Introduction
Hepatic fibrosis is caused by the destruction of the
architecture of the liver parenchyma and is a common wound-healing
response to hepatic diseases, including chronic hepatitis and liver
damage, which forms scars or fibrous tissues through excessive
fibrogenesis and insufficient fibrolysis. Hepatic stellate cells
(HSCs) are crucial to the development of hepatic fibrosis (1). During hepatic fibrosis, HSCs are
activated and change into myofibroblast-like cells, which are
characterized by increased proliferation and extracellular matrix
(ECM) synthesis (2). Fibrogenesis
is promoted by the activation and proliferation of HSCs and delayed
by the apoptosis of HSCs. Current antifibrosis strategies include
inhibition of the activation and proliferation of HSCs and
induction of the apoptosis of activated HSCs (3). HSCs are considered to be an
attractive target in antifibrosis strategies due to their essential
role in hepatic fibrosis.
Hydrogen sulfide (H2S) is a gaseous
messenger that displays many physiological and pathological
activities (4). The administration
of H2S has been reported to attenuate myocardial infarct
size (5), suppress the development
of hypertension (6) and alleviate
neuronal injury (7). The
mechanisms underlying the action of H2S predominantly
involve the inhibition of oxidative stress and inflammatory
responses and the activation of ATP-sensitive potassium (KATP)
channels (4).
H2S plays a regulatory role in hepatic
physiology and pathology (8). In
mammalian hepatic tissues, H2S is mainly produced by
cystathionine c-lyase (CSE) (9),
and activates KATP channels, leading to vasorelaxation of the
hepatic artery (10). Since
hepatic cirrhosis is associated with the development of a
hyperdynamic circulation caused by systemic vasodilation, it has
been postulated that H2S is involved in the pathogenesis
of the vascular abnormalities in cirrhosis (11). H2S displays
antioxidative, anti-inflammatory and cytoprotective activities;
therefore, we hypothesized that H2S may have a
protective effect against hepatic fibrosis.
The present study aimed to investigate the effects
of sodium hydrogen sulphide (NaHS), an H2S-releasing
molecule, on the proliferation, cell cycle, apoptosis,
intracellular reactive oxygen species (ROS) and free calcium levels
of activated HSC cells, and on fibrosis and ECM synthesis in rats
with carbon tetrachloride (CCl4)-induced hepatic
fibrosis.
Materials and methods
Cell culture
The rat HSC line (HSC-T6) was purchased from the
Institute of Biochemistry and Cell Biology, Shanghai Institutes for
Biological Sciences, Chinese Academy of Sciences (Shanghai, China).
The cells were cultured in DMEM (Invitrogen-Gibco, Carlsbad, CA,
USA), supplemented with 10% fetal bovine serum (FBS) (Sijichun
Bioengineering Materials, Inc., Hangzhou, China), 100 U/ml
penicillin and 100 μg/ml streptomycin, at 37°C in a humidified
incubator with 5% CO2.
Cell viability assay
The cell growth rate was determined by MTT
(Sigma-Aldrich, St. Louis, MO, USA) assay. Briefly, cells at the
logarithmic growth phase were seeded in 96-well culture plates at a
density of 1×103 cell/ml with 100 μl per well. The cells
were then cultured in DMEM and incubated with 500 μg/l ferric
nitrilotriacetate (Fe-NTA) (Sigma-Aldrich) and various
concentrations of NaHS (Sigma-Aldrich) (0, 100, 200 or 500 μmol/l)
for 24 h. For the cell viability assay, 10 μl MTT solution (5
mg/ml) was added to each well and the plates were incubated at 37°C
for 4 h. After centrifugation at 3,000 rpm for 10 min, the
supernatant was removed and the formazan pellet was dissolved
completely in 100 μl DMSO. The absorbance was measured with an
ELISA plate reader at a wavelength of 570 nm to determine the
amount of viable cells in the pellet.
