1. Introduction
Epigenetics constitutes an important mechanism
capable of regulating gene transcription, linking early life’s
events to adult morbidity. It entails heritable changes in
chromatin that alter gene expression without altering the DNA
sequence (1,2). It is the best-characterized
epigenetic modification. Evidence suggests that DNA methylation is
closely involved in the regulation of gene expression and that DNA
methylation patterns can be distorted during the pathogenetic
process of a disease (3). Findings
of previous reports suggest that DNA methylation is altered during
development and by environmental stress (4,5).
However, the mechanisms by which these epigenetic effects are
exerted remain to be clarified. In this review, we briefly present
the available evidence regarding the role of DNA methylation
patterns of the placenta on aberrant fetal growth.
2. DNA methylation
DNA methylation, which is accomplished by DNA-
methyltransferases, occurs on the cytosine residues of CG (also
designated CpG) dinucleotides. Enzymes known as DNA
methyltransferases (DNMTs) catalyse the addition of a methyl group
to the cytosine ring to form methyl cytosine, using
S-adenosylmethionine as a methyl donor (6). DNA methyltransferase-1 (DNMT1) is the
predominant mammalian DNA methylating enzyme responsible for the
restoration of hemi-methylated sites to full methylation, termed
maintenance methylation, which occurs after DNA replication. DNMT3A
and DNMT3B are mainly involved in the methylation of new sites,
known as de novo methylation (7). DNMT3L is postulated to play a
regulatory role in DNA methylation without DNA methyltransferase
activity in itself. In humans and other mammals, DNA modification
occurs predominantly on cytosines that precede a guanosine in the
DNA sequence (6). These
dinucleotides can be clustered in small stretches of DNA, termed
CpG islands, which are often associated with promoter regions. In
98% of the genome, CpGs are present approximately once per 80
dinucleotides. By contrast, CpG islands, which comprise 1–2% of the
genome, are approximately 200 base pairs (bp) to several kb in
length and have a frequency of CpGs approximately five times
greater than the genome as a whole (8,9).
Most CpG sites outside the CpG islands are methylated, suggesting a
role in the global maintenance of the genome, while most CpG
islands in gene promoters are unmethylated, which allows active
gene transcription (6,10). When a CpG becomes methylated in a
cell, it remains methylated in all its descendants (11). Generally, when a given stretch of
cytosines in a CpG island located in the promoter region of a gene
is methylated, that gene is silenced by methylation; such a CpG
island would be termed ‘hypermethylated’. Conversely, when a given
stretch of cytosines in a CpG island located in the promoter region
of a gene is not methylated, that gene is not silenced by
methylation; the CpG island in this case would be ‘hypomethylated’
(12). Methylation of promoters
inhibits their recognition by transcription factors and RNA
polymerase, as methylated cytosines preferentially bind to a
protein known as methyl cytosine binding protein, or MeCP. When a
promoter region normally recognized by an activating transcription
factor, is methylated, its transcription is inhibited (9).
3. DNA methylation in the developing embryo
and placenta
Methylation of gene promoters is probably one of the
foremost mechanisms responsible for cell differentiation during
embryogenesis: the transcription of unwanted genes is eliminated by
methylation of their promoters (13). As oocytes and spermatozoa are more
differentiated than the pluripotent cells of the early embryo, the
DNA of morula (16-cell embryo, third day post-conception) undergoes
global demethylation. CpGs are demethylated on a large scale, thus
reactivating the near-totality of the genome (a few genes escape
this demethylation, e.g., the genes subject to genomic imprinting).
Subsequently, as cells start differentiating, the gene promoters
involved in this differentiation become methylated according to a
strict sequence depending on each cell type (14).
On fertilisation a rapid paternal-specific
asymmetric loss of methylation is observed (15,16).
This process occurs in the absence of transcription or DNA
replication and is termed active demethylation. Thereafter, there
is a step-wise decline in methylation until the morula stage
(17,18). The initiation of the de novo
methylation occurs after the fifth cell cycle and coincides with
the time of the first differentiative event. The establishment of
the first two cell lineages results in a significant asymmetry. The
inner cell mass (ICM), which gives rise to all the tissues of the
adult, becomes hypermethylated, while the trophectoderm (TE), which
forms most of the structure of the placenta, is hypomethylated
(17,18). This differential methylation is
maintained and reflected in highly methylated somatic tissues and
the distinctively hypomethylated extra-embryonic tissues of the
placenta. This epigenetic inequality with higher overall DNA
methylation levels in the embryo compared with the placenta is
maintained throughout gestation (18).
4. Imprinting
Despite the genome-wide decline in DNA methylation,
certain sequences remain refractory to the general demethylation
during preimplantation development. Imprinted genes escape this
epigenetic reprogramming (15).
They are protected from demethylation because it is crucial that
the parental imprints are preserved in the developing embryo
(19).
