Introduction
Pancreatic carcinoma is one of the leading causes of
cancer mortality worldwide. Pancreatic cancer has been reported to
be associated with various environmental and lifestyle risk
factors. Although the molecular etiology of pancreatic carcinoma is
unclear, age and cigarette smoking are the unequivocal risk factors
(1).
Due to the aggressive nature of the disease and the
difficulties in diagnosis, the overall 5-year survival rate of
pancreatic carcinoma is less than 5% (2,3).
Novel approaches for the diagnosis and treatment of pancreatic
cancer are necessary to improve the survival rate.
Cancer is a genetic disease where alterations in
several genes accumulate and lead to a cancer cell growth
advantage. Cell-cycle progression is driven by cyclins and
cyclin-dependent kinases (CDKs). The activities of cyclin-CDK
complexes are modulated by two classes of CDK inhibitor (CDKI)
(4). The INK4 CDKI proteins
(p15INK4, p16INK4, p18INK4 and p19INK4) sequester CDKs and inhibit
the formation of CDK-cyclin complexes, whereas the Cip/Kip CDKIs
(p21Waf1/Cip1, p27Kip1 and p57Kip2) bind to cyclin-CDK complexes
(5). p21 (also known as waf1, cip1
or sdi1) is an important cellular checkpoint molecule for the
inhibition of a range of cyclin-CDK activities. In our previous
study, we demonstrated that association of p21cip1 with CDK2/cyclin
E blocks cell-cycle progression at multiple points (6,7). The
INK4A locus encodes two unrelated proteins, p14ARF and p16INK4a.
p16INK4a is a specific inhibitor of the cyclin D-dependent kinases
CDK4 and CDK6 (8) and antagonizes
their ability to phosphorylate the retinoblastoma (Rb) family of
proteins and so prevent exit from the G1 phase of the cell cycle
(9). INK4A is important in
mediating the signals that constrain the cell cycle in response to
hyperproliferative signals, and, furthermore, are the most
frequently inactivated tumor suppressor genes in human cancer
(10,11).
Promoter methylation is an alternative form of gene
silencing, which relies on epigenetic factors. Previous reports
have revealed that aberrant INK4a or Cip/Kip promoter
methylation is a frequent event in human tumors (12). Studies have indicated that
suppressed expression by aberrant promoter methylation may be an
alternative mechanism for inactivation of the tumor suppressor gene
in pancreatic cancer cases (13).
In the present study, we reported that reduced
levels of p15INK4b, p16INK4a, p21cip1 and p27kip1 CDKI proteins and
mRNA are prominent features of pancreatic carcinoma. We observed
that promoter hypermethylation of genes was partly correlated with
decreased p15INK4b, p16INK4a and p21cip1, but
not p27kip1, mRNA in the tumors from clinical patients.
Materials and methods
Study subjects
Five frozen fresh tumor specimens were surgically
isolated from patients with pancreatic carcinoma. All of these
patients were admitted into the third General Surgery Department,
Zhongshan Hospital. Ethical approval for this study from the
Zhongshan hospital and agreement by all patients were obtained.
Western blot analysis
The tumor and human normal tissues were prepared in
lysis buffer from the MC-CelLytics kit (Shenergy Biocolor,
Shanghai, China). The protein content was determined using the
Bradford calorimetric assay method (Shenergy Biocolor). The lysate
was resolved by 10% polyacrylamide-sodium lauryl sulfate gel
electrophoresis and Immobilon-P transfer membrane (Millipore, MA,
USA). Antibodies used for detection were p15INK4b (Santa Cruz),
p16INK4a (Santa Cruz), p21cip1 (Cell Signaling) and p27kip1 (Santa
Cruz). Then, the blot was incubated with a secondary antibody,
IRDye 800 conjugated affinity purified anti-mouse or anti-rabbit
IgG (Rockland Immunochemicals, Gilbertsville, PA, USA), and
detected with an Odyssey Infrared Imaging System (LI-COR
Bioscienceces, Lincoln, NE, USA).