Cell cycle analysis
The cells were harvested by trypsinization,
centrifuged at 2,000 rpm for 5 min, washed with PBS and resuspended
in cold 70% ethanol. Finally, 1 ml propidium iodide (PI) staining
solution (PI, 20 μg/ml; DNase free RNase A, 100 μg/ml) was added to
the samples which were analyzed using a FACScan (BD Biosciences,
San Francisco, CA, USA) within 30 min. Data on 10,000 cells were
acquired and processed using Lysis II software (BD
Biosciences).
Cell apoptosis assay
Cells at the logarithmic growth phase were randomly
divided into 4 groups: the normal control, NaHS (500 μmol/l),
Fe-NTA (500 μg/l) and Fe-NTA + NaHS groups. After 24 h,
2×105 cells were collected from each group and washed
with phosphate-buffered saline (PBS). The pellet was resuspended in
100 μl 1X binding buffer and combined with 2.5 μl Annexin V and 5
μl PI (final concentration, 10 μg/ml), followed by incubation for
15 min in the dark. Apoptosis was determined by flow cytometry. At
least 10,000 events were analyzed for each sample. The data were
analyzed using Lysis software.
Measurement of ROS generation
Intracellular ROS was quantified with a fluorescence
plate reader using 2,7-dichlorodihydrofluorescein diacetate
(DCFH-DA, Sigma-Aldrich). The cells on 96-well plates were treated
with NaHS (500 μmol/l) and/or Fe-NTA (500 μg/l) for 1, 3 or 6 h and
incubated with DCFH-DA at 37°C for 30 min. Following the removal of
the DCFH-DA, the cells were washed with PBS. The DCFH-DA-loaded
cells were read using a fluorescence plate reader (Tecan,
Crailsheim, Germany).
Measurement of intracellular free calcium
[Ca2+]i
Fura 2-acetoxymethyl ester (Fura 2-AM) (Dojindo
Laboratories, Kumamoto, Japan), a fluorescent
Ca2+-sensitive dye, was used to monitor
[Ca2+]i. The cells were cultured and treated
with NaHS (500 μmol/l) and/or Fe-NTA (500 μg/l) for 3 or 6 h and
preloaded with 1 μmol/l Fura 2-AM for 30 min in the dark at 37°C in
a humidified incubator. After loading with Fura 2-AM, the cells
were collected, washed 3 times with D-Hanks’ solution and
resuspended in D-Hanks’ solution containing 0.2% BSA at
1×106 cells/ml. Fluorescence intensity was measured at
an emission wavelength of 510 nm and excitation wavelengths of 340
and 380 nm using a Safire fluorescence plate reader (Tecan).
[Ca2+]i was estimated from the ratio of the
fluorescence intensities at 340 and 380 nm (F340/F380).
Western blot analysis
HSC-T6 cells were cultured and treated with NaHS
(500 μmol/l) and/or Fe-NTA (500 μg/l) for 24 h. The proteins were
isolated and their concentrations were determined. The proteins (50
μg) were separated on sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) gels (polyacrylamide concentration 100
g/l) and electrophoretically transferred onto a PVDF membrane. The
PVDF membrane was blocked with 3% BSA at 37°C for 1 h and probed
with the rabbit polyclonal antibody against collagen I (1:1,000)
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), followed by
horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1,000)
for 2 h at room temperature. The densities of the targeted bands
were visualized using the chemiluminescence method. β-actin was
used as the internal control.
Animal model of hepatic fibrosis
All studies were approved by the Animal Study
Committee of the Qinghai University School of Medicine. Male Wistar
rats (weighing 220–240 g) were supplied by the Animal Research
Center at the Affiliated Hospital of Qinghai University. They were
maintained on standard laboratory rat chow on a 12-h light/dark
cycle at a temperature of 22–23°C. Liver fibrosis was induced by
carbon tetrachloride (CCl4) injection. Briefly,
phenobarbital sodium (0.35 g/l) was administered to the rats with
their drinking water for 3 days. This was followed by an
intraperitoneal injection of 100 μl CCl4/100 g body
weight in an equal volume of paraffin oil twice a week for 6 weeks.
