Introduction
Atherosclerotic cardiovascular disease (ASCVD) is
highly prevalent and accounts for the majority of mortalities
worldwide (1). Characterized by a
chronic non-resolving low-grade sterile inflammation of the
arterial wall, atherosclerosis (AS) is mainly caused by low-density
lipoprotein (LDL) (2). The main
lesions in AS are characterized by LDL deposition in the intima,
the innermost layer of the artery wall. Activated by such stimuli,
endothelial cells express a leukocyte adhesion molecule, such as
vascular cell adhesion molecule-1, interacting with its cognate
ligands to promote the rolling and adherence of monocytes and
lymphocytes, accompanied by smooth muscle cells and fibrous matrix
proliferation, which gradually develop into the formation of an AS
plaque (3). Lipoproteins
sequestered in the arterial wall are susceptible to modifications,
such as oxidation, which render these particles pro-inflammatory
and immunogenic. Recruited monocytes mature into mononuclear
phagocytes, take in normal or modified lipoproteins and transform
into foam cells, a type of macrophage that promotes disease
progression. Above various cell types in AS plaques, macrophages
and foam cells are considered to be major contributors to
atherogenesis: Not only do they respond inflammatorily through the
secretion of pro-inflammatory mediators, matrix-degrading proteases
and final death, but also release lipid contents and tissue
factors, constituting a pro-thrombotic necrotic core after necrosis
or apoptosis (4). Thus, the
dynamics of macrophages should play a decisive part in
atherogenesis.
In contrast to traditional immunology, researchers
recently found the capacity of the innate immune system to build an
inflammatory memory named trained immunity. Trained immunity
possesses most prosperities from innate immunity and has the
ability to give a rapid response in reinfection, which is built on
epigenetic and metabolic reprogramming. The inflammatory memory
gifts the host both protection and susceptibility to
inflammation-driven diseases such as AS. As monocytes and
macrophages have a major role in atherogenesis and are widely
investigated in the field of trained immunity, the present review
will focus on them. After elaborating on trained immunity,
phenotypes of macrophages under signals in AS plaques will be
discussed. The mechanisms of this immune memory in trained AS
macrophages in aspects of metabolism, transcription and epigenetics
will then be presented, which may inspire future treatments of the
disease.
Trained immunity: A novel mechanism of
AS
Trained immunity
In the traditional cognation of immunology, in
contrast of adaptive immunity, innate immunity is unable to build
an immunological memory. However, prompted by evidence of
protection against reinfection in plants and invertebrates,
transplant rejection in invertebrates and cross-protection between
infections in mammals with dysfunctional T and B lymphocytes, Netea
et al (5) named the
inflammatory memory in innate immune cells ‘trained immunity’,
which represented hyper-responsiveness in the innate immune system
facing a second stimulus.
Like innate immunity, trained immunity is built on
pattern recognition receptors allowing recognition of different
kinds of, but not individual, pathogens (6) and induced by stimuli, including
pathogen-associated molecular patterns and damage-associated
molecular patterns (7). Also, its
properties have been described mainly in populations of innate
immune cells, including monocytes, macrophages and natural killer
(NK) cells. Studies have indicated the existence of trained
hematopoietic stem cells and progenitor cells (8), along with an inflammatory memory in
epidermal stem cells (9), showing
a broader scale of trained immunity both in time and space. The
innate immune memory lays its basis on epigenetic reprogramming in
the first stimulation (10), which
persists after the removal of the stimulus and leads to accessible
chromatin and rapid recruitment of RNA polymerase II (11) in the reinfection.
While trained immunity guarantees the host a rapid
response and better protection against restimulation, it brings the
potential danger of inflammatory disorders and cancer. For
instance, it was found that once infected, a pregnant individual
with higher levels of interleukin-6 (IL-6) passed on the
inflammatory memory to the fetal intestinal epithelium, which
persisted to adulthood. As a result, inflammatory-primed intestinal
stem cells showed both enhanced protection against
Salmonella infection and worsened pathology in a model of
colitis (12). Studies have been
centered widely around the maladaptation of the innate immune
memory and diseases, such as periodontitis (13) and Alzheimer's disease (14). AS, as a chronic inflammatory
disease, is no exception.
Trained immunity in AS
Trained immunity can be induced by endogenous
ligands, such as oxidized (ox)LDL (15), which has a major role in
atherogenesis, indicating the contribution of this proinflammatory
memory to AS. In fact, not only do traditional risk factors such as
dyslipidemia and hypertension predispose individuals to ASCVD via
trained immunity, but other factors, including lifestyle factors,
inflammatory conditions and acute adverse cardiovascular events,
were well classified in a review by Riksen et al (16).
Relevant research has also been conducted at the
cellular level. For instance, isolated human monocytes were shown
to mature into macrophages responding with enhanced production of
tumor necrosis factor-α (TNF-α) and IL-6 after brief stimulation by
oxLDL (15). Also, in a clinical
trial, isolated monocytes from patients with ASCVD had a higher
capacity of cytokine production than those from healthy subjects
(17). The property was indicated
to be maintained following conversion to macrophages. Furthermore,
trained immunity was shown to be induced not only in circulating
monocytes and tissue-resident macrophages, but also in
hematopoietic stem and progenitor cells, termed central trained
immunity, which guaranteed a long-term inflammatory memory
(18). Other immune and non-immune
cells related to AS, such as NK cells (19), neutrophils (20), dendritic cells (21) and endothelial cells (22), have also shown this potential, but
as monocytes and monocyte-derived macrophages make a predominant
contribution in all phases of AS (4), the present review will mainly focus
on them.
Phenotypes of macrophages in AS
Aside from functional reprogramming of trained
immunity, another form of immune cell adaption is differentiation
(23). Monocytes infiltrating the
sub-endothelium are exposed to multiple microenvironmental signals,
such as pro- and anti-inflammatory cytokines, irons, calcium,
lipids and their derivatives and heme from senescent erythrocytes.
Initially, they differentiate into M0 macrophages, also known as
resident-like macrophages, which can then become polarized into
several categories of macrophages (24). Previously, these cells were
classified by their activation states, such as classical,
alternative and innate activation and deactivation (25); while, nowadays, they are mostly
recognized by their secreta and the expression of various surface
markers.
Above all of the subtypes, M1 and M2 were first
described. M1 macrophages are classically activated by products of
type-1 T-helper lymphocytes, such as interferon-γ. In AS plaques,
observations suggested that cholesterol crystals (26), oxLDL (27,28)
and pro-inflammatory cytokines drive, alone or in combination, M1
activation. Trained M1 macrophages secrete pro-inflammatory
cytokines, such as IL-6, IL-1β, TNF, IL-23 and IL-12 (29), sustain inflammatory response and
cause tissue damage. At the opposite end, M2 macrophages, mainly
referred to as anti-inflammatory macrophages, are classically
activated by IL-4 and IL-13 cytokines produced by type 2 T-helper
cells (30,31). Activated by different signals, M2
macrophages have been classified into M2a, M2b and M2c, functioning
separately. While M2a macrophages are classically regarded as
‘wound healing macrophages’, M2b and M2c macrophages are considered
to be ‘regulatory macrophages’ (32). Studies on mice or humans have found
other types of macrophages: Mox, M4, M(Hb) and Mhem macrophages.
Mox macrophages found in mice are induced by oxLDL and act in a
proatherogenic way. M4 macrophages are induced by platelets, while
M(Hb) and Mhem macrophages are induced by the uptake of free
hemoglobin (33). Of note, those
polarized macrophages are able to depolarize and switch their
phenotypes in different microenvironments (34) (Table
I).
 | Table I.Phenotypes of macrophages in
atherosclerosis. |
Table I.
