (6,15). UCP2 is considered an important
neuroprotective element in many inflammatory and degenerative
states of the CNS (16). For more
than five years, our group has been dedicated to studying the role
of UCP2 and mitochondrial dysfunction in the pathophysiology of
sepsis. Previous studies by our team have demonstrated that the
total expression of UCP2 is increased in brain tissue (17), which is consistent with other
findings (11). However, the exact
function of UCP2 in astrocytes under experimental sepsis conditions
remains unknown. It is also unknown whether UCP2 has a positive
effect on astrocytes under experimental sepsis conditions by
maintaining the MMP and the production of ROS. We speculated that
UCP2 may regulate the MMP and mitochondrial function in
experimental sepsis models. In this study, synthesis of ROS and
ATP, the MMP level, and other related factors were investigated
following the knockdown of UCP2 by adenoviral transduction in
primary astrocytes that were used as an experimental sepsis
model.
Materials and methods
Chemicals and reagents
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS) and trypsin were purchased from Gibco; Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). The following
polyclonal primary antibodies were used in this study: Anti-UCP2
(1:1,000, 89326; Cell Signaling Technology, Inc.), anti-GFAP
(1:200, 80788; Cell Signaling Technology, Inc.) and anti-β-actin
(1:5,000, AP0060; Bioworld Technology, Inc.).
Cell culture
Neonatal rat cerebral cortical astrocytes were
prepared using enzymatic dissociation and culture from 1-day-old
newborn Sprague-Dawley rat brains from animals obtained from the
Laboratory Animal Center of Southern Medical University (Guangzhou,
China) according to a protocol described previously (6). This study was approval by the Nanfang
Hospital Ethics Committee (NFYY-2017-39). The animals used in the
study were provided by Southern Medical University Animal
Experiment Center [SCXK(Guangdong)2016-0041], and passed the
experimental animal quality inspection in Guangdong Province (no.
44002100020527).
Establishment of cell sepsis model. Astrocytes
isolated from neonatal rat cerebral codex were purified and
cultured, and then randomly allocated into 4 groups: i) Control
group, ii) LPS+IFN-γ+6 h group, iii) LPS+IFN-γ+12 h group and iv)
LPS+IFN-γ+24 h group. The cells were stimulated with LPS (150 ng/ml
from Escherichia coli O111:B4; Sigma-Aldrich; Merck KGaA)
and IFN-γ (200 U/ml; Thermo Fisher Scientific, Inc.)
Adenoviral transduction
The recombinant adenoviruses used to knockdown UCP2
(AD-UCP2) were designed and synthesized by Gene Chem Co. (Shanghai,
China). The cells were transfected by viruses according to the
manufacturer's instructions and stimulated with LPS (150 ng/ml from
Escherichia coli O111:B4; Sigma-Aldrich; Merck KGaA) and
IFN-γ (200 U/ml; Thermo Fisher Scientific, Inc.) at 24 h after
adenoviral transduction and for 48–72 h before the subsequent
detection.
Determination of the mRNA levels of
tumor necrosis factor (TNF)-α, interleukin (IL)-1β and UCP2
Total RNA was extracted from the cultured cells
using TRIzol reagent (Takara, Dalian, China). Reverse transcription
was conducted with a One-Step RNA-PCR Kit (Takara) according to the
manufacturer's instructions. β-actin was used as an internal
control for the real-time PCR amplification. The sequences of the
primers for real-time PCR analysis were as follows: β-actin
forward, 5′-AACACACGAGACGCTGAAGT-3′ and reverse primer,
5′-TCCAGTGAGTTCCGAAAGCC-3′; UCP2 forward, 5′-GCTGGTGGTGGTCGGAGAT-3′
and reverse primer, 5′-TGAAGTGGCAAGGGAGGT-3′; TNF-α forward,
5′-AACACACGAGACGCTGAAGT-3′ and reverse primer,
5′-TCCAGTGAGTTCCGAAAGCC-3′; IL-1β forward,
5′-CCTTGTCGAGAATGGGCAGT-3′ and reverse primer,
5′-TTCTGTCGACAATGCTGCCT-3′. Quantitative real-time PCR was
performed on a Roche Lightcycler 480 Real-Time PCR System using
SYBR Premix Ex Taq™ (Takara Shuzo Co., Kyoto, Japan). After adding
the corresponding primers and cDNA to the master mix, the PCR
thermal cycle parameters were as follows: 95°C for 10 min, 40
cycles at 60°C for 60 sec and 95°C for 15 sec, and a melting curve
from 60°C to 95°C, which was performed to ensure amplification of a
single product. The β-actin gene was used as an internal control to
normalize for differences in the amount of total RNA in each
sample. Calculations were based on the 2−ΔΔCq method
using the equation R (ratio) = 2−ΔΔCq and normalized to
β-actin expression in each sample (18).
Enzyme-linked immunosorbent assay
(ELISA) for the detection of IL-1β and TNF-α
ELISA kits (Cusabio, Wuhan, China) were used to
determine the protein levels of TNF-α and IL-1β in the cultured
supernatant. All procedures were performed according to the
manufacturer's instructions.
Transmission electron microscopy
(TEM)
After groups of astrocytes were treated, they were
collected and fixed in 2% glutaraldehyde at 4°C overnight. The
cells were dehydrated, embedded in epoxy resin (Piano) and
polymerized after fixation. The cells were then stained with uranyl
acetate and lead citrate and imaged using a Hitachi transmission
electron microscope (magnification ×20,000; H-7650; Hitachi, Tokyo,
Japan).