Rats receiving an intraperitoneal injection of 100 μl of saline/100
g body weight in an equal volume of paraffin oil were used as the
control group.
Animal grouping and experiment
A total of 24 rats with liver fibrosis receiving
CCl4 injections for 6 weeks were randomly assigned into
2 groups (each of 12 rats), and an intraperitoneal injection of 1
ml saline or NaHS solution (10 mmol/kg body weight) was
administered every 2 days for 6 weeks. The 12 rats in the control
group received an intraperitoneal injection of 1 ml saline for
every 2 days for 6 weeks. At the completion of the experiments, the
rats were sacrificed for pathological examination. Blood was
collected via cardiac puncture and then the liver tissue was
collected. Serum and liver samples were prepared and stored at
4°C.
Histological analysis
Formalin-fixed liver specimens were embedded in
paraffin, sectioned, stained with hematoxylin and eosin (H&E)
and examined under a light microscope. Cell numbers were quantified
by measuring the numbers of blue pixels in the images captured from
the H&E-stained sections. Ten images (magnification, ×400) were
randomly captured from each liver using a fixed exposure time and
conditions. The images were saved as Joint Photographic Experts
Group (JPEG) files and the numbers of blue pixels were counted
using the histogram function in Adobe Photoshop CS4.
Immunohistochemistry analysis
Liver sections (5 mm in thickness) were incubated at
4°C overnight with a primary antibody against collagen I (Santa
Cruz Biotechnology, Inc.) at a concentration of 1:100. The
secondary antibody was horseradish peroxidase-conjugated IgG
(Medical Biological Laboratory, Nagoya, Japan) and was used for 30
min at 37°C. After washing with Tris-buffered saline, the sections
were incubated with complex/horseradish peroxidase (1:200 dilution)
for 30 min at 37°C. Immunolocalization was performed by immersion