Phenotypes of macrophages in
atherosclerosis.
| Phenotype | Stimulus | Secreta |
|---|
| M1 macrophage | Cholesterol
crystals, lipopolysaccharide, proinflammatory cytokines, oxidized
LDL | IL-6, IL-1β, TNF-α,
IL-23, IL-12 |
| M2 macrophage |
|
|
|
M2a | IL-4, IL-13 | Fibronectin,
insulin-like growth factor, TGF-β |
|
M2b | Immune complexes,
IL-1β, lipopolysaccharide | IL-6, IL-1β, TNF-α,
IL-10, TGF-β1 |
|
M2c | IL-10,
glucocorticoids, TGF-β | IL-10, TGF-β,
Pentraxin-3, MERTKa |
| Mox macrophage | Oxidized
phospholipids | IL-1β,
cyclooxygenase 2 |
| M4 macrophage | C-X-C motif
chemokine ligand 4 | IL-6, TNF-α,
MMP-7 |
| M(Hb)
macrophage |
Haemoglobin/haptoglobin | IL-10 |
| Mhem
macrophage | Haem | IL-10 HMOX-1 |
In the context of AS, M1 and M2 macrophages were
shown to constitute 40 and 20% of total AS lesion macrophages,
respectively, in mice (35).
Furthermore, they possess distinct properties and separate tissue
localizations: While M1 macrophages localize near the lipid core
(36) and impair wound healing
(37), M2 macrophages are more
enriched in neo-angiogenic areas within the plaque (36) and more related to AS regression
(38). Since M1 and M2 macrophages
play a significant role in AS and have been widely studied, this
review will mainly focus on them.
Metabolic changes in trained
macrophages
Cellular metabolism not only follows and meets
energy demands, but also creates intermediate metabolites serving
important biological roles and responds to environmental cues by
regulating the functional state of cells (39,40).
The metabolism of macrophages in AS is no exception. Within AS
plaques, through external stimuli and corresponding signaling
pathways, intracellular metabolism reprograms and induces
epigenetic reprogramming, which mediates trained immunity and
macrophage phenotypes (16).
Furthermore, metabolic reprogramming occurs not only in macrophages
located in AS plaques, but also in circulating monocytes and their
bone marrow progenitors exposed to pro-atherogenic stimuli such as
lipoproteins (41), creating a
long-lasting inflammatory memory.
Via pathway analysis, genome-wide transcriptome and
histone modification profiling, Cheng et al (42) found that β-glucan-trained murine
monocytes showed elevated aerobic glycolysis with a reduced basal
respiration rate, as well as increased glucose consumption and
lactate production; further investigation suggested the
dectin-1/Akt/mTOR-hypoxia-inducible factor (HIF)-1α pathway
responsible for the metabolic shift. The results indicated aerobic
glycolysis (Warburg effect) as a metabolic basis of trained
immunity. Further metabolic pathways, such as fatty acid oxidation
(FAO) and fatty acid synthesis (FAS), also have important roles in
metabolic reprogramming, as will be discussed below (Fig. 1).
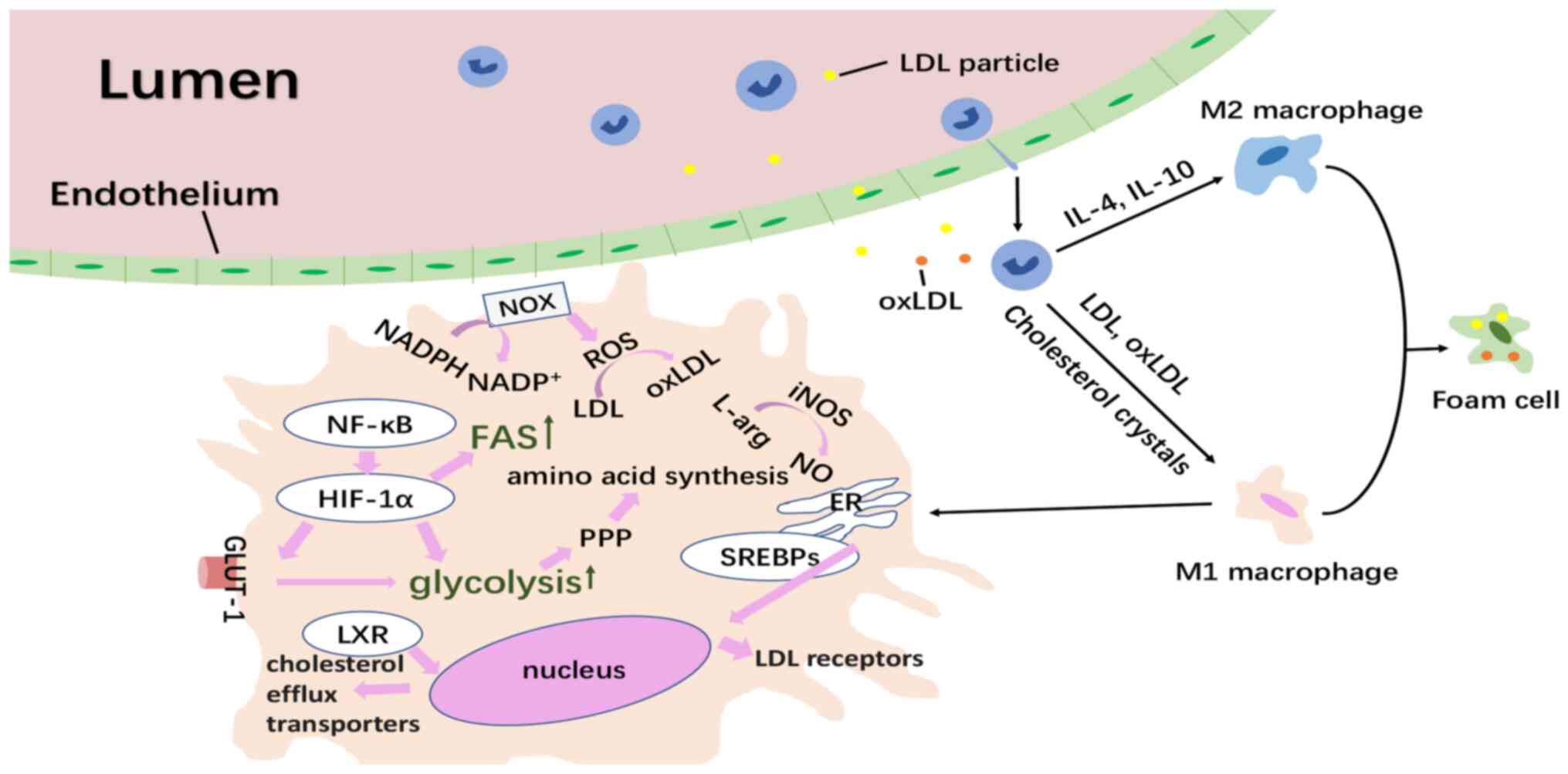 | Figure 1.Overview of the formation of trained
macrophages in atherosclerosis and their inner metabolic changes.