Measurement of intracellular ROS
levels
Intracellular ROS generation was assessed by
2,7-dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich;
Merck KGaA). Astrocytes (7.0×105 cells/well) were
treated with culture medium containing 10 µM DCFH-DA for 30 min at
37°C. After incubation, the cells were assessed using a
fluorescence microscope (magnification ×400; Olympus Corporation),
and the intensity of fluorescence was quantified by flow cytometry
(BD Biosciences).
Measurement of the MMP
The MMP was assessed in astrocytes using a JC-1 kit
(Beyotime, Jiangsu, China). After treatment, the cells
(1.5×105 cells/well) were incubated with JC-1 staining
solution at 37°C for 25 min and then washed twice with JC-1
staining buffer. MMP was detected by flow cytometry (BD
Biosciences), and cells were imaged using a confocal microscope
(magnification ×400; Olympus Corporation).
Measurement of intracellular ATP
levels
The ATP levels in astrocytes were assessed using an
ATP assay kit (Beyotime, Jiangsu, China) according to the
manufacturer's instructions. Data were normalized to the protein
concentration of the cell.
Immunofluorescence staining
Astrocytes (1.5×105 cells/well) were
fixed in 4% paraformaldehyde for 15 min and permeabilized in 0.2%
Triton X-100 for 15 min. Then, the cells were blocked with 5%
bovine serum albumin in TBS for 1 h and incubated with primary
antibodies (1:200) at 4°C overnight. Next, the cells were incubated
with the appropriate secondary antibody (1:5,000, ab150079) for 60
min at room temperature. Finally, the nuclei were stained with DAPI
for 15 min. After staining the cells were imaged using a
fluorescence microscope (magnification ×400; Olympus
Corporation).
Western blot analysis
Total protein was extracted using a Bradford Protein
Assay Kit (KeyGEN, Jiangsu, China). Each protein sample (30 µg) was
separated on an SDS-polyacrylamide gel (10%) and then transferred
onto a PVDF filter membrane (Millipore). The membranes were blocked
in 5% BSA for 60 min at room temperature and incubated with the
specified primary antibodies (1:1,000) at 4°C overnight.
Subsequently the membranes were incubated with HRP-conjugated