in 0.05% 3,3′-diaminobenzidine tetrahydrochloride as the chromagen.
The slides were counterstained with hematoxylin prior to
dehydration and mounting. Slides which did not undergo primary
antibody incubation were processed as a control for the background
staining. The brown positive cells throughout the entire section
were counted and the total counts in these sections were converted
into cell densities for quantification.
Statistical analysis
Statistical analysis was performed using
commercially available software (SPSS version 14.0). Data are
expressed as the means ± standard deviation. The Student’s t-test
(unpaired, two-tailed) was performed to compare the means of 2
groups. P<0.05 was considered as a statistically significant
result.
Results
H2S inhibits HSC
proliferation
The HSC-T6 cells were stimulated by incubation with
Fe-NTA (500 μmol/l). These cells were simultaneously treated with
various concentrations of NaHS (0, 100, 200 or 500 μmol/l) for 24
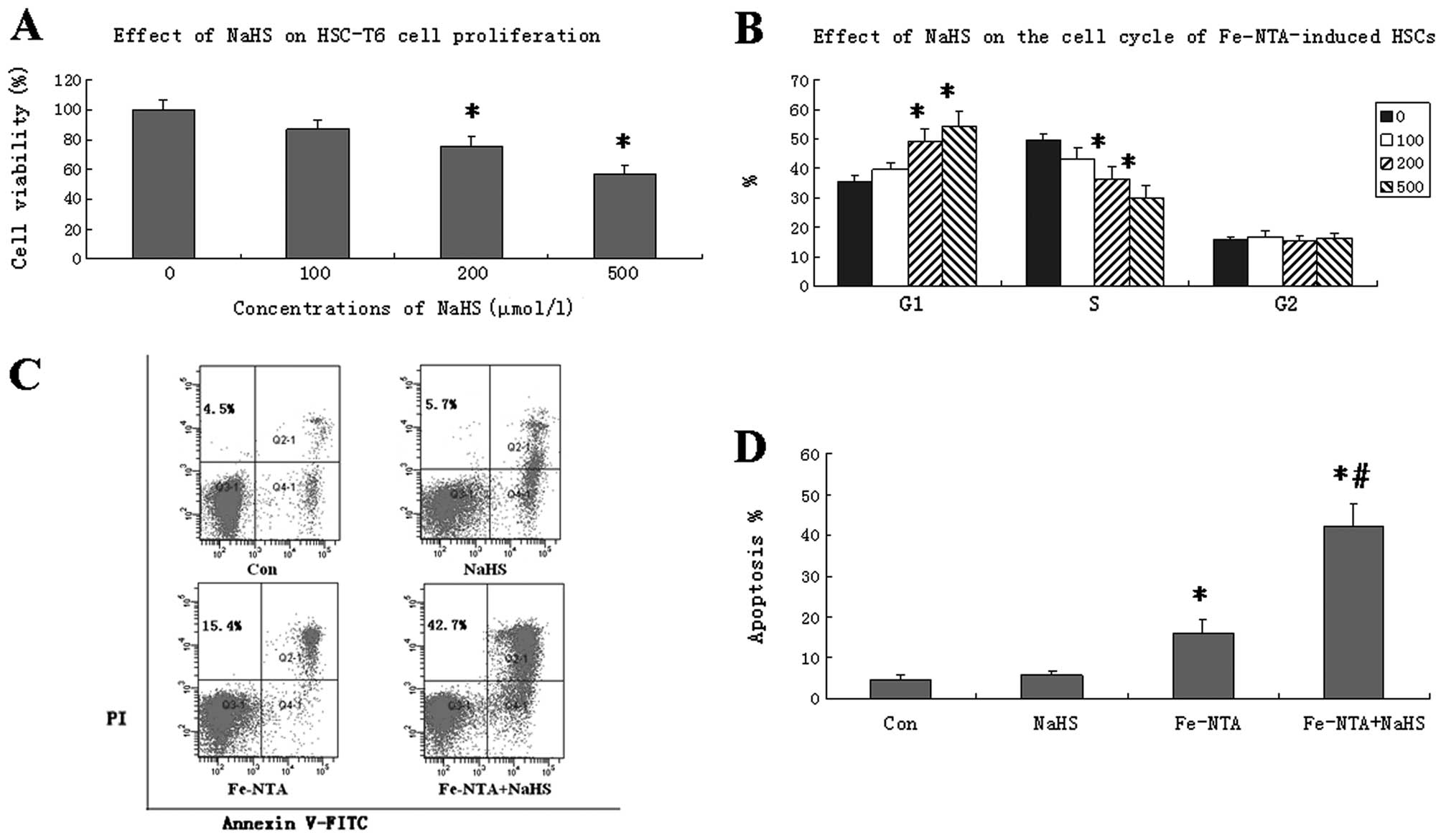
h. The MTT assay revealed that NaHS inhibited HSC proliferation in
a dose-dependent manner. Cell viability was significantly reduced
by the 200 and 500 μmol/l concentrations of NaHS (P<0.05;
Fig. 1A).
H2S induces G1 phase cell
cycle arrest in HSCs
To clarify the possible mechanism behind the
antiproliferative activity of NaHS, the cell cycle distribution in
the Fe-NTA-activated cells was determined following treatment with
0, 100, 200 or 500 μmol/l NaHS for 24 h. NaHS induced a significant
increase in the number of cells in the G1 phase, with a
corresponding decrease in the number of cells in the S phase.
However, no significant differences were found in the percentages
of the cells in the G2 phase following NaHS treatment (Fig. 1B). These results indicate that NaHS
inhibits HSC proliferation by inducing G1 phase arrest.