Real-time RT-PCR analysis
Total RNA was extracted using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) from tumor specimens and normal
tissues. RNA was reverse transcribed using a PrimeScript™ RT
reagent kit (DRR037A; Takara, Dalian, China). Real-time PCR
analysis was performed in a final volume of 25 μl, containing 2 μl
of cDNA template, 0.5 μl of each primer (10 μM) and 12.5 μl of a
SYBR-Green master mix (2X) according to the instructions of the
Real-time PCR kit (Takara, Japan) to evaluate the levels of mRNA
expression. The following primers were used: 5′-AAGCTGAGCCCAGGT
CTCCTA-3′ (forward) and 5′-CCACCGTTGGCCGTAAACT-3′ (reverse) for
p15INK4b; 5′-ACCCTTGTGCCTCGCTCAG-3′ (forward) and
5′-GGTCTGCCGCCGTTTTC-3′ (reverse) for p21cip1, and published
primers for p16INK4a (14) and
p27kip1 (15). The average amount
of the genes was normalized to the levels of GAPDH (16), an endogenous housekeeping gene.
Methylation analysis
The tumor and normal tissue DNA was extracted by
Tissue Genomic Isolation kits (Dingguo Bio-tech, Beijing, China).
For methylation-specific PCR (MSP), samples were prepared according
to the instructions of the CpGenome™ FAST DNA Modification kit
(S7824; Chemicon International, CA, USA). Bisulfite-treated DNA was
amplified using MSP primers specific for either methylated or
unmethylated DNA using published primers for p15INK4b (17), p16INK4a (17), p21cip1 (18) and p27kip1 (18). PCR was carried out with Ex-Taq Hot
Start DNA polymerase (Takara). The annealing temperatures used were
60˚C for p15 U/M, 65/60˚C for p16 U/M, 57˚C for p21 U/M and 55˚C
for p27 U/M.
Results
Reduced levels of p15INK4b, p16INK4a,
p21cip1 and p27kip1 proteins detected in pancreatic carcinoma
The events leading to pancreatic carcinoma
development remain largely unknown. Several studies have shown that
the Rb tumor-suppressive pathway is abrogated in almost all studied
cases of pancreatic carcinoma, and this disruption is caused
exclusively by inactivation of CDKI (19). p15INK4b, p16INK4a, p21cip1 and
p27kip1 are considered the most important CDKIs. This led us to ask
whether these CDKIs are involved in pancreatic tumor formation. We
examined p15INK4b, p16INK4a, p21cip1 and p27kip1 expression in 5
pancreatic carcinoma specimens and controls using western blotting.
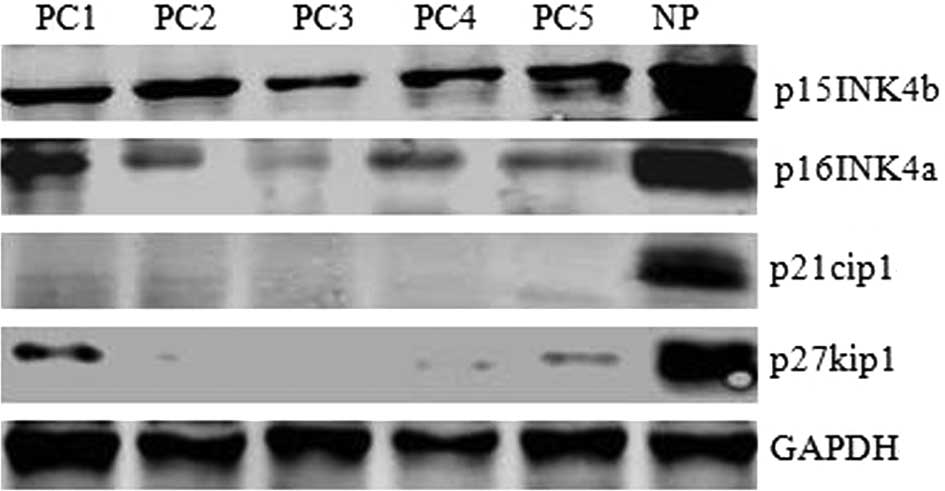
As shown in Fig. 1, p15INK4b,
p16INK4a, p21cip1 and p27kip1 protein levels in 5 pancreatic
carcinoma specimens (PC1-5) were much lower than in normal human
pancreatic tissue (NP). These data suggest that reduced levels of
p15INK4b, p16INK4a, p21cip1 and p27kip1, the negative regulators of
cell progression, may be causative of tumor formation in pancreatic
carcinoma.