Genomic imprinting refers to silencing of one
parental allele in the zygotes of gametes leading to monoallelic
expression of these genes in the offspring. During the process of
imprinting, the male and female germ line confer a gender-specific
mark (imprint) on certain chromosomal regions (20). Only one allele of the imprinted
genes, the maternal or paternal, can be active and expressed. Each
cluster is controlled by an imprinting control region (ICR) that
usually contains a stably maintained or developmentally changing
Differentially Methylated Region (DMR) (21,22).
Genomic imprinting arose during the mammalian evolution
(approximately 150 million years ago) and may be associated with
the evolution of intrauterine development that requires the
formation of a placenta (20).
The prevailing hypothesis on the evolution of
genomic imprinting is the ‘conflict hypothesis’ theory. This theory
suggests that paternally expressed genes strongly favor using
maternal resources to benefit offspring, while maternally expressed
genes attempt to preserve such maternal resources and thus, are in
direct conflict with one another. Many imprinted genes are involved
in fetal development and growth, and some affect behaviour
(20,23). Imprinting appears to be
particularly important for placental development (24,25).
Knockout studies of several paternally or maternally imprinted
genes result in intrauterine growth restriction (IUGR) and smaller
placental size or the overgrowth and hyperplasia of the placenta,
respectively (25–27). Certain maternal genes are required
for proper development of the embryo, whereas extraembryonic
tissues depend on the presence of active paternal genes.
Approximately 60 genes have been shown to be imprinted in humans,
two thirds of which are paternally expressed (maternally imprinted)
and one third maternally expressed (paternally imprinted) (28).
5. DNA methylation in the placenta
Throughout in utero development, the placenta
plays an important role in controlling growth and development
through the transfer of nutrients and waste, and in protecting the
fetus from insults (29). Findings
of recent studies have shown that placental genetic and epigenetic
profiles may serve as markers of the intrauterine and extrauterine
environment (30–32). Embryonic and fetal growth depends
on genetic, epigenetic and environmental factors, and the process
is the result of the interaction between these factors.
Approximately 7–9% of live-born infants have a birth weight below
the 10th percentile. Intrauterine growth restriction describes a
decrease in the fetal growth rate that prevents an infant from
obtaining his or her complete growth potential (33). IUGR infants are small for
gestational age (SGA) if their birth weight measures <10 to 3%
using standard growth curves (34,35).
Therefore, the terms IUGR and SGA are related but not synonymous.
The IUGR is a pathological condition, whereas SGA may reflect a
normal pattern in a given population. The placenta forms the
interface between the fetal and maternal circulations. For this
reason, fetal disease, maternal disease, primary placental disease,
and extrinsic factors could all interfere with the efficiency of
nutrient and waste exchange and result in growth restriction
(36,37). Fetal growth restriction is a
physical sign rather than a single disease. Improper placental
function accounts for the majority of IUGR cases.
Epigenetic modification in the placenta may provide
an attractive mechanism linking environmental cues to placental
pathology, with consequences for fetal growth and adult life.
Accumulating evidence suggests that the maternal nutritional status
is capable of altering the epigenetic state of the fetal genome and
imprinted gene expression. Epigenetic alterations in early embryos
may be carried forward to subsequent developmental stages (38). The placenta has been reported to
present high variability in overall DNA methylation compared to
other tissues (39), probably in
response to its role in mediating the conflicting demands of mother
and fetus (40). Methylation
patterns of several genes (imprinted and non-imprinted) in the
placenta have been investigated in an attempt to elucidate the
exact role of epigenetic modifications on fetal growth.
Administration of a DNA methyltransferase inhibitor to pregnant
rats at different gestational ages resulted in significantly
smaller placentas and histological evaluation showed the
labyrinthine part of the placenta to be severely reduced (41). In a similar study, a lack of the
labyrinth layer was observed with a strong proliferative activity
of the cells in the basal layer or complete disruption of the
placental structure (42).
Furthermore, administration of the same agent in human
choriocarcinoma-derived cell lines, resulted in disrupted
trophoblast migration (43).