secondary antibodies (1:10,000, BS13278; Bioworld Technology).
Densitometry was performed by using ImageJ software 1.8.0 (National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
SPSS 25.0 (IBM Corp., Armonk, NY, USA) was used to
analyze the experimental data, and values are expressed as the mean
± SD. Statistical analyses were performed using either one-way
analysis of variance (ANOVA) followed by post hoc pairwise
comparison (LSD) tests for analysis, or Student's t-tests and a
value of P<0.05 was considered statistically significant.
Results
Cell identification
To ensure the accuracy of the experiments, the
purity of the primary cells cultured were determined during the 4th
week of culture. We labeled the astrocytes with specific protein,
glial fibrillary acidic protein (GFAP), by immunochemical staining.
According to the observed results (Fig. 1A), the proportion of astrocytes in
the primary cultured cells was >95%, which met the requirements
of this experiment.
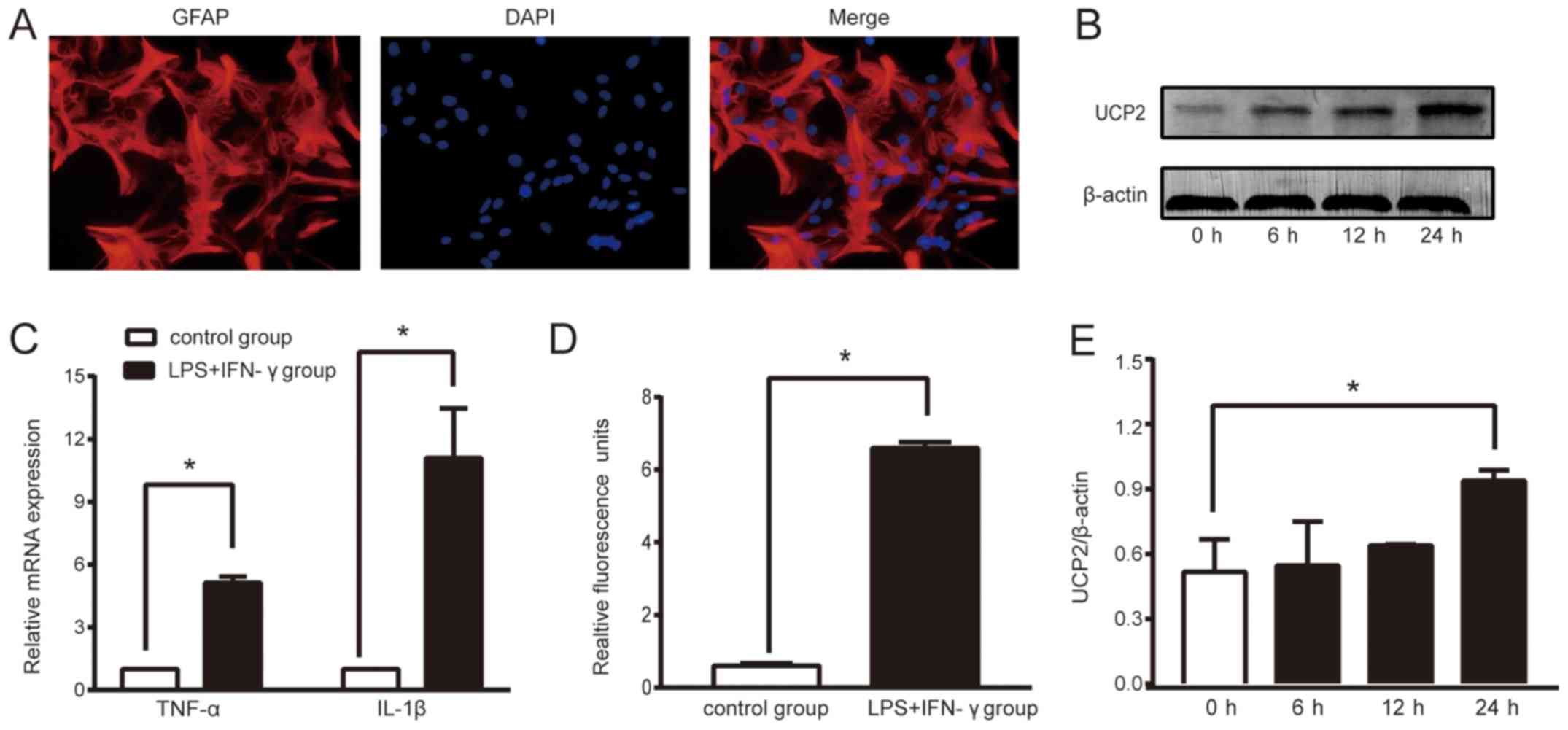 | Figure 1.LPS+IFN-γ treatment successfully
triggers sepsis and increases UCP2 expression in astrocytes. (A)
The proportion of astrocytes in primary cultured cells was more
than 95%, which met the requirements of this experiment (original
magnification ×400). (B) Relative fold change in the protein levels
of UCP2. Astrocytes treated with LPS+IFN-γ for 0, 6, 12, 24 h were
analyzed by western blot analysis. (C) Expression of TNF-α and
IL-1β mRNA in the two cell groups. (D) ROS level. Data are the
means ± SD (n=3–5 in each group). (E) Grayscale analysis of
relative fold change in the protein levels of UCP2. *P<0.05 vs.
the control or 0 h group. UCP2, uncoupling protein 2; LPS,
lipopolysaccharide; IFN-γ, interferon-γ; TNF-α, tumor necrosis
factor α; IL-1β, interleukin-1β; ROS, reactive oxygen species. |
Biomarkers of sepsis
Sepsis is caused by infection. During sepsis,
proinflammatory cytokine secretion and ROS generation are
increased, which damages the brain. After stimulation of astrocytes
with LPS (150 ng/ml) and IFN-γ (200 U/ml), the mRNA levels of TNF-α
and IL-1β were increased in the supernatant of cultured cells
compared to the control group (P<0.05; Fig. 1C). The ROS level was significantly