Reduced levels of p15INK4b, p16INK4a,
p21cip1 and p27kip1 mRNA expression detected in pancreatic
carcinoma
Subsequently, we were interested in whether aberrant
mRNA expression levels contributed to these CDKI alterations in
pancreatic carcinoma specimens from patients. We evaluated
p15INK4b, p16INK4a, p21cip1 and p27kip1
mRNA by real-time RT-PCR analysis to determine whether decreased
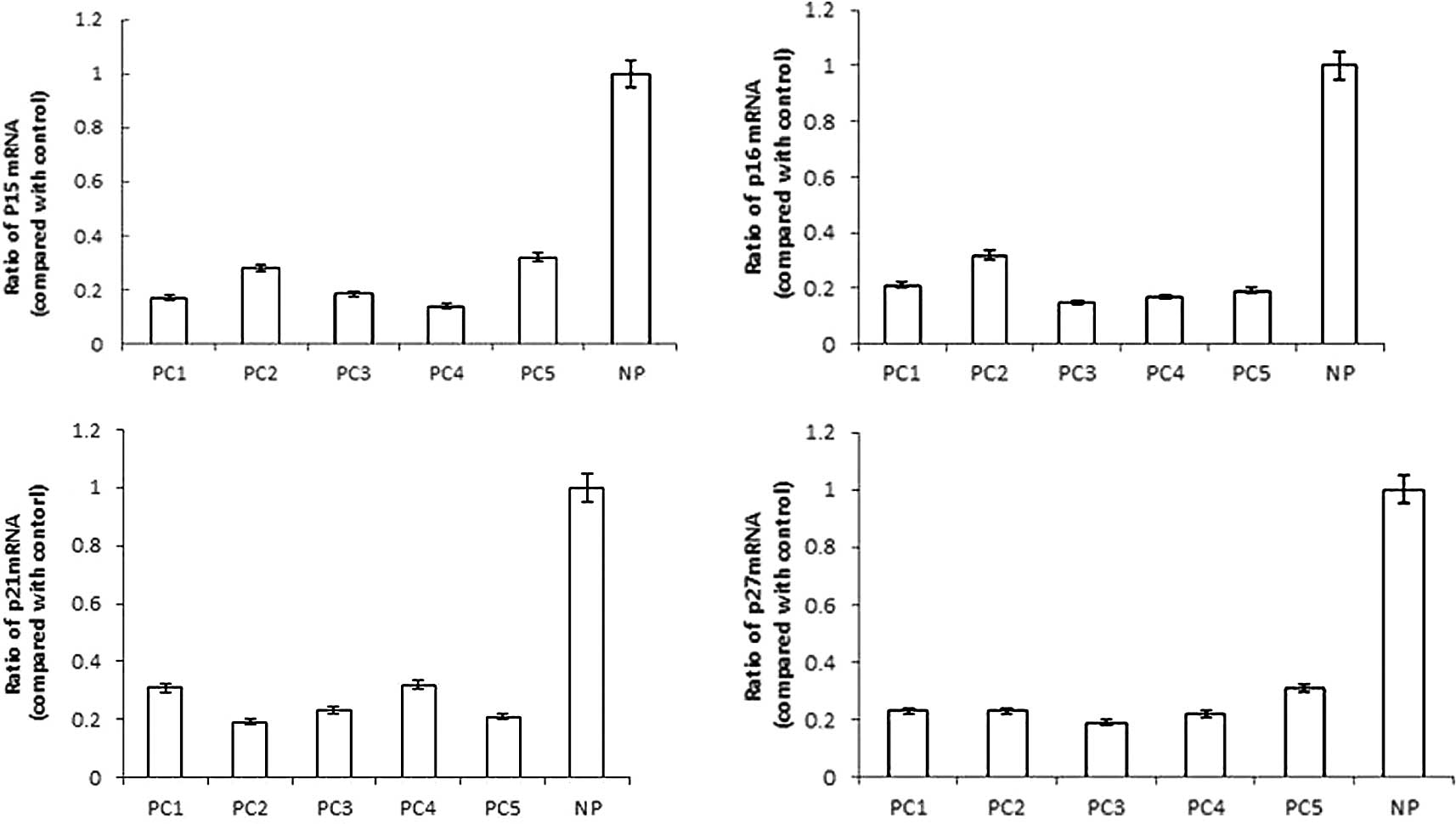
mRNA accumulation contributed to their protein levels. As shown in
Fig. 2, it was preceded by a
>3-fold reduction in p15INK4b mRNA expression, a
>4-fold reduction in p16INK4a mRNA expression, a
>3-fold reduction in p21cip1 mRNA expression and a
>4-fold reduction in p27kip1 mRNA expression of PC
specimens, as compared to human NP tissue. Our findings suggested
that the reduced levels of p15INK4b, p16INK4a, p21cip1 and p27kip1
proteins were likely due to a reduction in their mRNA synthesis,
stability or translation in pancreatic carcinoma.