DNA methylation and gene
transcription
Much of the recent research on placental epigenetics
has focused on imprinted genes that are known to affect growth,
such as insulin-like growth factor 2 (IGF2). IGF2 and
H19 are two oppositely expressed imprinted genes located
adjacent to each other at 11p15.5 that share the same transcription
regulatory epigenetic mechanisms and have an important role in
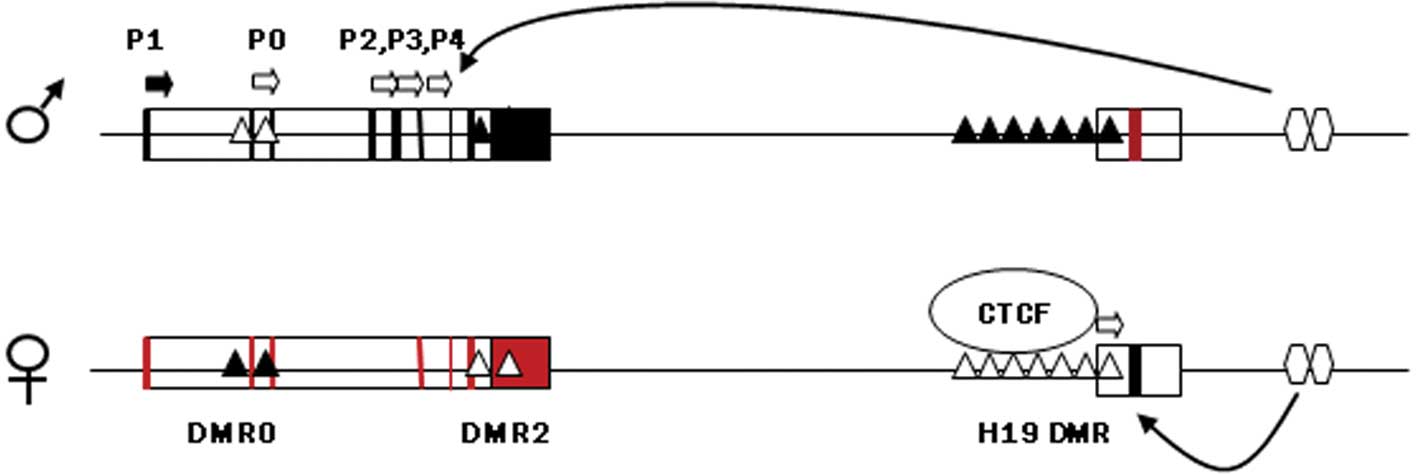
feto-placental development. The DMR upstream of H19, which harbors
sequences known to bind to the zinc finger protein CTCF, if
methylated on the paternal chromosome prevents binding with CTCF
and allows the IGF2 promoter to assess enhancers located
downstream of H19, thereby expressing IGF2 (Fig. 1). On the maternal chromosome the
non-methylated H19 DMR is bound to CTCF, thus insulating the
IGF2 promoter from the 3′ enhancers and allowing the H19
promoter unimpeded access to the enhancers. Maternal H19 is
subsequently transcribed (44).
IGF2 is highly expressed in normal mouse and human placenta
and affects the functional capacity of the placenta to transfer
nutrients to the fetus as well as placental size (45,46).
It is expressed in most tissues only from the paternal allele, with
the maternal allele being transcriptionally silent. The maternally
expressed H19 gene itself does not code a protein, but the
RNA has growth-suppressing functions, potentially through
inhibiting the translation of IGF2 RNA (47).
Specific regions of differential DNA methylation are
regarded as critical for the correct allelic expression of
IGF2/H19. Complete loss of methylation of the
H19 promoter is reported at all stages of placental
development (48). Hypomethylation
of IGF2 and H19 promoters as well as the ICR of those
genes has been reported in placentas derived from pregnancies
complicated with fetal growth restriction (49–51).
Since ICR controls the expression of both H19 and
IGF2 genes, which are known growth modulators, aberrant
methylation in that region may be a potential link between
epigenetic modifications and abnormal fetal and placental
growth.
Hypomethylation of the IGF2 and H19
promoters would indicate lower transcription levels of these genes
in placentas from pregnancies complicated with fetal growth
restriction. Underexpression of IGF2 is a repeated event in
growth-restricted placentas and it is postulated to be associated
with reduced diffusional capacity of the placenta, which in turn
affects fetal growth (52).
However, hypomethylation of the IGF2 promoter is in contrast
to the reduced transcription levels observed in placentas derived
from pregnancies with abnormal fetal growth. This discrepancy
suggests that there is no direct correlation between methylation
and imprinted gene expression in the placenta, and other mechanisms
may be involved in this sequence of molecular events. According to
recent studies, methylation of the IGF2/H19 promoters
is not prerequisite for the regulation of the imprinting domain
that controls transcription of the two genes in human placenta
(49,53). Moreover, hypomethylation of the
same regions does not have an impact on the expression pattern of
IGF2 and H19 (53).
Reduced methylation levels of the region that
controls the imprinting of IGF2/H19 (ICR) have been
reported in placentas derived from pregnancies with poor fetal
growth and those complicated with preeclampsia (49–51,54)
(Table I). ICR is hypomethylated
leading to the repression of IGF2 expression in
approximately one-third of patients with Silver Russell syndrome
(SRS), a syndrome associated with pre- and post-natal growth
deficiency (57). However, where
ICR is hypermethylated it leads to an increase in IGF2
expression in some cases of pre- and post-natal overgrowth
diagnosed as Beckwith-Wiedemann syndrome (BWS) (58). Methylation at ICR has been shown in
a number of studies to be particularly responsive to environmental
factors such as culture media (59), environmental toxins (60,61),
and prenatal ethanol exposure (61). The reduced methylation levels
associated with IUGR may reflect an adaptive process serving to
adjust placental and fetal growth in response to poor placental
perfusion.