LDL deposition in the intima activates endothelial cells to express
a leukocyte adhesion molecule promoting the rolling and adherence
of monocytes. Lipoproteins are modified occasionally and taken in
by recruited monocytes, which then polarize into diverse phenotypes
of macrophages according to their signals from the
microenvironment. Monocytes or macrophages encountering oxLDL
increase the expression of HIF-1α through the NF-κB pathway. The
HIF-1α increases the expression of the GLUT-1, enhancing the uptake
of glucose to support glycolysis, which then accelerates the PPP
for amino acid synthesis. Activated PPP produces NADPH, which can
further act as the electron donor in the production of oxygen
radicals, contributing to oxidative stress. Also, the HIF1-α
stimulates FAS. As for cholesterol metabolism, nuclear receptors
LXR have the ability to reverse cholesterol transport and the SREBP
family located within the ER can migrate into the nucleus and drive
the expression of LDL receptors. The trained pro-inflammatory M1
macrophages synthesize NO from arginine via iNOS. oxLDL, oxidized
low-density lipoprotein; arg, arginine; HIF-1α, hypoxia inducible
factor-1α; SREBP, sterol-regulatory element binding protein; PPP,
pentose phosphate pathway; GLUT1, glucose transporter 1; FAS, fatty
acid synthesis; NF-κB, nuclear factor κ light-chain-enhancer of
activated B cells; ROS, reactive oxygen species; ER, endoplasmic
reticulum; iNOS, inducible nitric oxide synthase; LXR, liver X
receptors; NOX, NADPH oxidase. |
Glucose metabolism
Monocytes or macrophages encountering
inflammation-induced stimuli such as oxLDL increase the expression
of HIF-1α through signaling pathways, including the nuclear factor
κ light-chain-enhancer of activated B cells (NF-κB) pathway. In
addition, hypoxia in regions rich in plaques is also an important
activator of the HIF-1α transcription factor (TF). HIF-1α increases
the expression of the glucose transporter 1 to enhance the uptake
of glucose. Furthermore, the TF initiates glycolytic metabolism, as
well as increases the expression of key glycolytic enzymes, such as
hexokinase II (HK-II) and
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3),
resulting in increased glycolytic flux (43). Research has shown that monocytes
from patients with symptomatic AS expressed higher levels of
glycolysis-related genes, including HK-II, PFKFB3 and pyruvate
kinase M (PKM)1 (44).
Though less efficient, the enhancement of glucose
uptake and glycolysis accelerate ATP production and become the
major pathway for energy production (45). In addition, certain relevant
enzymes contribute to the inflammatory states. For instance, PKM2,
shown to be upregulated in pro-inflammatory macrophages,
phosphorylates the TF STAT3, which consequently enhances the
production of pro-inflammatory cytokines IL-6 and IL-1β (17). Apart from fueling the inflammation
in atherogenesis, glycolysis also accelerates the pentose phosphate
pathway (PPP) to synthesize amino acids needed for the increased
protein, RNA and DNA synthesis burden of trained macrophages. An
observation of only trained macrophages with an activated PPP
(42) suggests that PPP is vital
to the increased cellular dynamics of trained macrophages and takes
part in the inflammatory process of atherogenesis (46). In addition, activated PPP produces
NADPH. NADPH can further act as the electron donor in the
production of oxygen radicals, which contribute to the oxidative
stress within the AS plaque.
The pro-inflammatory macrophages with aerobic
glycolysis also maintain a tricarboxylic acid cycle, despite two
blockades after citrate and succinate leading to the accumulation
of the two metabolites (17,47).
Citrate is crucial for cholesterol and phospholipid synthesis,
leading to the formation of new membranes. As for succinate,
together with α-ketoglutarate, it is critical for the activity of
two families of enzymes controlling epigenetic modifications: The
JMJ family of lysine demethylases and the TET family of
methyl-cytosine hydroxylases (48,49),
mediating the epigenetic reprogramming in innate immune memory.
Succinate is also able to activate HIF-1α and induces IL-1β
production (46), contributing to
the inflammatory states of pro-inflammatory M1 macrophages.
Cholesterol metabolism
A major risk factor for the development of AS is the
high levels of oxLDL within AS plaques, which is taken up by
macrophages, resulting in cholesterol-loaded macrophage foam cells.
Foam cells secrete pro-inflammatory cytokines and chemokines, as
well as produce matrix metalloproteinases to degrade the
extracellular matrix of the plaque, leading to rupture (50,51).
Furthermore, oxLDLs are highly pro-inflammatory and can induce a
trained immune macrophage phenotype, making the control of
cholesterol influx vs. efflux vital for atherogenesis.
This regulation extensively relies on nuclear
receptors liver X receptors (LXRs) including LXR-α and -β.
Activated by oxysterols, LXRs promote reverse cholesterol transport
and have potent anti-inflammatory effects (52). Studies have indicated that LXRs are
able to suppress the expression of inflammatory genes in
macrophages, such as lipopolysaccharide (LPS)-stimulated cultured
macrophages (52–55) and foam cells (56,57).
It has also been reported that LXR agonists are able to reduce AS
plaque formation (50). A recent
study also showed that myeloid LXR deficiency led to a marked
increase in AS with increased monocyte entry, foam-cell formation
and plaque inflammation (58),
marking the importance of LXRs in atherogenesis.
LXRs are also vital to the expression of
sterol-regulatory element binding protein (SREBP)-1c, which is
upregulated in the pro-inflammatory M1 macrophages and turns on the
FAS (59). SREBP-1c belongs to the
SREBP family of TFs, which also include SREBP-1a and SREBP-2 and
contribute to the regulation of cholesterol and fatty acid
biosynthetic gene expression (60). The family is located within the
endoplasmic reticulum, where they are retained by cholesterols,
oxysterols and desmosterols. When the intracellular concentration
of these metabolites declines, the SREBPs are released, migrate
into the nucleus and drive the expression of LDL receptors and
genes involved in the synthesis pathway of both cholesterol and
fatty acid (61). Studies have
shown that cholesterol synthesis is upregulated in macrophages
trained with β-glucan or Bacillus Calmette-Guérin, and
β-glucan-induced trained immunity both in vivo and in
vitro can be abrogated by statins inhibiting cholesterol
synthesis (62); however, the role
of cholesterol synthesis in oxLDL-induced trained macrophages
remains to be elucidated.
Lipid metabolism
While glycolysis predominantly energizes the
pro-inflammatory M1 macrophages, FAO is the major source of energy
production in M2 anti-inflammatory macrophages (63,64).
Thanks to the overexpression of carnitine palmitoyl transferase 1
transporting long-chain fatty acids into the mitochondria, rates of
FAO are predictably increased, accompanied with decreased
production of inflammatory cytokines in these cells (65).
By contrast, FAS is generally associated with a
pro-inflammatory macrophage phenotype (66). In this pathway, genes involved in
the multi-complex enzyme, such as fatty acid synthase (FASN), are
upregulated (59), and observation
has shown that macrophage-targeted deletion of FASN reduced
AS plaque formation and foam cell formation in Apolipoprotein
E−/− mice (67).
Hypoxia, together with NF-κB, activates the expression of HIF1-α,
which stimulates stearoyl-coenzyme A desaturase (SCD), an important
enzyme in FAS. That way, hypoxia enhances FAS, while suppressing
FAO (68). SCD also drives the
synthesis of monounsaturated fatty acids from palmitic acid.
Increased intracellular levels of unsaturated fatty acids stimulate
a pro-inflammatory phenotype by upregulating IL-1α production in
foam cells (69).