Methylation status of the p15INK4b,
p16INK4a, p21cip1 and p27kip1 promoter region in pancreatic
carcinoma
To explore the mechanism associated with the
transcriptional silencing of the p15INK4b, p16INK4a,
p21cip1 and p27kip1 genes, we examined 5 PC and human
NP tissue samples to see whether there was methylation alteration
in a CpG-rich region of the transcription initiation site of the
these genes. The presence or absence of methylation in the
promoters of the p15INK4b, p16INK4a, p21cip1
and p27kip1 genes was determined by MSP. MSP distinguishes
unmethylated from methylated alleles based on sequence changes
produced following bisulfite treatment of DNA, which converts
unmethylated cytosine to uracil, and subsequent PCR using primers
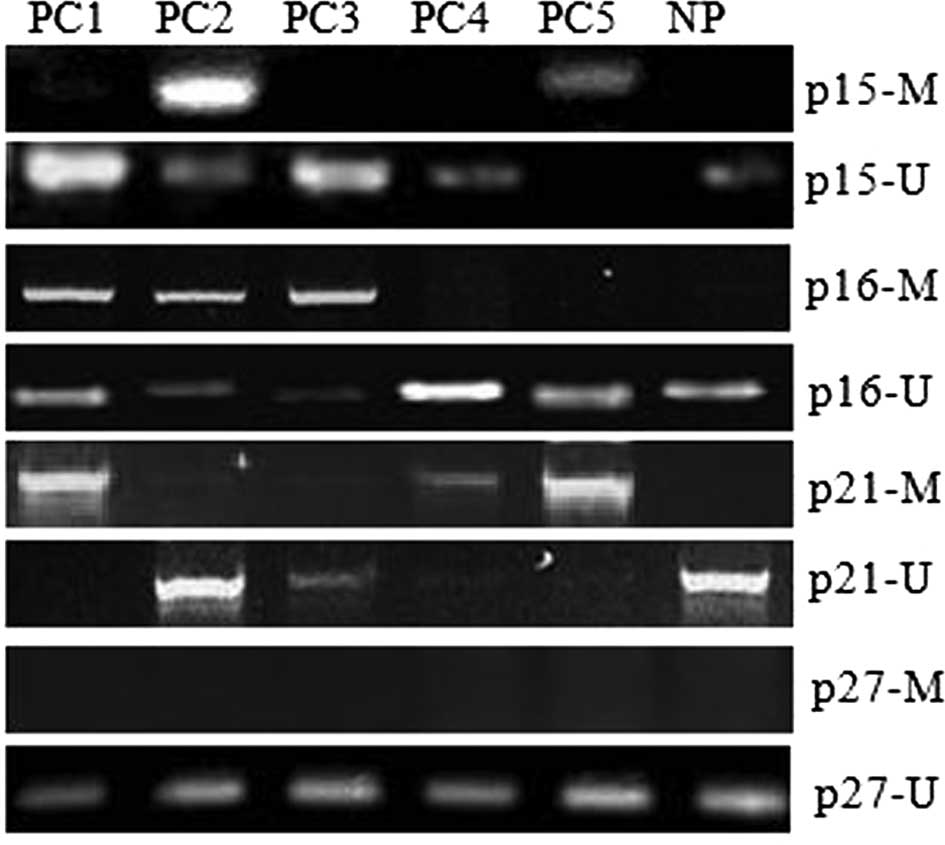
designed for either methylated or unmethylated DNA. As shown in
Fig. 3, promoter hypermethylation
was detected in PC2 and PC5 samples in the p15INK4b gene,
PC1, 2 and 3 samples in the p16INK4a gene or PC1, 4 and 5
samples in the p21cip1 gene, while no detectable promoter
hypermethylation of the p27kip1 gene was found in all 5 PC
samples. No detectable promoter hypermethylation of the
p15INK4b, p16INK4a, p21cip1 and p27kip1 genes
was found in human NP tissue samples (Fig. 3). Thus, aberrant DNA promoter
hypermethylation of the p15INK4b, p16INK4a and
p21cip1 genes was thought to play a role in several cases of
pancreatic carcinoma that had markedly decreased expression of
p15INK4b, p16INK4a and p21cip1 mRNA,
concomitant with loss of p15INK4b, p16INK4a and p21cip1 proteins,
but not p27kip1 protein (Figs. 2
and 3). Thus, we suggest that DNA
hypermethylation associated with transcriptional silencing of the
p15INK4b, p16INK4a and p21cip1 genes may
partly contribute to pancreatic carcinoma progression.