higher in the LPS+IFN-γ group than that noted in the control group
(P<0.05, Fig. 1D). Thus,
compared to the control group, treatment with LPS+IFN-γ induced a
6.59-fold increase in the ROS level, a 5.13-fold increase in the
TNF-α mRNA level and a 11.09-fold increase the IL-1β mRNA level.
These results showed that an astrocyte sepsis model had been
successfully created.
Sepsis increases the expression levels
of UCP2 in astrocytes
To assess the expression of UCP2 in an experimental
sepsis model in astrocytes, we performed a time course study of
UCP2. The western blot results showed that UCP2 protein levels
increased following sepsis induction and reached a peak expression
level at 24 h. Compared to the control group, sepsis caused a
1.82-fold increase in UCP2 protein expression (P<0.05; Fig. 1B and E).
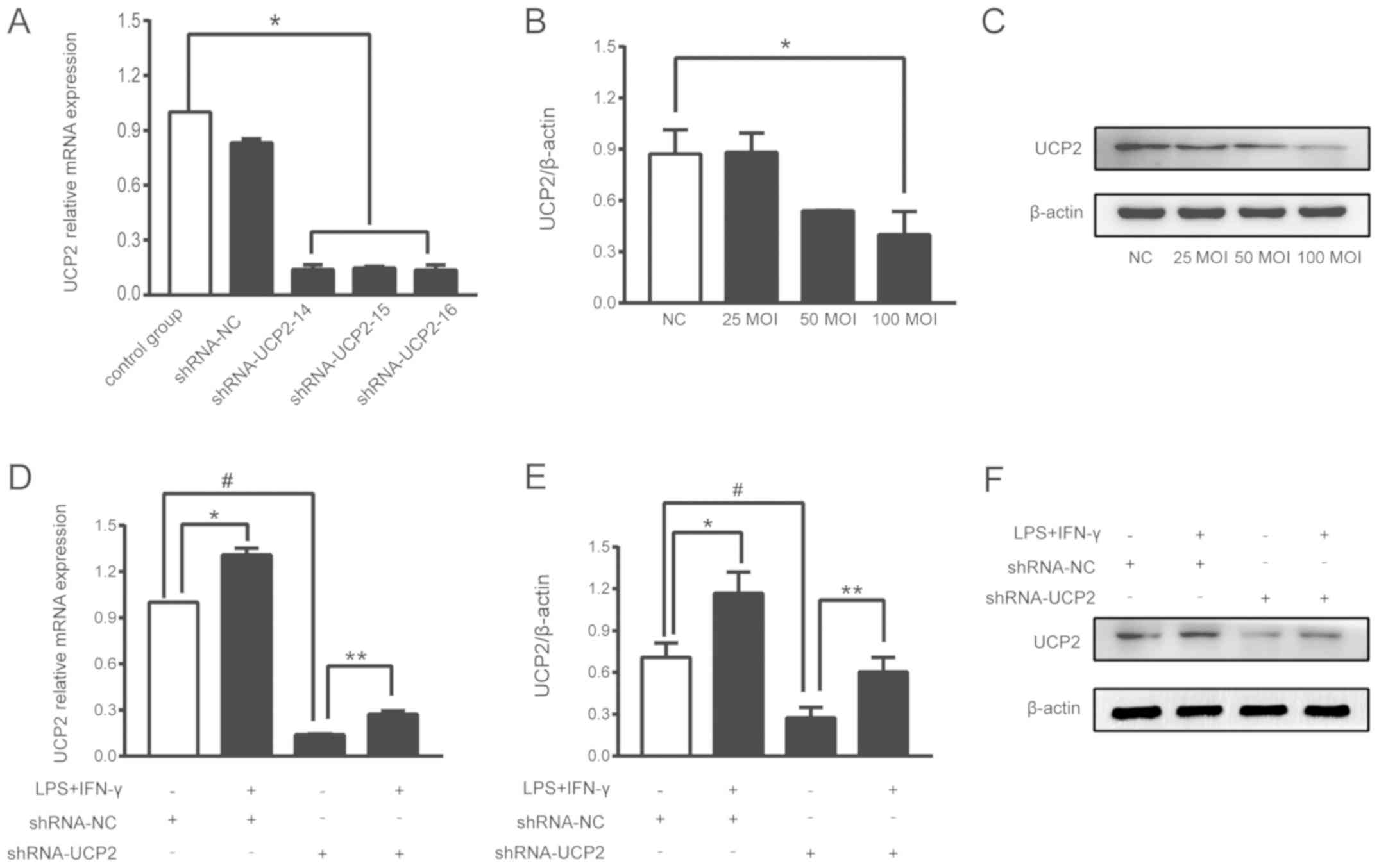
Transfection efficiency of UCP2
UCP2 expression in astrocytes was successfully
knocked down by transfection with adenoviral-shUCP2, which was
performed according to the manufacturer's instructions. The mRNA
levels of UCP2, which represent the knockdown efficiency of UCP2,
were measured by RT-qPCR at 72 h after transfection. After
transfection with shRNA-UCP2-14, the mRNA level of UCP2 was reduced
by 88%, and transfection with shRNA-UCP2-15 and shRNA-UCP2-16
resulted in reductions of 85% compared to the control group
(P<0.05; Fig. 2A).
shRNA-UCP2-14 was thus selected for use in the subsequent
experiments. Western blot analysis revealed that an MOI of 25 to
100 for each astrocyte was able to reduce the expression of UCP2,
and an MOI of 100 showed a higher inhibition efficiency (P<0.05;
Fig. 2B and C).
UCP2 mRNA and protein levels are
increased in transfected astrocytes in an experimental sepsis
model
To assess the role of UCP2 in the astrocyte sepsis
model after UCP2 knockdown, astrocytes were transfected with
adenoviral-shUCP2 (shRNA-UCP2) to suppress UCP2 expression. After
adenoviral transduction for 48 h, the cells were treated with
LPS+IFN-γ or DMEM for 2 h. The total RNA and protein levels of UCP2
were analyzed. Compared to the shRNA-NC group, shRNA-UCP2 induced a