Amino acid metabolism
Another classic example of metabolic reprogramming
is amino acid metabolism, particularly that of arginine. While M2
macrophages catabolize arginine via arginase and finally break it
down into molecules that support cell growth and division, as well
as a building block for collagen production, the trained
pro-inflammatory M1 macrophages synthesize nitric oxide (NO) from
arginine via inducible-NO synthase (iNOS) (70). NO acts not only as a signal of
important cues including vasodilation, insulin secretion and
angiogenesis, but also as an important microbicidal agent, which is
vital for the early stages of AS (71). Enhanced expression of the iNOS has
been used to characterize activated M1 macrophages, and the
expression of iNOS is demonstrated to be elevated compared to
normal arterial tissue by in situ hybridization experiments
(72,73).
Experiments also found that glutamine had pro-AS
effects on a murine macrophage-like cell line (74). Other research has shown the
importance of glutamine in the induction of IL-1 by macrophages in
response to LPS stimulation (75),
highlighting the nonnegligible role of glutamine metabolism in
trained immunity.
Transcriptional and epigenetic regulation in
trained plaque macrophages
In the pathogenesis of AS, the cellular phenotype
and function of macrophages, including differentiation and
activation, are of great importance. The robust execution of this
cascade of biological reactions is guaranteed by multilayered
regulation, such as TF and co-factor binding, the epigenetic
landscape, long-range interactions, DNA methylation, RNA editing
and long non-coding (lnc)RNAs (76). Specifically, in trained macrophages
in AS, studies have found that epigenetic processes, including
histone modification, DNA methylation, modulation of microRNA and
lncRNA expression (8), are at the
basis of the innate immune memory (10,77)
(Fig. 2).
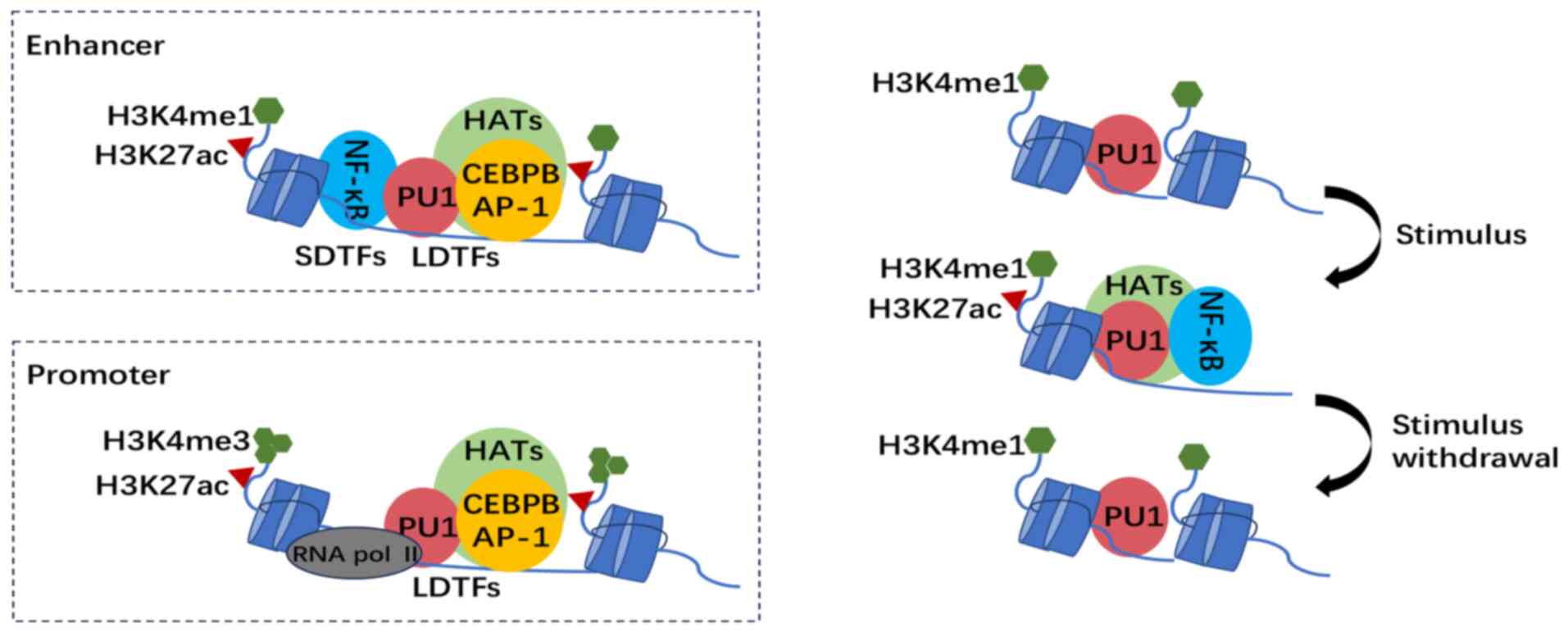 | Figure 2.Epigenetic landscape and
transcription factors binding in trained macrophages. Top left
panel: Distal genetic elements are set up by LDTFs PU-1, CEBPB and
AP-1 via deposition of H3K4me1 by HMEs. Signal-dependent TFs such
as NF-κB recruit histone acetyltransferases, leading to histone
acetylation, including H3K27ac and the formation of active
chromatin. Bottom left panel: H3K4me3 accumulates on immune gene
promoters, where RNA pol II, instead of SDTFs, binds and initiates
gene transcription. Right-hand panel: During myeloid lineage
differentiation, LDTF PU-1 and the accumulation of H3K4me1 prime
the enhancers. Upon stimulation with factors such as oxLDL and
cholesterol crystals, NF-κB binds pre-accessible chromatin and
recruits HATs, leading to histone acetylation such as H3K27ac and
the formation of active chromatin. The SDTF leaves and the H3K27ac
marks are lost gradually after the removal of the initial stimulus,
while H3K4me1 remains, keeping the enhancer in a primed state for
subsequent activation. LDTFs, lineage-determining TFs; HATs,
histone acetyltransferases; CEBPB, CCAAT/enhancer binding protein
beta; AP-1, activator protein 1; H3K27ac, histone 3 lysine 27
acetylation; H3K4me3, histone 3 lysine 4 trimethylation; H3K4me1,
histone 3 lysine 4 methylation; HME, histone-modifying enzyme;
SDTF, signal-dependent TF; PU-1, Spi-1 proto-oncogene; NF-κB,
nuclear factor κ light-chain-enhancer of activated B cells; TF,
transcription factor; pol, polymerase. |
Histone modification
Histone-modifying enzymes (HMEs) with
histone-binding domains have the ability to recognize histone tails
extended out of the octamers and catalyze the addition or removal
of different histone modifications. Several modifications, such as
methylation, acetylation, phosphorylation and ubiquitination,
together with their role in supporting transcriptional processes,
have been well-studied. Amid a vast repertoire of histone
modifications, acetylation (lysine residues) and methylation
(arginine and lysine residues) are the most broadly studied and
extensively characterized (78).
In the context of a trained immune response,
epigenetic reprogramming takes place in different types of immune
cells at a large number of immune genes and their distal genetic
elements called enhancers. Upon primary stimulation, histone 3
lysine 4 trimethylation (H3K4me3) accumulates on immune gene
promoters, with histone 3 lysine 4 methylation (H3K4me1) on
enhancers and histone 3 lysine 27 acetylation (H3K27ac) marking
active promoters and enhancers (79,80).
H3K27ac marks are gradually lost after removal of the initial
stimulus, while H3K4me1 and H3K4me3 modifications remain (49), suggesting that H3K27ac appears to
function more as a mark of changes in promoter activity than
H3K4me3, and H3K4me1 provides an epigenetic memory function in
macrophages (81).