 | Table IMethylation patterns of genes
expressed in the placenta of pregnancies that delivered a
growth-restricted or small for gestational age neonate.a |
Table I
Methylation patterns of genes
expressed in the placenta of pregnancies that delivered a
growth-restricted or small for gestational age neonate.a
| Gene | Imprinting | Tissue | Methylation | Expression | Author/Year
(Refs.) |
|---|
|
IGF2/H19 (ICR) | Yes | IUGR and control
placentas |
Hypomethylation | Decreased
(IGF2) | Bourque et
al, 2010 (54) |
|
IGF2/H19 (ICR) | Yes | IUGR + PET and
control placentas | No difference | | |
| CDKN1C (ICR) | Yes | IUGR and control
placentas | No difference | No difference | |
| H19 | Yes | IUGR and control
placentas | No difference | | |
| CDKN1C | Yes | IUGR and control
placentas | No difference | | |
| PEG10 | Yes | IUGR and control
placentas | No difference | | |
| PLAGL1 | Yes | IUGR and control
placentas | No difference | | |
| SNRPN | Yes | IUGR and control
placentas | No difference | Increased | |
| MEST | Yes | IUGR and control
placentas | No difference | Increased | |
| PHLDA2 | Yes | IUGR and control
placentas | | Increased | McMinn et
al, 2006 (55) |
| MEST | Yes | IUGR and control
placentas | No difference | Decreased | |
| SERPINA3 | No | IUGR and control
placentas |
Hypomethylation | Increased | Chelbi et
al, 2007 (56) |
| SERPINA3 | No | IUGR+ preeclampsia
and control placentas |
Hypomethylation | Increased | |
| IGF2 | Yes | SGA placentas and
neonatal blood |
Hypomethylation | | Guo et al,
2008 (49) |
| H19 | Yes | SGA placentas and
neonatal blood |
Hypomethylation | | |
| IGF2 | Yes | SGA and control
placentas | - | Decreased | |
| H19 | Yes | SGA and control
placentas | - | No difference | |
| H19 | Yes | IUGR and control
placentas |
Hypomethylation | Increased | Koukoura et
al, 2011 (51) |
|
IGF2/H19 (ICR) | Yes | IUGR and control
placentas | No difference | | |
| IGF2 | Yes | IUGR and control
placentas |
Hypomethylation | Decreased | Koukoura et
al, 2001 (50) |
Hypomethylation of the imprinted gene promoters is
not a universal finding in cases where fetal growth is compromised.
In their study, Lambertini et al demonstrated a slight
tendency towards hypermethylation of the DMRs of all known
imprinted genes identified to be expressed in growth-restricted
placentas (62). These authors
suggested that differential methylation changes in
growth-restricted placentas occur throughout the genomic regions,
encompassing genes actively expressed in the placenta. Analysis of
other imprinted genes from the placentas of pregnancies complicated
with IUGR, revealed a lack of altered DNA methylation at their
imprinting centers (55), although
they demonstrated differences in their transcription levels
(Table I).
The contradictory results that stem from different
studies regarding imprinted gene methylation patterns in the
placenta highlight the already reported DNA methylation variation
at the imprinted genes. Inter-individual, tissue-specific variation
in DNA-methylation level is widespread in the human genome, with
implications on phenotypic variation and disease (63). Several genes have been described to
exhibit this polymorphic pattern of DNA methylation in the human
placenta (64). Therefore, the
exact epigenetic defects in the human placenta, which control
imprinted gene expression and affect fetal development, remain to
be determined.
The hypothesis that variation in the DNA methylation
profile of human term placenta can serve as a marker of growth has
been confirmed by Banister et al who demonstrated a pattern
of methylation of 22 critical loci in human term placentas.
Specific methylation alterations of these genes were highly
predictive of IUGR or SGA (65). A
significant association has also been shown between the
differential methylation of the glucocorticoid receptor gene in the
placenta and Large for Gestational Growth (LGA) infants (31). Placental gene serine protease
inhibitor A3 (SERPINA3), whose expression is known to be
affected by placental pathologies such as preeclampsia, has been
shown to exhibit hypomethylation of its promoters in IUGR
placentas. Hypomethylation coincided with increased transcription
levels of the same gene in placentas derived from IUGR pregnancies
as well as preeclamptic IUGR cases. The hypomethylated CpGs were
found to be located at putative binding sites for developmental and
stress response (hypoxia and inflammation) factors (56).
Recent studies have demonstrated significant
associations between infant growth, in utero exposures and
repetitive element methylation in placental tissue (66). These DNA repetitive elements are
made up of interspersed and tandem repeats and comprise at least
half of the human genome (67).
Interspersed repeats are composed of long interspersed nuclear
elements (LINEs) and short interspersed nuclear elements (SINEs). A
significant correlation was found between methylation levels and
the birthweight percentile. A 10% methylation increase in
LINE-1 mean levels caused the birthweight percentile to
significantly increase by 9.7. Similarly, a 10% methylation
increase in AluYb8 mean levels caused the birthweight
percentile to significantly increase by 14.5. Furthermore, mean
AluYb8 levels differed significantly due to maternal tobacco
use during pregnancy; whereas, mean LINE-1 levels only
significantly differed due to maternal alcohol use during
pregnancy. Authors of these studies concluded that the alterations
may reflect underlying functional epigenetic alterations to genes
important in placental growth and development.