H2S increases Fe-NTA-induced
apoptosis in HSCs
To investigate whether apoptosis is involved in the
reduction in the number of viable cells when HSCs are treated with
NaHS, the cells were cultivated in the presence of Fe-NTA and/or
NaHS for 24 h. Cell apoptosis was analyzed by Annexin V-FITC and PI
double staining. NaHS treatment (500 μmol/l) alone did not increase
the apoptotic rate. Fe-NTA increased the apoptotic rate of the HSCs
moderately. However, treatment with Fe-NTA and NaHS resulted in a
significantly higher apoptotic rate than treatment with Fe-NTA
alone (P<0.05; Fig. 1C and
D).
NaHS decreases ROS generation in HSC-T6
cells
To determine whether NaHS is able to influence ROS
generation in the Fe-NTA-induced HSC-T6 cells, the fluorescent
probe, DCFH-DA, was used to measure the levels of ROS. The
intracellular ROS level was significantly higher in the HSC-T6
cells treated with Fe-NTA for 1, 3 (peak) and 6 h than in the
control cells (Fig. 2A). NaHS
alone did not influence ROS levels; however, it significantly
reduced ROS levels in the Fe-NTA-treated cells at all time points
(P<0.05).
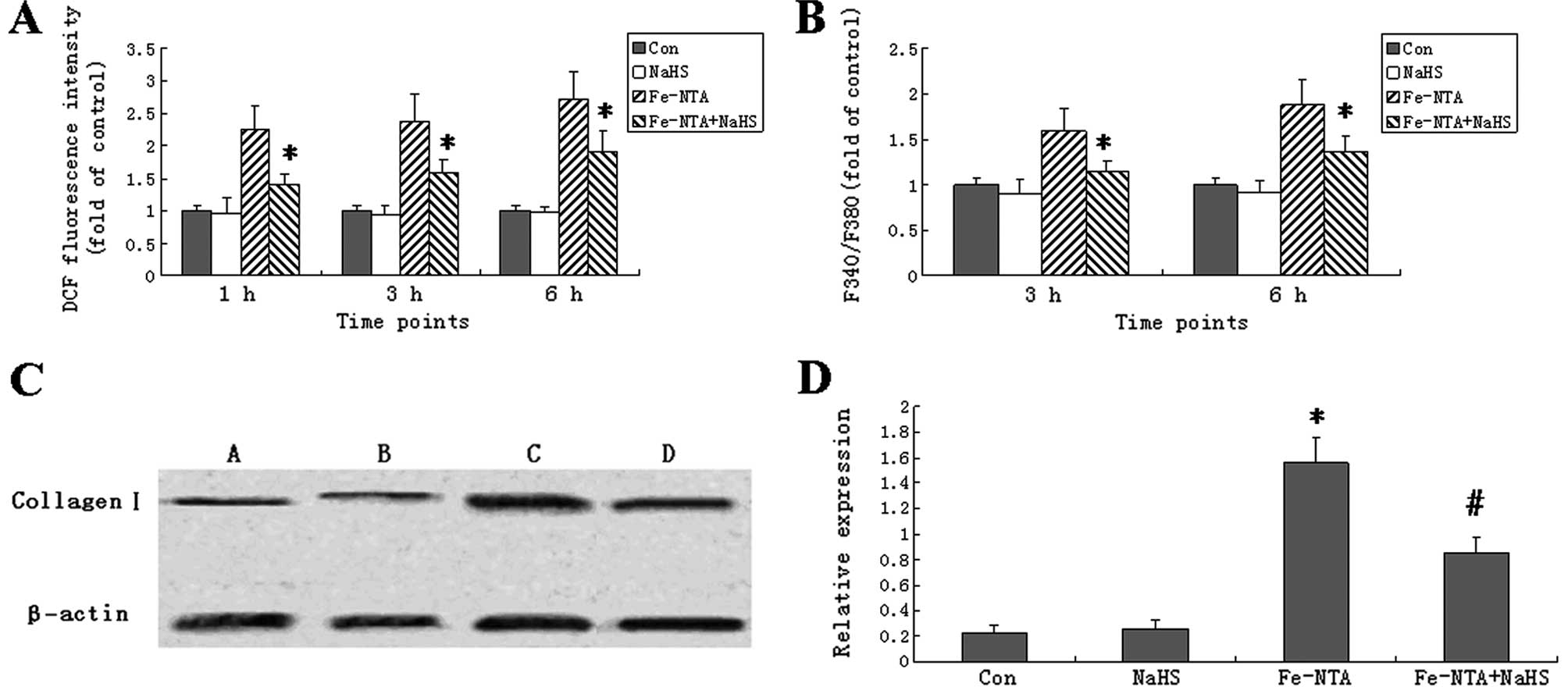 | Figure 2Sodium hydrogen sulfide (NaHS)
decreased reactive oxygen species (ROS), intracellular
Ca2+ [Ca2+]i and collagen I
expression in ferric nitrilotriacetate (Fe-NTA)-treated HSC-T6
cells. (A) Cells were treated with Fe-NTA and/or NaHS for 1, 3 or 6
h, followed by a 30-min incubation at 37°C for ROS detection using
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). (B) Cells
were treated with Fe-NTA and/or NaHS for 3 or 6 h.
[Ca2+]i was detected from the fura
2-acetoxymethyl ester (Fura 2-AM) 380/340 nm fluorescence ratio
(F340/F380). In (A and B), data are expressed as fold increase over
those of control cells. Bars represent the mean ± SD.
*Significant difference between the Fe-NTA and