0.87-fold decrease in UCP2 mRNA and a 0.39-fold decrease in UCP2
protein expression suggesting that UCP2 expression was successfully
suppressed (#P<0.01; Fig. 2D-F). Compared to the shRNA-UCP2
group, sepsis caused a 2.08-fold increase in UCP2 mRNA and a
2.22-fold increase in UCP2 protein expression as noted in the
LPS+IFN-γ+shRNA-UCP2 group (**P<0.05; Fig. 2D-F). The results showed that UCP2
expression was enhanced at the mRNA and protein levels after
stimulation with LPS+IFN-γ.
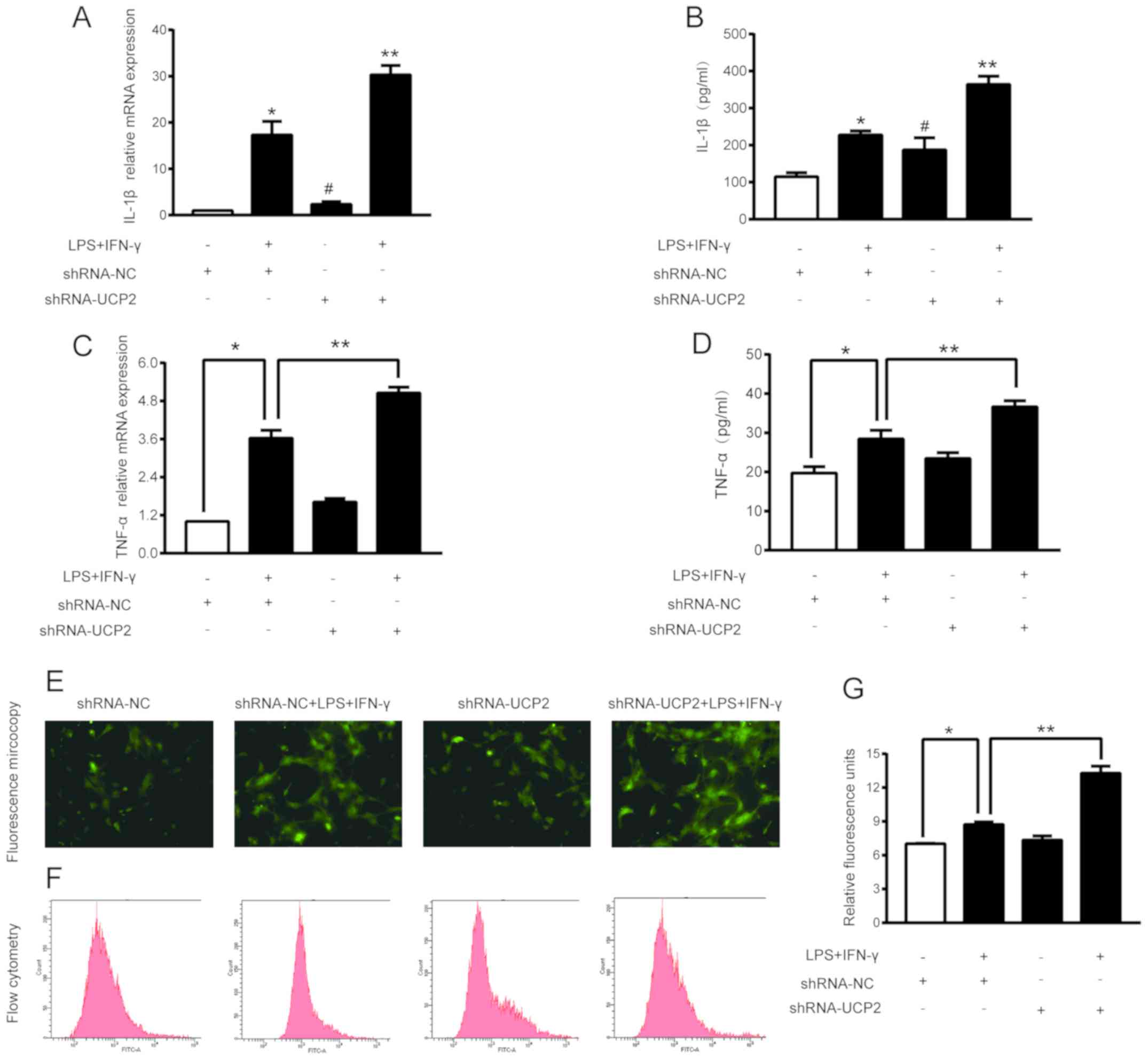
UCP2 knockdown enhances TNF-a and
IL-1β levels in an experimental sepsis model in astrocytes
TNF-α and IL-1β are important indicators of sepsis.
Treatment with shRNA-UCP2 slightly increased the TNF-α and IL-1β
mRNA and protein expression levels. As shown in Fig. 3A-D, compared to the shRNA-NC group,
treatment with shRNA-UCP2 induced a 1.62-fold increase in the TNF-α
mRNA level (P<0.05) resulting in a TNF-α concentration in the
culture medium of 23.44±1.5 (pg/ml), and a 3.15-fold increase the
IL-1β mRNA level (P<0.05) resulting in a IL-1β concentration in
the culture medium of 187.46±32.19 (pg/ml). Subsequently, we
explored the effects of shRNA-UCP2 on astrocytes under septic
conditions. mRNA and supernatant from cells treated with
LPS+IFN-γ+shRNA-UCP2 had significantly higher IL-1β and TNF-α
concentrations and mRNA levels (all P<0.05) than the
LPS+IFN-γ+shRNA-NC group. This significant increase indicated that
UCP2 knockdown enhances the TNF- α and IL-1β levels in astrocytes
in an experimental sepsis model.
UCP2 knockdown exacerbated ROS
elevation in astrocytes under septic conditions
To investigate the role of UCP2 in mitochondrial
function, the intracellular ROS level of each group was measured by
using DCFH-DA probes. Compared to the shRNA-NC group, treatment
with LPS and IFN-γ induced a 1.24-fold increase in ROS generation
in the LPS+IFN-γ+shRNA-NC group (P<0.05). Compared to the
LPS+IFN-γ+shRNA-NC group, shRNA-UCP2 caused a 1.52-fold-increase in
ROS generation in the LPS+IFN-γ+shRNA-UCP2 group (P<0.05;
Fig. 3E-G). The results indicated
that UCP2 knockdown significantly increased the ROS generation in
astrocytes after treatment with LPS+IFN-γ for 24 h.