Previous investigations emphasized marked
similarities between the proliferative, migratory and invasive
properties of placental cells and those of cancer cells.
Alterations in the expression of tumour suppressor gene expression
profiles have been identified in placentas from preeclamptic
pregnancies (68). A distinct
pattern of tumour-associated methylation, linking a coordinated
series of epigenetic silencing events, similar to those associated
with some tumours, in the distinct, features of normal human
placental invasion and function has been observed (69). A genome-wide methylation analysis
revealed reduced methylation levels of trophoblastic tissues
derived from chorionic villous sampling during the first trimester
of pregnancy. The highly proliferative and invasive nature of early
placenta may explain this relative hypomethylation as a requirement
for an intensively active transcriptional state. Trophoblasts and
cancer cells may use common epigenetic modifications to facilitate
their proliferative, migratory and invasive properties (70). However, no data are currently
available that may indicate a correlation between the epigenetic
modification of tumor-associated genes and fetal growth. In a study
where the methylation status of genes regulating vitamin D
bioavailability and activity in the placenta was investigated, the
CYP24A1 gene was methylated in human placenta, purified
cytotrophoblasts, and primary and cultured chorionic villus
sampling tissue, whereas vitamin D receptor (VDR) and
CYP27B1 genes were non-methylated. All three genes were
hypermethylated in choriocarcinoma cell lines, emphasizing the role
of vitamin D deregulation in this type of cancer. The promoter
methylation of the CYP24A1 gene, directly downregulated
basal promoter activity and abolished vitamin D-mediated feedback
activation. This event resulted in maximizing active vitamin D
bioavailability at the fetomaternal interface suggesting a role in
pregnancy progression (71).
Environmental impact on DNA methylation
in the placenta
There is a critical window, at some stage in
intrauterine life, during which balanced homeostasis is essential
for normal fetal growth and development. Adverse effects during
that period alter the structure and function of distinct cells,
organ systems or homeostatic pathways, thereby ‘programming’ the
individual for an increased risk of developing diseases in adult
life. Placental phenotype is responsive to environmental conditions
and may help predict the risk of adult disease programmed in
utero. The placenta responds to and is potentially marked in an
epigenetic context by environmental insults, suggesting that the
placental epigenome serves, not only as a record of in utero
exposure, but also as a mediator and/or modulator of disease
pathogenesis.
Accumulating evidence suggests that the maternal
nutritional status is capable of altering the epigenetic state of
the fetal genome and imprinted gene expression. Ethanol-exposed
midgestation placentas and embryos were severely growth retarded
when compared with the controls. The relationship between placental
weight and ethanol treatment suggested that this was partially
dependent on DNA methylation at the CCCTC-binding factor (CTCF)
site on the paternal allele in placentas (61). Preimplantation embryo culture has
been shown to affect the methylation and expression of imprinted
genes in several animal models. One particularly favoured
explanation for the association between the environmental impact in
early life and long-term physiological functions lies with the
epigenetic modification of gene expression (71). Although there is strong evidence to
demonstrate that the environment affects the pattern of DNA
methylation during fetal development, the direct association
between environmental conditions, methylation alterations and gene
expression is difficult to verify (Fig. 2).
6. Conclusion
Numerous links have been made between infant growth
restriction and specific epigenetic alterations, including changes
to the gene imprinting status and to DNA methylation. Fetal growth
is affected by the proper function of many imprinted and
non-imprinted genes which are subject to epigenetic control through
methylation of their promoters. DNA methylation has a critical role
in placenta development, and alterations to its methylation pattern
can lead to adverse placental morphology and birth outcome.
However, since DNA methylation represents a delicate molecular
mechanism that is easily affected by various factors, data that
associate methylation patterns with placental pathology or abnormal
fetal growth, should be interpreted with caution.