DNA methylation
CpG islands at promoters and enhancers can be
recognized by DNA methyltransferases (DNMTs) and are generally
related to transcriptional repression. DNA methylation has been
shown to be more associated with monocyte-to-macrophage
differentiation than subsequent activation (82,83).
Epigenome-wide analyses have demonstrated a general loss of 5mc
during ex vivo monocyte-to-macrophage differentiation
(84). The role of DNA methylation
in trained immunity is still in need of further investigation, and
different combinations of histone and DNA modifications should
determine the accessibility of DNA undoubtedly.
TF binding
TFs usually bind to specific DNA motifs, in
requirement of chromatin accessibility, which is modulated by
epigenetic modification. The macrophage epigenetic and chromatin
accessibility landscape is established by the divergent binding of
lineage-determining TFs (LDTFs) and signal-dependent TFs (SDTFs).
LDTFs act as master regulators of the cell-specific epigenetic
landscape by binding to closed chromatin, setting up a
cell-specific regulatory landscape and driving cell-specific
transcription programs (85).
SDTFs can activate a stimulus-specific regulatory program (86,87)
and can also activate epigenetically unmarked elements termed
latent enhancers, which can remain marked after stimulus removal
and ready for rapid response to a second activation, acting as
cellular memory (81). In the
context of AS, LDTFs such as PU-1 and CCAAT/enhancer binding
protein β (C/EBPβ) instruct macrophage differentiation, while SDTFs
such as NF-κB trigger macrophage activation regulated by cytokine
stimulation (86).
NF-κB, for instance, is in the cytoplasm under basal
conditions. Upon stimulation with substances such as oxLDL or
cholesterol crystals, it translocates to the nucleus (88) and binds pre-accessible chromatin.
Prior to this, classic enhancers are set up by LDTFs PU-1 and
C/EBPβ (89) via deposition of
H3K4me1 by HMEs. Upon binding, NF-κB recruits the histone
acetyltransferase EP300, leading to histone acetylation such as
H3K27ac and formation of active chromatin (90). Subsequently, the translation of
genes such as HIF-1α is permitted and then causes changes in
cellular metabolic and consequent biologic behavior. After removal
of the stimulus, the SDTF leaves, and activating histone marks are
removed under recruitment of histone deacetylases (HDACs), whereas
H3K4me1 remains and keeps the enhancers in a primed state for
subsequent activation.
Besides the above epigenetic and chromatin
accessibility landscape, long-range interactions and ncRNAs are
also found to be critical modulators of macrophage activation. For
instance, an immune gene-priming lncRNA called upstream master
lncRNA of the inflammatory chemokine locus is able to direct
histone methyltransferase to immune-related genes topologically,
leading to increased local H3K4me3, which is a ubiquitous
epigenetic modification in trained immunity (91). The deposition of this mark and its
persistence through the inhibition of histone demethylases provide
specificity for trained immunity (16). Recent findings of epigenetic
mechanisms and metabolic processes from the molecular basis of
trained immunity were summarized in a previous review (92).
In an AS plaque environment, macrophages are exposed
to a variety of signals and differentiate into complex phenotypes
(93). Studies have shown that the
identified pro-inflammatory and anti-inflammatory macrophage
subsets in the plaque do not resemble the classical M1 and M2
macrophages and express both pro- and anti-inflammatory macrophage
markers at the same time (94).
Indeed, an epigenetic and transcriptional crosstalk between pro-
and anti-inflammatory signaling in macrophages is suggested in
several studies, highlighting the complexity of inflammatory
signaling in an AS plaque in vivo (95,96).
Therapeutic approaches
AS is an inflammation-driven disease and macrophages
have a central role in the modulation of inflammation. Due to the
need to respond in a rapid and specific way, macrophages adopt a
unique, permissive epigenetic landscape that is established and
controlled by sequential binding of LDTFs and SDTFs, which shed
light on therapy targeting epigenetic enzymes. Indeed,
macrophage-specific genetic ablation or pharmacological inhibition
of HMEs holds promise for the treatment of inflammatory diseases
(97). For instance, HDAC3 is able
to deactivate key factors needed for IL-4-dependent
anti-inflammatory activation (98). Macrophage-specific knockout of
HDAC3 in mice promotes plaque stability and steers the cells
into a more wound-healing, fibrotic and anti-inflammatory phenotype
(99). Thus, the deletion of
HDAC3 may attenuate AS. However, owing to the broad action
of epigenetic enzymes, strategies to increase specificity would be
beneficial and possibly even required. There is a great demand to
characterize the epigenetic landscape and pinpoint TFs, as well as
upstream signaling pathways regulating cell type-specific gene
expression programs.
Furthermore, due to the reprogramming of metabolism
in trained macrophages in AS, new drugs interfering with key
metabolic pathways such as glycolysis, which is involved in the
activation and differentiation of monocytes, should be taken into
consideration. Indeed, the observation that trained immunity is
completely prevented by pharmacological blockers of glycolysis
(42) not only validates the
causal role of glycolysis in trained immunity but also enlightens
future treatments of AS. Also, several animal studies have reported
that LXR agonists can reduce AS plaque formation (50), making cholesterol metabolism a key
pathway in AS treatment. For future employment, a targeted therapy
would be required likewise. Also, it may be worthwhile
investigating to what extent the metabolic pathways determine the
destiny of macrophages.
Conclusions
AS, a major threat to global public health, has its
basis in chronic inflammation of the artery wall. Above all cells
involved in atherogenesis, monocytes and macrophages have a major
role in the pathogenesis, highlighting the importance of their
biological activities. Trained immunity, the memory of innate
immunity, has aroused the interest of investigators and may be a
novel mechanism of atherogenesis. In the microenvironment of AS
plaque, monocytes are activated and differentiate into various
phenotypes of macrophages in response to a cascade of signals,
mainly pro-inflammatory M1 and anti-inflammatory M2 cells. During
this process, metabolic changes take place, such as enhanced
glycolysis in trained pro-inflammatory macrophages. Epigenetic and
transcriptional regulation acts as initiators in these metabolic
changes.
Recent discoveries of trained immunity not only
refresh the concept of immunology, but also inspire researchers to
reconsider inflammatory processes in AS. In that way, researchers
find heterogeneity of macrophages in AS plaque and further
elucidate mechanisms that drive the differentiation. To meet the
need for more experiments and findings, research on potential
therapies for AS based on metabolism and gene expression is
underway.
Acknowledgements
Not applicable.
Funding
This work is supported by grants from the National Natural
Science Foundation of China (grant nos. 82273011 and 82072648), the
Natural Science Foundation of Jiangsu Province (grant no.
BK20211508) and the Fundamental Research Funds for the Central
Universities (grant no. 021414380472).
Availability of data and materials
Not applicable.
Authors' contributions
TL and WF collected the references and were involved
in the initial conception of this review. TL wrote most parts of
the manuscript. TW and WY provided writing guidance and revisions.
All authors have read and approved the final version of the
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Libby P: The changing landscape of
atherosclerosis. Nature. 592:524–533. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boren J, Chapman MJ, Krauss RM, Packard
CJ, Bentzon JF, Binder CJ, Daemen MJ, Demer LL, Hegele RA, Nicholls
SJ, et al: Low-density lipoproteins cause atherosclerotic
cardiovascular disease: Pathophysiological, genetic, and
therapeutic insights: A consensus statement from the european
atherosclerosis society consensus panel. Eur Heart J. 41:2313–2330.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han
X, Tang D and Chen R: Research progress on the relationship between
atherosclerosis and inflammation. Biomolecules. 8:802018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moore KJ, Sheedy FJ and Fisher EA:
Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol.