Fe-NTA+NaHS groups (P<0.05). (C) HSC-T6 cells were treated with
NaHS, Fe-NTA or Fe-NTA+NaHS for 24 h. Untreated cells served as
healthy controls. Whole hepatic cell extracts were immunoblotted
with the indicated antibodies. The expression of collagen I was
detected in cells from healthy controls (lane A) and cells treated
with NaHS (lane B), Fe-NTA (lane C) or Fe-NTA+NaHS (lane D) by
western blot analysis. (D) The density of each band was measured
and compared to that of the internal control, β-actin. Results are
expressed as the means ± SD. Collagen I expression was
significantly higher in the Fe-NTA group than in the normal group
(P<0.05, n=12). Treatment with NaHS decreased the Fe-NTA-induced
collagen I expression (P<0.05, n=12). Each bar represents the
mean ± SD for 12 animals. *P<0.05 vs. control group;
#P<0.05 vs. the Fe-NTA group (n=12). |
NaHS increases cytoplasmic
Ca2+ levels in HSC-T6 cells
To determine whether NaHS influences the levels of
[Ca2+]i, Fura 2-AM staining was performed.
The Fe-NTA treatment increased the fluorescence ratios (F340/F380)
of the HSC-T6 cells to 159.7±24.5% (3 h) and 187.7±28.7% (6 h) of
those of the control cells (Fig.
2B). Moreover, NaHS decreased cytoplasmic Ca2+ in
the Fe-NTA-induced cells. NaHS alone did not change cytoplasmic
Ca2+.
NaHS decreases collagen I protein
expression in HSC-T6 cells treated with Fe-NTA
To further explore the effects of NaHS on collagen
degradation in HSCs, the collagen I protein levels were determined
by western blot analysis. Compared with the control group, the
Fe-NTA treatment significantly increased the levels of collagen I
protein in the HSC-T6 cells. In the cells treated with Fe-NTA and
NaHS together, the collagen I protein levels were significantly
lower than in the cells treated with Fe-NTA alone (P<0.05;
Fig. 2C and D).