References
|
1
|
M NakaoEpigenetics: interaction of DNA
methylation and
chromatinGene2782531200110.1016/S0378-1119(01)00721-111707319
|
|
2
|
R HollidayThe inheritance of epigenetic
defectsScience238163170198710.1126/science.33102303310230
|
|
3
|
J Van VlietNA OatesE WhitelawEpigenetic
mechanisms in the context of complex diseasesCell Mol Life
Sci6415311538200717458502
|
|
4
|
V BollatiA BaccarelliL HouChanges in DNA
methylation patterns in subjects exposed to low-dose benzeneCancer
Res67876880200710.1158/0008-5472.CAN-06-299517283117
|
|
5
|
MD AnwayMK SkinnerEpigenetic programming
of the germ line: effects of endocrine disruptors on the
development of transgenerational diseaseReprod Biomed
Online162325200810.1016/S1472-6483(10)60553-618252044
|
|
6
|
JG HermanSB BaylinGene silencing in cancer
in association with promoter hypermethylationN Engl J
Med34920422054200310.1056/NEJMra02307514627790
|
|
7
|
PW LairdThe power and the promise of DNA
methylation markersNat Rev
Cancer3253266200310.1038/nrc104512671664
|
|
8
|
AP BirdCpG-rich islands and the function
of DNA methylationNature321209213198610.1038/321209a02423876
|
|
9
|
JF CostelloC PlassMethylation mattersJ Med
Genet38285303200110.1136/jmg.38.5.28511333864
|
|
10
|
M WeberD SchubelerGenomic patterns of DNA
methylation: targets and function of an epigenetic markCurr Opin
Cell Biol19273280200710.1016/j.ceb.2007.04.01117466503
|
|
11
|
AP BirdAP WolffeMethylation-induced
repression-belts, braces, and
chromatinCell99451454199910.1016/S0092-8674(00)81532-910589672
|
|
12
|
MA MaccaniCJ MarsitEpigenetics in the
placentaAm J Reprod
Immunol627889200910.1111/j.1600-0897.2009.00716.x
|
|
13
|
LL OlignyHuman molecular embryogenesis: an
overviewPediatr Dev
Pathol4324343200110.1007/s10024001-0033-211441334
|
|
14
|
F SantosW DeanEpigenetic reprogramming
during early development in
mammalsReproduction127643651200410.1530/rep.1.0022115175501
|
|
15
|
W MayerA NiveleauJ WalterR FundeleT
HaafDemethylation of the zygotic paternal
genomeNature403501502200010.1038/3500065610676950
|
|
16
|
W DeanF SantosW ReikEpigenetic
reprogramming in early mammalian development and following somatic
nuclear transferSemin Cell Dev
Biol1493100200310.1016/S1084-9521(02)00141-612524012
|
|
17
|
W DeanF SantosM StojkovicConservation of
methylation reprogramming in mammalian development: aberrant
reprogramming in cloned embryosProc Natl Acad Sci
USA981373413738200110.1073/pnas.24152269811717434
|
|
18
|
F SantosB HendrichW ReikW DeanDynamic
reprogramming of DNA methylation in the early mouse embryoDev
Biol241172182200210.1006/dbio.2001.050111784103
|
|
19
|
KD TremblayJR SaamRS IngramSM TilghmanMS
BartolomeiA paternal-specific methylation imprint marks the alleles
of the mouse H19 geneNat
Genet9407413199510.1038/ng0495-4077795647
|
|
20
|
W ReikJ WalterGenomic imprinting: parental
influence on the genomeNat Rev
Genet22132200110.1038/3504755411253064
|
|
21
|
AJ WoodRJ OakeyGenomic imprinting in
mammals: emerging themes and established theoriesPLoS
Genet2e147200610.1371/journal.pgen.002014717121465
|
|
22
|
CA EdwardsAC Ferguson-SmithMechanisms
regulating imprinted genes in clustersCurr Opin Cell
Biol19281289200710.1016/j.ceb.2007.04.01317467259
|
|
23
|
JF WilkinsD HaigWhat good is genomic
imprinting: the function of parent-specific gene expressionNat Rev
Genet4359368200310.1038/nrg106212728278
|
|
24
|
M ConstanciaM HembergerJ
HughesPlacental-specific IGF-II is a major modulator of placental
and fetal growthNature417945948200210.1038/nature0081912087403
|
|
25
|
D FrankW FortinoL ClarkPlacental
overgrowth in mice lacking the imprinted gene IplProc Natl Acad Sci
USA9974907495200210.1073/pnas.12203999912032310
|
|
26
|
L LefebvreS VivilleSC BartonF IshinoEB
KeverneMA SuraniAbnormal maternal behaviour and growth retardation
associated with loss of the imprinted gene MestNat
Genet20163169199810.1038/24649771709
|
|
27
|
K TakahashiT KobayashiN Kanayamap57(Kip2)
regulates the proper development of labyrinthine and
spongiotrophoblastsMol Hum
Reprod610191025200010.1093/molehr/6.11.101911044465
|
|
28
|
RL GlaserJP RamsayIM MorisonThe imprinted
gene and parent-of-origin effect database now includes parental
origin of de novo mutationsNucleic Acids Res34Database
issueD29D31200610.1093/nar/gkj10116381868
|
|
29
|
JC RobinsCJ MarsitJF PadburySS