13:709–721. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Netea MG, Quintin J and van der Meer JW:
Trained immunity: A memory for innate host defense. Cell Host
Microbe. 9:355–361. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Netea MG: Training innate immunity: The
changing concept of immunological memory in innate host defence.
Eur J Clin Invest. 43:881–884. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dominguez-Andres J, Santos JC, Bekkering
S, Mulder WJM, van der Meer JWM, Riksen NP, Joosten LAB and Netea
MG: Trained immunity: Adaptation within innate immune mechanisms.
Physiol Rev. 103:313–346. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Netea MG, Joosten LA, Latz E, Mills KH,
Natoli G, Stunnenberg HG, O'Neill LA and Xavier RJ: Trained
immunity: A program of innate immune memory in health and disease.
Science. 352:aaf10982016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Naik S, Larsen SB, Gomez NC, Alaverdyan K,
Sendoel A, Yuan S, Polak L, Kulukian A, Chai S and Fuchs E:
Inflammatory memory sensitizes skin epithelial stem cells to tissue
damage. Nature. 550:475–480. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van den Burg HA and Takken FL: Does
chromatin remodeling mark systemic acquired resistance? Trends
Plant Sci. 14:286–294. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Foster SL, Hargreaves DC and Medzhitov R:
Gene-specific control of inflammation by TLR-induced chromatin
modifications. Nature. 447:972–978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lim AI, McFadden T, Link VM, Han SJ,
Karlsson RM, Stacy A, Farley TK, Lima-Junior DS, Harrison OJ, Desai
JV, et al: Prenatal maternal infection promotes tissue-specific
immunity and inflammation in offspring. Science. 373:eabf30022021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Y, Chen Y, Cai G, Ni Q, Geng Y, Wang T,
Bao C, Ruan X, Wang H and Sun W: Roles of trained immunity in the
pathogenesis of periodontitis. J Periodontal Res. 58:864–873. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wendeln AC, Degenhardt K, Kaurani L,
Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A,
et al: Innate immune memory in the brain shapes neurological
disease hallmarks. Nature. 556:332–338. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bekkering S, Quintin J, Joosten LA, van
der Meer JW, Netea MG and Riksen NP: Oxidized low-density
lipoprotein induces long-term proinflammatory cytokine production
and foam cell formation via epigenetic reprogramming of monocytes.
Arterioscler Thromb Vasc Biol. 34:1731–1718. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Riksen NP, Bekkering S, Mulder WJM and
Netea MG: Trained immunity in atherosclerotic cardiovascular
disease. Nat Rev Cardiol. 20:799–811. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shirai T, Nazarewicz RR, Wallis BB, Yanes
RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC,
Assimes TL, et al: The glycolytic enzyme PKM2 bridges metabolic and
inflammatory dysfunction in coronary artery disease. J Exp Med.
213:337–354. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mitroulis I, Hajishengallis G and Chavakis
T: Bone marrow inflammatory memory in cardiometabolic disease and
inflammatory comorbidities. Cardiovasc Res. 119:2801–2812. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kleinnijenhuis J, Quintin J, Preijers F,
Joosten LA, Jacobs C, Xavier RJ, van der Meer JW, van Crevel R and
Netea MG: BCG-induced trained immunity in NK cells: Role for
non-specific protection to infection. Clin Immunol. 155:213–219.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moorlag SJCFM, Rodriguez-Rosales YA,
Gillard J, Fanucchi S, Theunissen K, Novakovic B, de Bont CM,
Negishi Y, Fok ET, Kalafati L, et al: BCG vaccination induces
long-term functional reprogramming of human neutrophils. Cell Rep.
33:1083872020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hole CR, Wager CML, Castro-Lopez N,
Campuzano A, Cai H, Wozniak KL, Wang Y and Wormley FL Jr: Induction
of memory-like dendritic cell responses in vivo. Nat Commun.
10:29552019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sohrabi Y, Lagache SMM, Voges VC, Semo D,
Sonntag G, Hanemann I, Kahles F, Waltenberger J and Findeisen HM:
OxLDL-mediated immunologic memory in endothelial cells. J Mol Cell
Cardiol. 146:121–132. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Netea MG, Domínguez-Andrés J, Barreiro LB,
Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM,
Mhlanga MM, Mulder WJM, et al: Defining trained immunity and its
role in health and disease. Nat Rev Immunol. 20:375–388. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bonetti J, Corti A, Lerouge L, Pompella A
and Gaucher C: Phenotypic modulation of macrophages and vascular
smooth muscle cells in atherosclerosis-nitro-redox
interconnections. Antioxidants (Basel). 10:5162021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Duewell P, Kono H, Rayner KJ, Sirois CM,
Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr
M, et al: NLRP3 inflammasomes are required for atherogenesis and
activated by cholesterol crystals. Nature. 464:1357–1361. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chavez-Sanchez L, Garza-Reyes MG,
Espinosa-Luna JE, Chávez-Rueda K, Legorreta-Haquet MV and
Blanco-Favela F: The role of TLR2, TLR4 and CD36 in macrophage
activation and foam cell formation in response to oxLDL in humans.
Hum Immunol. 75:322–329. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hirose K, Iwabuchi K, Shimada K, Kiyanagi
T, Iwahara C, Nakayama H and Daida H: Different responses to
oxidized low-density lipoproteins in human polarized macrophages.
Lipids Health Dis. 10:12011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Verreck FA, de Boer T, Langenberg DM,
Hoeve MA, Kramer M, Vaisberg E, Kastelein R, Kolk A, de
Waal-Malefyt R and Ottenhoff TH: Human IL-23-producing type 1
macrophages promote but IL-10-producing type 2 macrophages subvert
immunity to (myco)bacteria. Proc Natl Acad Sci USA. 101:4560–4565.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stein M, Keshav S, Harris N and Gordon S:
Interleukin 4 potently enhances murine macrophage mannose receptor
activity: A marker of alternative immunologic macrophage
activation. J Exp Med. 176:287–292. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gordon S: Alternative activation of
macrophages. Nat Rev Immunol. 3:23–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jinnouchi H, Guo L, Sakamoto A, Torii S,
Sato Y, Cornelissen A, Kuntz S, Paek KH, Fernandez R, Fuller D, et
al: Diversity of macrophage phenotypes and responses in
atherosclerosis. Cell Mol Life Sci. 77:1919–1932. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chinetti-Gbaguidi G, Colin S and Staels B:
Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 12:10–17.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kadl A, Meher AK, Sharma PR, Lee MY, Doran
AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, et al:
Identification of a novel macrophage phenotype that develops in
response to atherogenic phospholipids via Nrf2. Circ Res.
107:737–746. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stoger JL, Gijbels MJ, van der Velden S,
Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E and de
Winther MP: Distribution of macrophage polarization markers in
human atherosclerosis. Atherosclerosis. 225:461–468. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stewart CR, Stuart LM, Wilkinson K, van
Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA,
et al: CD36 ligands promote sterile inflammation through assembly
of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol.
11:155–161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Feig JE, Vengrenyuk Y, Reiser V, Wu C,
Statnikov A, Aliferis CF, Garabedian MJ, Fisher EA and Puig O:
Regression of atherosclerosis is characterized by broad changes in
the plaque macrophage transcriptome. PLoS One. 7:e397902012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
O'Neill LA, Kishton RJ and Rathmell J: A
guide to immunometabolism for immunologists. Nat Rev Immunol.