Pancreatic carcinoma patients with
smoking habits displayed methylation of 2 CDKI genes
According to our results, pancreatic carcinoma
patients showed reduced levels of p15, p16, p21 and p27 mRNA or
proteins. To investigate the correlation between smoking habit and
the methylation status of the promoter region in pancreatic
carcinoma, these 5 randomly selected pancreatic carcinomas removed
from the patients were further studied. As shown in Table I, the patients having smoked for
>15 years (PC1, 2 and 5) displayed methylation of 2 genes in
their pancreatic carcinoma specimens. On the other hand, the other
2 pancreatic carcinoma specimens (PC3 and 4), with only 1
methylation region in the CDKI genes, occurred in the
patients without a cigarette smoking record (Table I). Thus, we suggest that cigarette
smoking may be associated with pancreatic tumorigenesis by inducing
the methylation of the promoter regions of the CDKI
genes.
 | Table IAssociation between smoking and
methylation status. |
Table I
Association between smoking and
methylation status.
| No. | Gender | Age (years) | Cigarette
smoking | p15a | p16a | p21a |
|---|
| 1 | M | 42 | Yes | × | ✓ | ✓ |
| 2 | F | 62 | Yes | ✓ | ✓ | × |
| 3 | F | 49 | No | × | ✓ | × |
| 4 | F | 60 | No | × | × | ✓ |
| 5 | M | 56 | Yes | ✓ | × | ✓ |
Discussion
In this study, we revealed that the reduced levels
of two classes of CDKI protein, including p15INK4b, p16INK4a,
p21cip1 and p27kip1, are a prominent feature of pancreatic
carcinomas. Moreover, a reduced amount of p15INK4b,
p16INK4a, p21cip1 and p27kip1 mRNA expression
was found in all of the pancreatic carcinoma samples. We observed
that hypermethylation of the p15INK4b, p16INK4a and
p21cip1 promoters, but not the p27kip1 promoter, was
partly correlated with markedly decreased mRNA expression. Thus, we
suggest that hypermethylation associated with transcriptional
silencing of the p15INK4b, p16INK4a and
p21cip1 gene may contribute to the progression of certain
pancreatic carcinomas.
Due to the few treatment options for pancreatic
carcinoma, understanding of the molecular pathology is a
prerequisite for identifying potential molecular targets for drug
therapy. CDKIs are negative regulators of cell-cycle progression at
the G1-S restriction point. In this study, we showed that the
reduced levels of p15INK4b, p16INK4a, p21cip1 and p27kip1 proteins
were a prominent feature in the 5 pancreatic carcinoma specimens
analyzed (Fig. 1), indicating that
the reduced levels of p15INK4b, p16INK4a, p21cip1 and p27kip1, the
negative regulators of cell progression, are involved in the tumor
formation in pancreatic carcinoma.
Moreover, a reduced amount of p15INK4b,
p16INK4a, p21cip1 and p27kip1 mRNA normal
expression was found in 5 pancreatic carcinoma samples (Fig. 2). We observed patients with a
markedly decreased >3-fold reduction in p15INK4b mRNA, a
4-fold reduction in p16INK4a mRNA, a 3-fold reduction in
p21cip1 mRNA and a 4-fold reduction in p27kip1 mRNA,
concomitant with reduced levels of these CDKI proteins in sporadic
pancreatic carcinoma (Figs. 1 and
2). The methylation status of the
p15INK4b, p16INK4a, p21cip1 and p27kip1
promoter region was detected using MSP. The DNA hypermethylation
promoter was found in 40% (2 of 5) of the p15INK4b genes,
60% (3 of 5) of the p16INK4a genes, 60% (3 of 5) of the
p21cip1 genes and 0% (0 of 5) of the p27kip1 genes,
and markedly correlated with decreased mRNA expression in
pancreatic carcinoma patients (Figs.