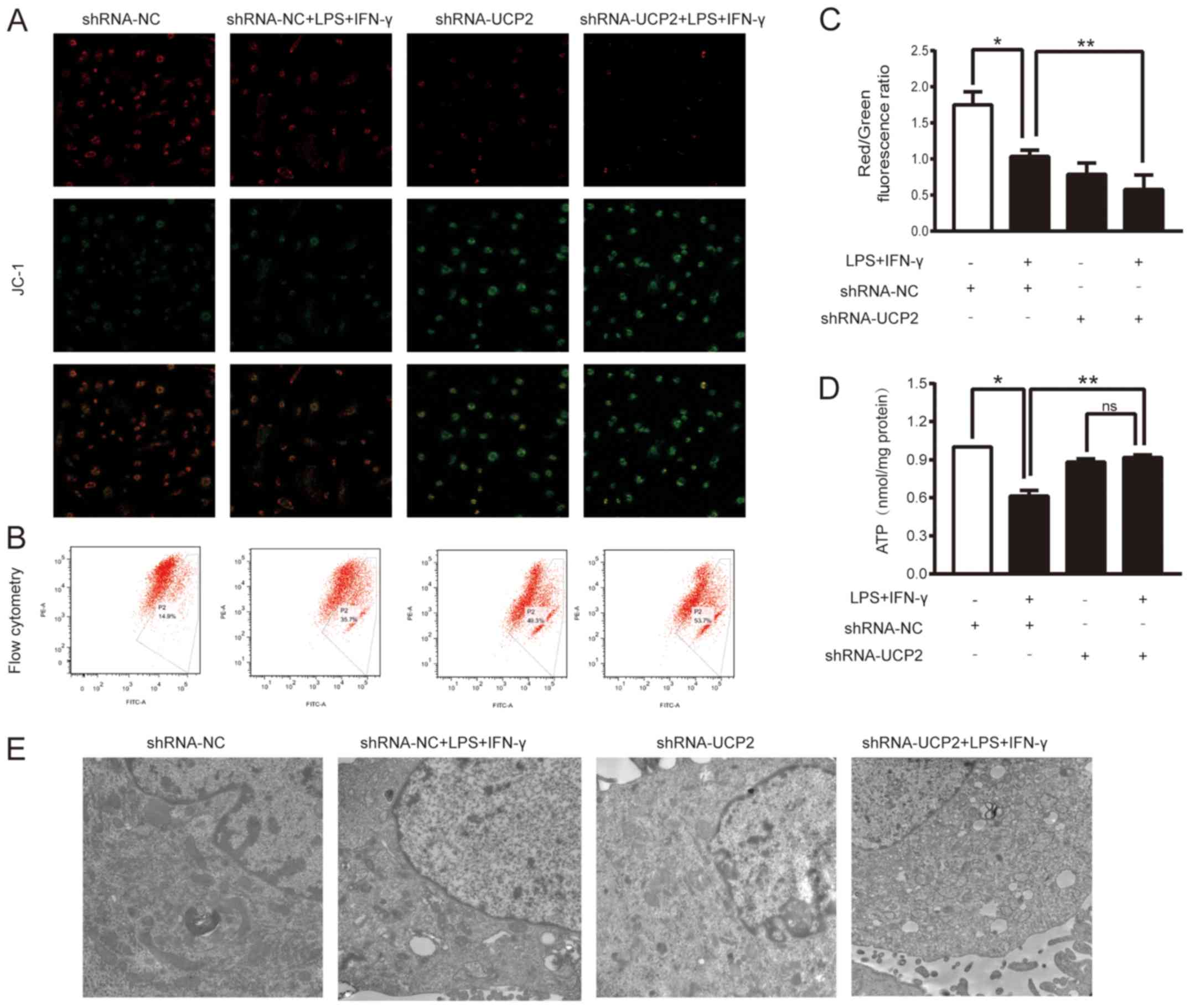
UCP2 knockdown aggravates
mitochondrial dysfunction in an experimental sepsis model in
astrocytes
To further evaluate the effects of UCP2 knockdown on
LPS+IFN-γ-induced injury, we analyzed the MMP and the ATP levels in
astrocytes after various treatments. The ratio of red to green
fluorescence was determined to indicate the MMP value. As shown in
Fig. 4A, the red fluorescence
intensity in the LPS+IFN-γ+shRNA-UCP2 group was weaker than that in
the LPS+IFN-γ+shRNA-NC group. However, the opposite results were
observed for the green fluorescence intensity. To further determine
the change in the MMP, the ratio of red fluorescence to green
fluorescence as shown by flow cytometry was assessed (Fig. 4B and C). In the shRNA-NC group, the
ratio was 1.589±0.429, while the LPS+IFN-γ+shRNA-NC group showed a
lower ratio (1.033±0.089; P<0.05 vs. the shRNA-NC group, n=3),
indicating a decrease in the MMP in the astrocytes under
experimental sepsis. Astrocytes treated with LPS+IFN-γ+shRNA-UCP2
exhibited a further decline, with a ratio of 0.578±0.202,
(P<0.05 vs. LPS+IFN-γ+shRNA-NC group, n=3). The results
indicated that knockdown of UCP2 exacerbated the decrease in the
MMP as detected by confocal microscopy and flow cytometry. Cellular
ATP detection showed that after 24 h of exposure to LPS+IFN-γ, the
cellular ATP content was markedly reduced in the shRNA-NC+LPS+IFN-γ
group compared to the shRNA-NC group and the shRNA-UCP2+LPS+IFN-γ
group (P<0.05; Fig. 4D).
However, compared to that of the shRNA-UCP2 group, the cellular ATP
content in the shRNA-UCP2+LPS+IFN-γ group was not significantly
(ns) different (P>0.05; Fig.
4D).
UCP2 knockdown increases mitochondrial
ultrastructure damage in astrocytes under septic conditions
The mitochondrial ultrastructure was examined by TEM
(Fig. 4E), which showed that most
mitochondria of astrocytes in the shRNA-NC group had little injury,
while damage was increased in the shRNA-NC+LPS+IFN-γ group, with
some swelling and even vacuolization in mitochondria. Compared to
the shRNA-NC group, knockdown of UCP2 significantly enhanced
mitochondrial swelling damage. Furthermore, after stimulation of
UCP2 knockdown in astrocytes with LPS+IFN-γ, the mitochondrial
ultrastructure was most severely damaged; increased mitochondrial
vacuolization and rupture of mitochondrial membranes were
observed.
Discussion
Our study findings demonstrated that astrocytes
subjected to an in vitro model of sepsis-associated
encephalopathy (SAE) induced by lipopolysaccharide (LPS) and
interferon (IFN)-γ exhibited increased expression of uncoupling
protein 2 (UCP2) during the early stage. Adenoviral transduction
successfully decreased UCP2 mRNA and protein expression in cells
during SAE. In addition, astrocytes with adenoviral transduction of
shUCP2 that were stimulated with LPS+IFN-γ showed an increase in
reactive oxygen species (ROS) production, marked by damage to the
mitochondrion, higher UCP2 protein levels, and decreases in the
mitochondrial membrane potential (MMP) and cellular adenosine
triphosphate (ATP) levels. When astrocytes with UCP2 knocked down
were stimulated by LPS+IFN-γ, we observed an increase in
proinflammatory marker expression and damage to the mitochondrial
ultrastructure.