16:553–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stienstra R, Netea-Maier RT, Riksen NP,
Joosten LAB and Netea MG: Specific and complex reprogramming of
cellular metabolism in myeloid cells during innate immune
responses. Cell Metab. 26:142–156. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Groh L, Keating ST, Joosten LAB, Netea MG
and Riksen NP: Monocyte and macrophage immunometabolism in
atherosclerosis. Semin Immunopathol. 40:203–214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cheng SC, Quintin J, Cramer RA, Shepardson
KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao
NA, Aghajanirefah A, et al: mTOR- and HIF-1α-mediated aerobic
glycolysis as metabolic basis for trained immunity. Science.
345:12506842014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tawakol A, Singh P, Mojena M,
Pimentel-Santillana M, Emami H, MacNabb M, Rudd JH, Narula J,
Enriquez JA, Través PG, et al: HIF-1α and PFKFB3 mediate a tight
relationship between proinflammatory activation and anerobic
metabolism in atherosclerotic macrophages. Arterioscler Thromb Vasc
Biol. 35:1463–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bekkering S, van den Munckhof I, Nielen T,
Lamfers E, Dinarello C, Rutten J, de Graaf J, Joosten LA, Netea MG,
Gomes ME and Riksen NP: Innate immune cell activation and
epigenetic remodeling in symptomatic and asymptomatic
atherosclerosis in humans in vivo. Atherosclerosis. 254:228–236.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Riksen NP and Netea MG: Immunometabolic
control of trained immunity. Mol Aspects Med. 77:1008972021.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tannahill GM, Curtis AM, Adamik J,
Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ,
Kelly B, Foley NH, et al: Succinate is an inflammatory signal that
induces IL-1β through HIF-1α. Nature. 496:238–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jha AK, Huang SC, Sergushichev A,
Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart
KM, Ashall J, Everts B, et al: Network integration of parallel
metabolic and transcriptional data reveals metabolic modules that
regulate macrophage polarization. Immunity. 42:419–430. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Benit P, Letouzé E, Rak M, Aubry L,
Burnichon N, Favier J, Gimenez-Roqueplo AP and Rustin P:
Unsuspected task for an old team: Succinate, fumarate and other
Krebs cycle acids in metabolic remodeling. Biochim Biophys Acta.
1837:1330–1337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Saeed S, Quintin J, Kerstens HH, Rao NA,
Aghajanirefah A, Matarese F, Cheng SC, Ratter J, Berentsen K, van
der Ent MA, et al: Epigenetic programming of monocyte-to-macrophage
differentiation and trained innate immunity. Science.
345:12510862014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tall AR and Yvan-Charvet L: Cholesterol,
inflammation and innate immunity. Nat Rev Immunol. 15:104–116.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Khokha R, Murthy A and Weiss A:
Metalloproteinases and their natural inhibitors in inflammation and
immunity. Nat Rev Immunol. 13:649–665. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ito A, Hong C, Rong X, Zhu X, Tarling EJ,
Hedde PN, Gratton E, Parks J and Tontonoz P: LXRs link metabolism
to inflammation through Abca1-dependent regulation of membrane
composition and TLR signaling. Elife. 4:e080092015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Thomas DG, Doran AC, Fotakis P, Westerterp
M, Antonson P, Jiang H, Jiang XC, Gustafsson JÅ, Tabas I and Tall
AR: LXR suppresses inflammatory gene expression and neutrophil
migration through cis-repression and cholesterol efflux. Cell Rep.
25:3774–3785. e42018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ghisletti S, Huang W, Ogawa S, Pascual G,
Lin ME, Willson TM, Rosenfeld MG and Glass CK: Parallel
SUMOylation-dependent pathways mediate gene- and signal-specific
transrepression by LXRs and PPARgamma. Mol Cell. 25:57–70. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bories G, Colin S, Vanhoutte J, Derudas B,
Copin C, Fanchon M, Daoudi M, Belloy L, Haulon S, Zawadzki C, et
al: Liver X receptor activation stimulates iron export in human
alternative macrophages. Circ Res. 113:1196–1205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Spann NJ, Garmire LX, McDonald JG, Myers
DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D,
et al: Regulated accumulation of desmosterol integrates macrophage
lipid metabolism and inflammatory responses. Cell. 151:138–152.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang X, McDonald JG, Aryal B,
Canfrán-Duque A, Goldberg EL, Araldi E, Ding W, Fan Y, Thompson BM,
Singh AK, et al: Desmosterol suppresses macrophage inflammasome
activation and protects against vascular inflammation and
atherosclerosis. Proc Natl Acad Sci USA. 118:e21076821182021.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Endo-Umeda K, Kim E, Thomas DG, Liu W, Dou
H, Yalcinkaya M, Abramowicz S, Xiao T, Antonson P, Gustafsson JÅ,
et al: Myeloid LXR (Liver X Receptor) deficiency induces
inflammatory gene expression in foamy macrophages and accelerates
atherosclerosis. Arterioscler Thromb Vasc Biol. 42:719–731. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ecker J, Liebisch G, Englmaier M, Grandl
M, Robenek H and Schmitz G: Induction of fatty acid synthesis is a
key requirement for phagocytic differentiation of human monocytes.
Proc Natl Acad Sci USA. 107:7817–7822. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rong S, Cortés VA, Rashid S, Anderson NN,
McDonald JG, Liang G, Moon YA, Hammer RE and Horton JD: Expression
of SREBP-1c requires SREBP-2-mediated generation of a sterol ligand
for LXR in livers of mice. Elife. 6:e250152017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Im SS, Yousef L, Blaschitz C, Liu JZ,
Edwards RA, Young SG, Raffatellu M and Osborne TF: Linking lipid
metabolism to the innate immune response in macrophages through
sterol regulatory element binding protein-1a. Cell Metab.
13:540–549. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Arts RJ, Novakovic B, Horst RT, Carvalho
A, Bekkering S, Lachmandas E, Rodrigues F, Silvestre R, Cheng SC,
Wang SY, et al: Glutaminolysis and fumarate accumulation integrate
immunometabolic and epigenetic programs in trained immunity. Cell
Metab. 24:807–819. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Huang SC, Everts B, Ivanova Y, O'Sullivan
D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY,
O'Neill CM, et al: Cell-intrinsic lysosomal lipolysis is essential
for alternative activation of macrophages. Nat Immunol. 15:846–855.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Van den Bossche J, O'Neill LA and Menon D:
Macrophage immunometabolism: Where are we (Going)? Trends Immunol.
38:395–406. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Malandrino MI, Fucho R, Weber M,
Calderon-Dominguez M, Mir JF, Valcarcel L, Escoté X, Gómez-Serrano
M, Peral B, Salvadó L, et al: Enhanced fatty acid oxidation in
adipocytes and macrophages reduces lipid-induced triglyceride
accumulation and inflammation. Am J Physiol Endocrinol Metab.
308:E756–E769. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Feingold KR, Shigenaga JK, Kazemi MR,
McDonald CM, Patzek SM, Cross AS, Moser A and Grunfeld C:
Mechanisms of triglyceride accumulation in activated macrophages. J
Leukoc Biol. 92:829–839. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Schneider JG, Yang Z, Chakravarthy MV,
Lodhi IJ, Wei X, Turk J and Semenkovich CF: Macrophage fatty-acid
synthase deficiency decreases diet-induced atherosclerosis. J Biol
Chem. 285:23398–23409. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bostrom P, Magnusson B, Svensson PA,
Wiklund O, Borén J, Carlsson LM, Ståhlman M, Olofsson SO and Hultén
LM: Hypoxia converts human macrophages into triglyceride-loaded
foam cells. Arterioscler Thromb Vasc Biol. 26:1871–1876. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Freigang S, Ampenberger F, Weiss A,
Kanneganti TD, Iwakura Y, Hersberger M and Kopf M: Fatty
acid-induced mitochondrial uncoupling elicits
inflammasome-independent IL-1α and sterile vascular inflammation in
atherosclerosis. Nat Immunol. 14:1045–1053. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rath M, Müller I, Kropf P, Closs EI and
Munder M: Metabolism via Arginase or nitric oxide synthase: Two
competing arginine pathways in macrophages. Front Immunol.