SharmaEndocrine disruptors, environmental oxygen, epigenetics and
pregnancyFront Biosci (Elite Ed)3690700201110.2741/e27921196344
|
|
30
|
R SoodJL ZehnderML DruzinPO BrownGene
expression patterns in human placentaProc Natl Acad Sci
USA10354785483200610.1073/pnas.050803510316567644
|
|
31
|
AC FilibertoMA MaccaniD
KoestlerBirthweight is associated with DNA promoter methylation of
the glucocorticoid receptor in human
placentaEpigenetics6566572201110.4161/epi.6.5.1523621521940
|
|
32
|
EC NelissenAP van MontfoortJC DumoulinJL
EversEpigenetics and the placentaHum Reprod
Update17397417201110.1093/humupd/dmq05220959349
|
|
33
|
R ResnikIntrauterine growth
restrictionObstet
Gynecol99490496200210.1016/S0029-7844(01)01780-X11864679
|
|
34
|
D BrodskyH ChristouCurrent concepts in
intrauterine growth restrictionJ Intensive Care
Med19307319200410.1177/088506660426966315523117
|
|
35
|
LO LubchencoC HansmanM DresslerE
BoydIntrauterine growth as estimated from liveborn birth-weight
data at 24 to 42 weeks of gestationPediatrics327938001963
|
|
36
|
RJ SnijdersC SherrodCM GosdenKH
NicolaidesFetal growth retardation: associated malformations and
chromosomal abnormalitiesAm J Obstet
Gynecol168547555199310.1016/0002-9378(93)90491-Z8438926
|
|
37
|
RA OdegardLJ VattenST NilsenKA SalvesenR
AustgulenPreeclampsia and fetal growthObstet
Gynecol96950955200010.1016/S0029-7844(00)01040-1
|
|
38
|
RA WaterlandRL JirtleEarly nutrition,
epigenetic changes at transposons and imprinted genes, and enhanced
susceptibility to adult chronic
diseasesNutrition206368200410.1016/j.nut.2003.09.01114698016
|
|
39
|
EA HousemanBC ChristensenRF YehModel-based
clustering of DNA methylation array data: a recursive-partitioning
algorithm for high-dimensional data arising as a mixture of beta
distributionsBMC
Bioinformatics9365200810.1186/1471-2105-9-36518782434
|
|
40
|
M ConstanciaG KelseyW ReikResourceful
imprintingNature43253572004
|
|
41
|
M VlahovicF Bulic-JakusG Juric-LekicA
FucicS MaricD SermanChanges in the placenta and in the rat embryo
caused by the demethylating agent 5-azacytidineInt J Dev
Biol43843846199910707910
|
|
42
|
L SermanM VlahovicM SijanThe impact of
5-azacytidine on placental weight, glycoprotein pattern and
proliferating cell nuclear antigen expression in rat
placentaPlacenta28803811200710.1016/j.placenta.2007.04.00117509679
|
|
43
|
F RahnamaF ShafieiPD GluckmanMD MitchellPE
LobieEpigenetic regulation of human trophoblastic cell migration
and
invasionEndocrinology14752755283200610.1210/en.2006-028816887905
|
|
44
|
S KurukutiVK TiwariG TavoosidanaE
PugachevaA MurrellZ ZhaoV LobanenkovW ReikR OhlssonCTCF binding at
the H19 imprinting control region mediates maternally inherited
higher-order chromatin conformation to restrict enhancer access to
IGF2Proc Natl Acad Sci
USA1031068410689200610.1073/pnas.0600326103
|
|
45
|
AL FowdenC SibleyW ReikM
ConstanciaImprinted genes, placental development and fetal
growthHorm Res65Suppl 35058200610.1159/00009150616612114
|
|
46
|
R RandhawaP CohenThe role of the
insulin-like growth factor system in prenatal growthMol Genet
Metab868490200510.1016/j.ymgme.2005.07.02816165387
|
|
47
|
CJ PetryKK OngBJ BarrattCommon
polymorphism in H19 associated with birthweight and cord blood
IGF-II levels in humansBMC
Genet622200510.1186/1471-2156-6-2215885138
|
|
48
|
Y JinnoY IkedaK YunEstablishment of
functional imprinting of the H19 gene in human developing
placentaeNat Genet1031824199510.1038/ng0795-3187670470
|
|
49
|
L GuoS ChoufaniJ FerreiraAltered gene
expression and methylation of the human chromosome 11 imprinted
region in small for gestational age (SGA) placentaeDev
Biol3207991200810.1016/j.ydbio.2008.04.02518550048
|
|
50
|
O KoukouraS SifakisG SouflaLoss of
imprinting and aberrant methylation of IGF2 in placentas from
pregnancies complicated with fetal growth restrictionInt J Mol
Med28481487201121805044
|
|
51
|
O KoukouraS SifakisA
ZaravinosHypomethylation along with increased H19 expression in
placentas from pregnancies complicated with fetal growth
restrictionPlacenta325157201110.1016/j.placenta.2010.10.01721129773
|
|
52
|
CP SibleyPM CoanAC
Ferguson-SmithPlacental-specific insulin-like growth factor 2
(IGF2) regulates the diffusional exchange characteristics of the
mouse placentaProc Natl Acad Sci
USA10182048208200410.1073/pnas.040250810115150410