2 and 3). Moreover, all 5
pancreatic carcinoma patients (100%; 5/5) displayed methylation of
1 or more genes, 60% (3/5) displayed methylation of 2 genes, while
control tissue did not display any methylation. It has been shown
that aberrant methylation is the most prominent feature of
pancreatic carcinoma, causing alterations in the expressions of
genes (13). Thus, we demonstrated
that the epigenetic modification of p15INK4b,
p16INK4a and p21cip1 via hypermethylation represents
a critical mechanism for, at least in part, the inactivation of
these genes in pancreatic carcinoma. In all 5 of our pancreatic
carcinoma patients with markedly decreased expression of
p27kip1 mRNA, we were unable to detect hypermethylation in
the promoter region, suggesting an alternative mechanism of the
p27kip1 gene in these patients.
Pancreatic carcinoma has been reported to be
associated with various environmental and lifestyle risk factors,
occupational exposures and medical conditions; however, the only
risk factors consistently reported are age and smoking status, and
the etiology of the disease remains largely unknown (1). In this study, we analyzed the
correlation between smoking status and biological events in 5
randomly selected sporadic pancreatic carcinomas. Indeed, the
patients smoking cigarettes for more than 15 years (PC1, 2 and 5)
displayed methylation of 2 genes in their pancreatic carcinoma
specimens. On the other hand, the other 2 pancreatic carcinoma
specimens (PC3 and 4) with only 1 methylation region in the
CDKI genes were surgically isolated from the patients
without a cigarette smoking record (Table I). Thus, we suggest that cigarette
smoking may be associated with pancreatic tumorigenesis by inducing
the methylation of the promoter regions of the CDKI
genes.
Our previous data suggested that expression of
p21cip1 stops cell growth progression by inactivation of CDK
activity, which in turn blocks the cell cycle at the G1 and G2
phases (20,21). Consistent with our previous finding
that heightened CDK/cyclin signal transduction concomitant with
loss of p27kip1 (22), the present
study indicates that the reduced levels of the CDKI protein are a
prominent feature of pancreatic carcinoma. These data suggest that
the reduced levels of p15INK4b, p16INK4a, p21cip1 and p27kip1 may
lead to defects in cell-cycle regulation and confer a selective
growth advantage for pancreatic cancer cells. Thus, we showed that
reduced levels of p15INK4b, p16INK4a, p21cip1 and p27kip1, the
negative regulators of cell progression, may be causative of tumor
formation in pancreatic cancer cells.
Taken together, reduced levels of p15INK4b,
p16INK4a, p21cip1 and p27kip1 are a fundamental event in tumor
formation in the clinical pancreatic carcinoma patients. The CDK
inhibitors may interact to mediate signals that are critical growth
inhibitors. This growth regulatory circuit would be disrupted, such
as in pancreatic carcinoma. Thus, our observations have significant
implications for understanding the importance of p15INK4b,
p16INK4a, p21cip1 and p27kip1; reduced levels of these CDKI
proteins contribute to tumorigenesis in pancreatic carcinoma and
developing ways to target aberrantly active parts of this growth
regulatory pathway may lead to increased survival.
Acknowledgements
This study was supported by the Natural Science
Foundation of China (81071740) and the Shanghai Science Foundation
(10ZR1406300).