5:5322014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Napoli C, de Nigris F, Williams-Ignarro S,
Pignalosa O, Sica V and Ignarro LJ: Nitric oxide and
atherosclerosis: An update. Nitric Oxide. 15:265–279. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Luoma JS and Yla-Herttuala S: Expression
of inducible nitric oxide synthase in macrophages and smooth muscle
cells in various types of human atherosclerotic lesions. Virchows
Arch. 434:561–568. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Esaki T, Hayashi T, Muto E, Yamada K,
Kuzuya M and Iguchi A: Expression of inducible nitric oxide
synthase in T lymphocytes and macrophages of cholesterol-fed
rabbits. Atherosclerosis. 128:39–46. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Rom O, Grajeda-Iglesias C, Najjar M,
Abu-Saleh N, Volkova N, Dar DE, Hayek T and Aviram M:
Atherogenicity of amino acids in the lipid-laden macrophage model
system in vitro and in atherosclerotic mice: A key role for
triglyceride metabolism. J Nutr Biochem. 45:24–38. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wallace C and Keast D: Glutamine and
macrophage function. Metabolism. 41:1016–1020. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kuznetsova T, Prange KHM, Glass CK and de
Winther MPJ: Transcriptional and epigenetic regulation of
macrophages in atherosclerosis. Nat Rev Cardiol. 17:216–228. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Conrath U: Molecular aspects of defence
priming. Trends Plant Sci. 16:524–531. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
van der Heijden C, Noz MP, Joosten LAB,
Netea MG, Riksen NP and Keating ST: Epigenetics and trained
immunity. Antioxid Redox Signal. 29:1023–1040. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Heintzman ND, Hon GC, Hawkins RD,
Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW,
et al: Histone modifications at human enhancers reflect global
cell-type-specific gene expression. Nature. 459:108–112. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Rada-Iglesias A, Bajpai R, Swigut T,
Brugmann SA, Flynn RA and Wysocka J: A unique chromatin signature
uncovers early developmental enhancers in humans. Nature.
470:279–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ostuni R, Piccolo V, Barozzi I, Polletti
S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S
and Natoli G: Latent enhancers activated by stimulation in
differentiated cells. Cell. 152:157–171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Vento-Tormo R, Company C, Rodríguez-Ubreva
J, de la Rica L, Urquiza JM, Javierre BM, Sabarinathan R, Luque A,
Esteller M, Aran JM, et al: IL-4 orchestrates STAT6-mediated DNA
demethylation leading to dendritic cell differentiation. Genome
Biol. 17:42016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Dekkers KF, Neele AE, Jukema JW, Heijmans
BT and de Winther MPJ: Human monocyte-to-macrophage differentiation
involves highly localized gain and loss of DNA methylation at
transcription factor binding sites. Epigenetics Chromatin.
12:342019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Novakovic B, Habibi E, Wang SY, Arts RJW,
Davar R, Megchelenbrink W, Kim B, Kuznetsova T, Kox M, Zwaag J, et
al: β-glucan reverses the epigenetic state of LPS-induced
immunological tolerance. Cell. 167:1354–1368. e142016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zaret KS and Mango SE: Pioneer
transcription factors, chromatin dynamics, and cell fate control.
Curr Opin Genet Dev. 37:76–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Glass CK and Natoli G: Molecular control
of activation and priming in macrophages. Nat Immunol. 17:26–33.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Schmidt SV, Krebs W, Ulas T, Xue J, Baßler
K, Günther P, Hardt AL, Schultze H, Sander J, Klee K, et al: The
transcriptional regulator network of human inflammatory macrophages
is defined by open chromatin. Cell Res. 26:151–170. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Liu T, Zhang L, Joo D and Sun SC: NF-κB
signaling in inflammation. Signal Transduct Target Ther.
2:170232017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Heinz S, Benner C, Spann N, Bertolino E,
Lin YC, Laslo P, Cheng JX, Murre C, Singh H and Glass CK: Simple
combinations of lineage-determining transcription factors prime
cis-regulatory elements required for macrophage and B cell
identities. Mol Cell. 38:576–589. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ghisletti S, Barozzi I, Mietton F,
Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A,
Wei CL, et al: Identification and characterization of enhancers
controlling the inflammatory gene expression program in
macrophages. Immunity. 32:317–328. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Fanucchi S, Fok ET, Dalla E, Shibayama Y,
Börner K, Chang EY, Stoychev S, Imakaev M, Grimm D, Wang KC, et al:
Immune genes are primed for robust transcription by proximal long
noncoding RNAs located in nuclear compartments. Nat Genet.
51:138–150. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Fanucchi S, Domínguez-Andrés J, Joosten
LAB, Netea MG and Mhlanga MM: The intersection of epigenetics and
metabolism in trained immunity. Immunity. 54:32–43. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Colin S, Chinetti-Gbaguidi G and Staels B:
Macrophage phenotypes in atherosclerosis. Immunol Rev. 262:153–166.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Cochain C, Vafadarnejad E, Arampatzi P,
Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE and Zernecke A:
Single-cell RNA-seq reveals the transcriptional landscape and
heterogeneity of aortic macrophages in murine atherosclerosis. Circ
Res. 122:1661–1674. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Piccolo V, Curina A, Genua M, Ghisletti S,
Simonatto M, Sabò A, Amati B, Ostuni R and Natoli G: Opposing
macrophage polarization programs show extensive epigenomic and
transcriptional cross-talk. Nat Immunol. 18:530–540. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Czimmerer Z, Daniel B, Horvath A, Rückerl
D, Nagy G, Kiss M, Peloquin M, Budai MM, Cuaranta-Monroy I, Simandi
Z, et al: The transcription factor STAT6 mediates direct repression
of inflammatory enhancers and limits activation of alternatively
polarized macrophages. Immunity. 48:75–90. e62018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Neele AE, Van den Bossche J, Hoeksema MA
and de Winther MP: Epigenetic pathways in macrophages emerge as
novel targets in atherosclerosis. Eur J Pharmacol. 763:79–89. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mullican SE, Gaddis CA, Alenghat T, Nair
MG, Giacomin PR, Everett LJ, Feng D, Steger DJ, Schug J, Artis D
and Lazar MA: Histone deacetylase 3 is an epigenomic brake in
macrophage alternative activation. Genes Dev. 25:2480–2488. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hoeksema MA, Gijbels MJ, Van den Bossche
J, van der Velden S, Sijm A, Neele AE, Seijkens T, Stöger JL,
Meiler S, Boshuizen MC, et al: Targeting macrophage Histone
deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med.
6:1124–1132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Chistiakov DA, Kashirskikh DA, Khotina VA,
Grechko AV and Orekhov AN: Immune-inflammatory responses in
atherosclerosis: The role of myeloid cells. J Clin Med. 8:17982019.
View Article : Google Scholar : PubMed/NCBI
|