Our group has reported the mechanisms of
sepsis-induced mitochondrial dysfunction in various organs; these
mechanisms are gradually assuming an important role in sepsis
research and its potential treatment (6,19–21).
To establish the mechanisms underlying the sepsis-induced brain
inflammatory process, we have observed numerous cellular and
molecular changes that, with time and increased understanding,
broaden the list of new molecules that may contribute to the
pathological state of the disease. While UCP2-based neuroprotection
has been widely reported in many species (22,23)
its participation in SAE requires further investigation. Increased
expression of UCP2 was observed after LPS+IFN-γ was added to the
primary astrocytes, reaching a peak at 24 h after stimulation.
Combined stimulation with LPS+IFN-γ previously showed greater
efficiency in activating astrocytes in vitro than
stimulation with LPS alone (6).
The silencing of UCP2 gene expression is a molecular
protocol that targets the mRNA of this molecule, limiting its
availability by annealing with complementary nucleotide sequences.
Lu et al (22) demonstrated
that UCP2 deficiency increased intracellular ROS generation and
elevated oxidative stress response l-methyl-4-phenylpyridinium
treatment in primary astrocytes. Moreover, Deng et al
(14) reported that overexpression
of UCP2 in A549 cells inhibited ROS accumulation and apoptosis
under hypoxic conditions. The authors assumed that the induction of
UCP2 expression in the selected cells could be considered an
endogenous protective mechanism that might effectively reduce the
stimulation of inflammation. Our observation that knockdown of UCP2
in primary astrocytes made them more susceptible to the
mitochondrial dysfunction might be evidence of the neuroprotection
elicited by UCP2.
The MMP is formed by the asymmetric distribution of
protons and other ions on both sides of the membrane. Stabilization
of the MMP is conducive to maintaining the normal physiological
function of cells. As previously demonstrated, a decrease in the
MMP is closely related to cell damage (24). In the present study, stimulation
with LPS+IFN-γ led to a decrease in the MMP in an experimental
sepsis model in astrocytes, and knockdown of UCP2 markedly
aggravated mitochondrial damage (12), which is consistent with our
previous study that analyzed LPS-induced sepsis in neonatal rat
cardiomyocytes (unpublished data). UCP2 regulates the proton
concentration of the mitochondrial inner and outer membranes.
Therefore, UCP2 is strongly associated with the MMP, as indicated
by the fact that in LPS-induced cardiomyocyte injury,
overexpression of UCP2 can markedly change the MMP and alter ROS
generation. Many studies have shown an interaction between the MMP
and ROS (19,25). UCP2 is considered an important
factor in increasing neuronal survival after stroke and brain
trauma, and its activation can reduce ROS production and cellular
stress (10).
In the present study, the ROS level was increased
when UCP2 was knocked down under experimental septic conditions.
This is consistent with the result observed for MMP, and could
explain the protective role of UCP2 on a different level.
Cellular ATP, which is the special energy currency
in the cell, is also an important indicator of mitochondrial
function as it is generated from the coupling of electron transport
and proton dynamics through the electron transport chain (ETC).
UCP2 decouples oxidation and phosphorylation in mitochondria by
reducing the proton concentration difference on both sides of the
mitochondrial inner membrane and ATP synthesis. Our present study
found that sepsis damaged astrocytes and led to low levels of ATP,
while UCP2 silencing rescued the ATP levels. This finding of the
function of UCP2 was previously identified.
Our experimental results showed that mitochondrial
dysfunction aggravated oxidative stress, leading to a decrease in
the MMP and ATP levels under septic conditions. Knockdown of UCP2
aggravated the destruction of mitochondrial ultrastructure, caused
more severe edema and damage to mitochondrial inner membrane, and
increased MMP disruption, suggesting that UCP2 may play a positive
role under SAE conditions. However, the neuroprotective effect of
UCP2 in sepsis-associated encephalopathy remains to be determined.
The role of UCP2 in neurons in the brain requires further
study.
In conclusion, our study showed that co-stimulation
with LPS+IFN-γ triggered mitochondrial disorder in astrocytes and
that knockdown of UCP2 by adenoviral transfection exacerbated
mitochondrial injury, indicating that UCP2 has a positive effect in
astrocytes under septic conditions.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (81272070,81601664) and the Guangzhou
Key Laboratory of Inflammatory and Immune Diseases
[(2012)224-226].
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
QZ and WP conceived and designed this study. WP, JH,
YZ, YD, SL and JZ performed the experiments and analyzed the data.
QZ, and WP wrote, edited and reviewed the manuscript. JL was
responsible for the planning of experimental scheme and the
arrangement of experimental tasks. WP and JH contributed equally to
this work. All authors read and approved the final submitted
version of the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
This study was approval by the Nanfang Hospital
Ethics Committee (NFYY-2017-39). The animals used in the study were
provided by Southern Medical University Animal Experiment Center
[SCXK(Guangdong)2016-0041], and passed the experimental animal
quality inspection in Guangdong Province (no. 44002100020527).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there have no competing
interests.
References
|
1
|
Hawiger J, Veach RA and Zienkiewicz J: New
paradigms in sepsis: From prevention to protection of failing
microcirculation. J Thromb Haemost. 13:1743–1756. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chaudhry N and Duggal AK: Sepsis
associated encephalopathy. Adv Med. 2014:7623202014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Helbing DL, Bohm L and Witte OW:
Sepsis-associated encephalopathy. CMAJ. 190:E10832018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eyenga P, Roussel D, Morel J, Rey B,
Romestaing C, Gueguen-Chaignon V, Sheu SS and Viale JP: Time course
of liver mitochondrial function and intrinsic changes in oxidative
phosphorylation in a rat model of sepsis. Intensive Care Med Exp.