References
|
1
|
P GhadirianHT LynchD KrewskiEpidemiology
of pancreatic cancer: an overviewCancer Detect
Prev278793200310.1016/S0361-090X(03)00002-312670518
|
|
2
|
AF HezelAC KimmelmanBZ StangerN BardeesyRA
DepinhoGenetics and biology of pancreatic ductal
adenocarcinomaGenes
Dev2012181249200610.1101/gad.141560616702400
|
|
3
|
A JemalR SiegelE WardCancer statisticsCA
Cancer J Clin5871962008
|
|
4
|
SJ ElledgeJ WinstonJW HarperA question of
balance: the role of cyclin-kinase inhibitors in development and
tumorigenesisTrends Cell
Biol6388392199610.1016/0962-8924(96)10030-115157521
|
|
5
|
JW HarperGR AdamiN WeiK KeyomarsiSJ
ElledgeThe p21 Cdk-interacting protein Cip1 is a potent inhibitor
of G1 cyclin-dependent
kinasesCell75805816199310.1016/0092-8674(93)90499-G8242751
|
|
6
|
JJ TangC ShenYJ LuRequirement for
pre-existing of p21 to prevent doxorubicin-induced apoptosis
through inhibition of caspase-3 activationMol Cell
Biochem291139144200610.1007/s11010-006-9206-716909308
|
|
7
|
Y LuN YamagishiT YagiH TakebeMutated
p21(WAF1/CIP1/SDI1) lacking CDK-inhibitory activity fails to
prevent apoptosis in human colorectal carcinoma
cellsOncogene16705712199810.1038/sj.onc.12015859488034
|
|
8
|
M SerranoGJ HannonD BeachA new regulatory
motif in cell-cycle control causing specific inhibition of cyclin
D/CDK4Nature366704707199310.1038/366704a08259215
|
|
9
|
CJ SherrJM RobertsCDK inhibitors: positive
and negative regulators of G1-phase progressionGenes
Dev1315011512199910.1101/gad.13.12.150110385618
|
|
10
|
P HainautT SoussiB ShomerDatabase of p53
gene somatic mutations in human tumors and cell lines: updated
compilation and future prospectsNucleic Acids
Res25151157199710.1093/nar/25.1.1519016527
|
|
11
|
M HallG PetersGenetic alterations of
cyclins, cyclin-dependent kinases, and Cdk inhibitors in human
cancerAdv Cancer
Res6867108199610.1016/S0065-230X(08)60352-88712071
|
|
12
|
F PerroneS TabanoF Colombop15INK4b,
p14ARF, and p16INK4a inactivation in sporadic and neurofibromatosis
type 1-related malignant peripheral nerve sheath tumorsClin Cancer
Res941324138200314519636
|
|
13
|
B KlumpCJ HsiehO NehlsMethylation status
of p14ARF and p16INK4a as detected in pancreatic secretionsBr J
Cancer88217222200310.1038/sj.bjc.660073412610506
|
|
14
|
Y KuriharaK EgawaS KunimotoT TakeuchiK
NoseInduction of p16/INK4a gene expression and cellular senescence
by toyocamycinBio Pharm
Bull2512721276200210.1248/bpb.25.127212392077
|
|
15
|
B CenH LiIB WeinsteinHistidine triad
nucleotide-binding protein 1 up-regulates cellular levels of
p27KIP1 by targeting ScfSKP2 ubiquitin ligase and SrcJ Biol
Chem28452655276200919112177
|
|
16
|
AB WestG KapatosC O'FarrellN-myc regulates
parkin expressionJ Biol Chem2792889628902200415078880
|
|
17
|
JG HermanJR GraffS MyöhänenBD NelkinSB
BaylinMethylation-specific PCR: a novel PCR assay for methylation
status of CpG islandsProc Natl Acad Sci
USA939821982619968790415
|
|
18
|
K BrakensiekF LängerH KreipeU
LehmannAbsence of p21CIP1, p27KIP1 and p57KIP2 methylation in MDS
and AMLLeuk Res2913571360200515936816
|
|
19
|
G SchneiderRM SchmidGenetic alterations in
pancreatic carcinomaMol Cancer2215200310.1186/1476-4598-2-15
|
|
20
|
H WuY ChenZY WangInvolvement of p21 (waf1)
in merlin deficient sporadic vestibular
schwannomasNeuroscience170149155201020600642
|
|
21
|
Y JiSL MaYP ZhangA combined treatment
TNF-α/Gefitinib alleviates the resistance to Gefitinib in PC-9
cells with acquired resistance to GefitinibAnticancer
Drug208328372009
|
|
22
|
Z WangY LuJ TangH WangH WuThe
phosphorylation status of merlin in sporadic vestibular
SchwannomasMol Cell Biochem324201206200919142715
|