|
|
53
|
S TabanoP ColapietroI CetinEpigenetic
modulation of the IGF2/H19 imprinted domain in human embryonic and
extra-embryonic compartments and its possible role in fetal growth
restrictionEpigenetics5313324201010.4161/epi.5.4.1163720418667
|
|
54
|
DK BourqueL AvilaM PenaherreraP von
DadelszenWP RobinsonDecreased placental methylation at the H19/IGF2
imprinting control region is associated with normotensive
intrauterine growth restriction but not
preeclampsiaPlacenta31197202201010.1016/j.placenta.2009.12.003
|
|
55
|
J McMinnM WeiN SchupfUnbalanced placental
expression of imprinted genes in human intrauterine growth
restrictionPlacenta27540549200610.1016/j.placenta.2005.07.00416125225
|
|
56
|
ST ChelbiF MondonH JammesExpressional and
epigenetic alterations of placental serine protease inhibitors:
SERPINA3 is a potential marker of
preeclampsiaHypertension497683200710.1161/01.HYP.0000250831.52876.cb17088445
|
|
57
|
C GicquelS RossignolS CabrolEpimutation of
the telomeric imprinting center region on chromosome 11p15 in
Silver-Russell syndromeNat
Genet3710031007200510.1038/ng162916086014
|
|
58
|
R WeksbergC ShumanAC
SmithBeckwith-Wiedemann syndromeAm J Med Genet C Semin Med
Genet137C1223200510.1002/ajmg.c.3005816010676
|
|
59
|
AS DohertyMR MannKD TremblayMS
BartolomeiRM SchultzDifferential effects of culture on imprinted
H19 expression in the preimplantation mouse embryoBiol
Reprod6215261535200010.1095/biolreprod62.6.152610819752
|
|
60
|
Q WuS OhsakoR IshimuraJS SuzukiC
TohyamaExposure of mouse preimplantation embryos to
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the methylation
status of imprinted genes H19 and IGF2Biol
Reprod7017901797200410.1095/biolreprod.103.02538714960483
|
|
61
|
PC HaycockM RamsayExposure of mouse
embryos to ethanol during preimplantation development: effect on
DNA methylation in the H19 imprinting control regionBiol
Reprod81618627200910.1095/biolreprod.108.07468219279321
|
|
62
|
L LambertiniAI DiplasMJ LeeR SperlingJ
ChenJ WetmurA sensitive functional assay reveals frequent loss of
genomic imprinting in human
placentaEpigenetics3261269200810.4161/epi.3.5.675518769151
|
|
63
|
D MonkR SanchesP ArnaudImprinting of IGF2
P0 transcript and novel alternatively spliced INS-IGF2 isoforms
show differences between mouse and humanHum Mol
Genet1512591269200610.1093/hmg/ddl04116531418
|
|
64
|
RK YuenL AvilaMS PenaherreraHuman
placental-specific epipolymorphism and its association with adverse
pregnancy outcomesPLoS
One4e7389200910.1371/journal.pone.000738919838307
|
|
65
|
CE BanisterDC KoestlerMA MaccaniJF
PadburyEA HousemanCJ MarsitInfant growth restriction is associated
with distinct patterns of DNA methylation in human
placentasEpigenetics6920927201110.4161/epi.6.7.1607921758004
|
|
66
|
CS Wilhelm-BenartziEA HousemanMA MaccaniIn
Utero Exposures, Infant Growth, and DNA Methylation of Repetitive
Element and Developmentally Related Genes in Human PlacentaEnviron
Health Perspect201110.1289/ehp.110392722005006
|
|
67
|
N ZamudioD Bourc’hisTransposable elements
in the mammalian germline: a comfortable niche or a deadly
trap?Heredity (Edinb)10592104201010.1038/hdy.2010.5320442734
|
|
68
|
A HeikkiläT TuomistoSK HäkkinenL
Keski-NisulaS HeinonenS Yla-HerttualaTumor suppressor and growth
regulatory genes are overexpressed in severe early-onset
preeclampsia-an array study on case-specific human preeclamptic
placental tissueActa Obstet Gynecol Scand846796892005
|
|
69
|
B NovakovicV RakyanHK NgSpecific
tumour-associated methylation in normal human term placenta and
first-trimester cytotrophoblastsMol Hum
Reprod14547554200810.1093/molehr/gan04618708652
|
|
70
|
C FerrettiL BruniV Dangles-MarieAP
PeckingD BelletMolecular circuits shared by placental and cancer
cells, and their implications in the proliferative, invasive and
migratory capacities of trophoblastsHum Reprod
Update13121141200710.1093/humupd/dml04817068222
|
|
71
|
B NovakovicM SibsonHK NgPlacenta-specific
methylation of the vitamin D 24-hydroxylase gene: implications for
feedback autoregulation of active vitamin D levels at the
fetomaternal interfaceJ Biol
Chem2841483814848200910.1074/jbc.M80954220019237542
|
|
72
|
A RazinCpG methylation, chromatin
structure and gene silencing-a three-way connectionEMBO
J1749054908199810.1093/emboj/17.17.49059724627
|