6:312018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dutra MR, Feliciano RD, Jacinto KR,
Gouveia TL, Brigidio E, Serra AJ, Morris M, Naffah-Mazzacoratti MD
and Silva JA Jr: Protective role of UCP2 in oxidative stress and
apoptosis during the silent phase of an experimental model of
epilepsy induced by pilocarpine. Oxid Med Cell Longev.
6:67367212018.
|
|
6
|
Chen XL, Wang Y, Peng WW, Zheng YJ, Zhang
TN, Wang PJ, Huang JD and Zeng QY: Effects of interleukin-6 and
IL-6/AMPK signaling pathway on mitochondrial biogenesis and
astrocytes viability under experimental septic condition. Int
Immunopharmacol. 59:287–294. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Molofsky AV and Deneen B: Astrocyte
development: A guide for the perplexed. Glia. 63:1320–1329. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Donadelli M, Dando I, Fiorini C and
Palmieri M: UCP2, a mitochondrial protein regulated at multiple
levels. Cell Mol Life Sci. 71:1171–1190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moukdar F, Robidoux J, Lyght O, Pi J,
Daniel KW and Collins S: Reduced antioxidant capacity and
diet-induced atherosclerosis in uncoupling protein-2-deficient
mice. J Lipid Res. 50:59–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haines B and Li PA: Overexpression of
mitochondrial uncoupling protein 2 inhibits inflammatory cytokines
and activates cell survival factors after cerebral ischemia. PLoS
One. 7:e317392012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vogler S, Pahnke J, Rousset S, Ricquier D,
Moch H, Miroux B and Ibrahim SM: Uncoupling protein 2 has
protective function during experimental autoimmune
encephalomyelitis. Am J Pathol. 168:1570–1575. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen W, Luo S, Xie P, Hou T, Yu T and Fu
X: Overexpressed UCP2 regulates mitochondrial flashes and reverses
lipopolysaccharide-induced cardiomyocytes injury. Am J Transl Res.
10:1347–1356. 2018.PubMed/NCBI
|
|
13
|
Singer M: The role of mitochondrial
dysfunction in sepsis-induced multi-organ failure. Virulence.
5:66–72. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deng S, Yang Y, Han Y, Li X, Wang X, Li X,
Zhang Z and Wang Y: UCP2 inhibits ROS-mediated apoptosis in A549
under hypoxic conditions. PLoS One. 7:e307142012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao YZ, Gao ZY, Ma LQ, Zhuang YY and Guan
FL: Research on biogenesis of mitochondria in astrocytes in
sepsis-associated encephalopathy models. Eur Rev Med Pharmacol Sci.
21:3924–3934. 2017.PubMed/NCBI
|
|
16
|
Lu M, Zhao FF, Tang JJ, Su CJ, Fan Y, Ding
JH, Bian JS and Hu G: The neuroprotection of hydrogen sulfide
against MPTP-induced dopaminergic neuron degeneration involves
uncoupling protein 2 rather than ATP-sensitive potassium channels.
Antioxid Redox Signal. 17:849–859. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang T, Chen Y, Zhang J, Chen G, Zhang J,
Huang J and Zeng Q: Protective effects and mechanism of insulin on
brain in septic rats. Chin J Appl Clin Pediatr. 32:856–860.
2017.
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lyu J, Zheng G, Chen Z, Wang B, Tao S,
Xiang D, Xie M, Huang J, Liu C and Zeng Q: Sepsis-induced brain
mitochondrial dysfunction is associated with altered mitochondrial
Src and PTP1B levels. Brain Res. 1620:130–138. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen GD, Zhang JL, Chen YT, Zhang JX, Wang
T and Zeng QY: Insulin alleviates mitochondrial oxidative stress
involving upregulation of superoxide dismutase 2 and uncoupling
protein 2 in septic acute kidney injury. Exp Ther Med.
15:3967–3975. 2018.PubMed/NCBI
|
|
21
|
Zheng G, Lyu J, Liu S, Huang J, Liu C,
Xiang D, Xie M and Zeng Q: Silencing of uncoupling protein 2 by
small interfering RNA aggravates mitochondrial dysfunction in
cardiomyocytes under septic conditions. Int J Mol Med.
35:1525–1536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu M, Sun XL, Qiao C, Liu Y, Ding JH and
Hu G: Uncoupling protein 2 deficiency aggravates astrocytic
endoplasmic reticulum stress and nod-like receptor protein 3
inflammasome activation. Neurobiol Aging. 35:421–430. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Normoyle KP, Kim M, Farahvar A, Llano D,
Jackson K and Wang H: The emerging neuroprotective role of
mitochondrial uncoupling protein-2 in traumatic brain injury.
Transl Neurosci. 6:179–186. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Braun RJ: Mitochondrion-mediated cell
death: Dissecting yeast apoptosis for a better understanding of
neurodegeneration. Front Oncol. 2:1822012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiong W, Hua J, Liu Z, Cai W, Bai Y, Zhan
Q, Lai W, Zeng Q, Ren H and Xu D: PTEN induced putative kinase 1
(PINK1) alleviates angiotensin II-induced cardiac injury by
ameliorating mitochondrial dysfunction. Int J Cardiol. 266:198–205.
2018. View Article : Google Scholar : PubMed/NCBI
|