H2S attenuates
CCl4-induced liver fibrosis
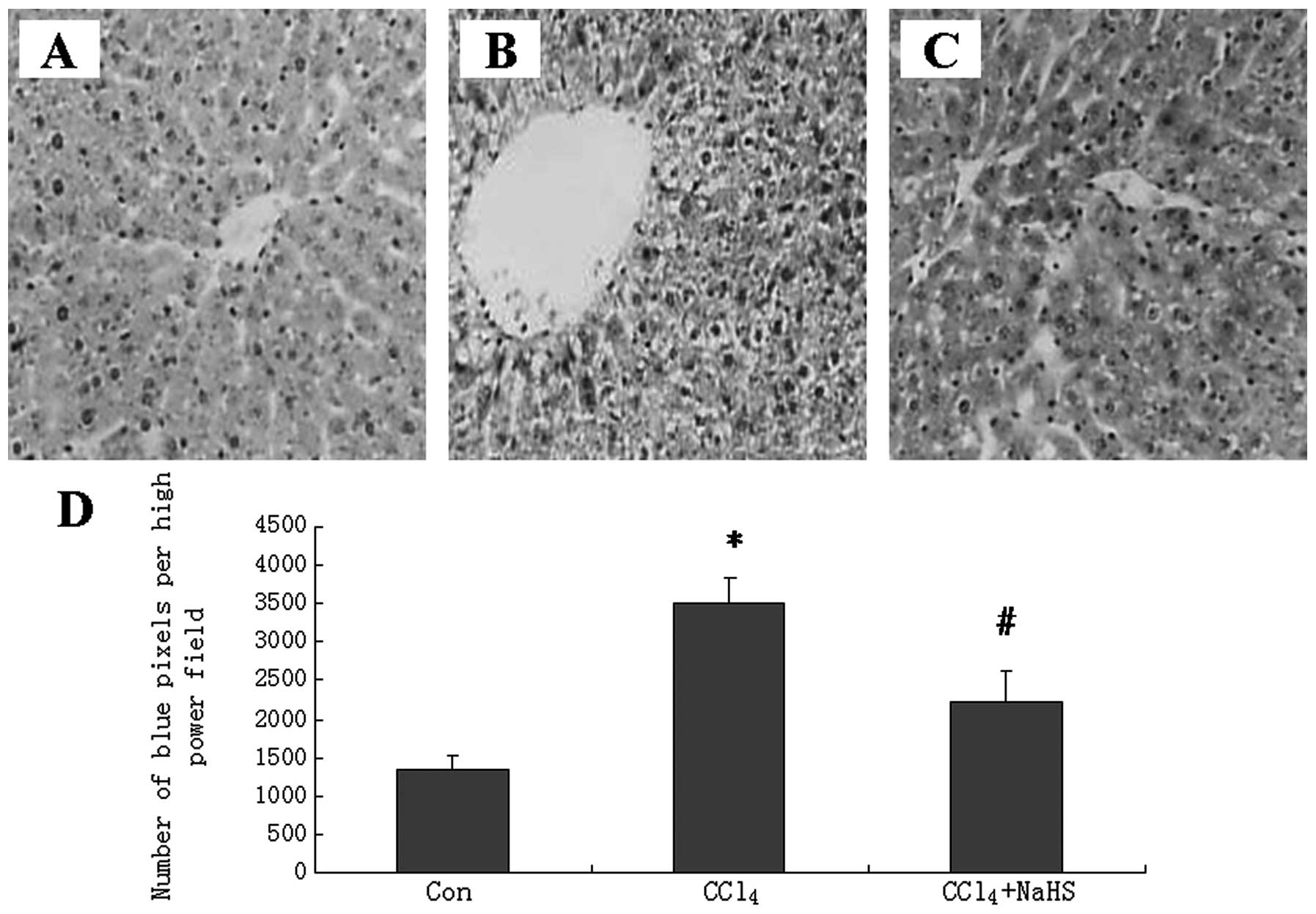
The blue pixels in the images of the liver sections
from the rats with CCl4-induced fibrosis that were
injected with saline were significantly stronger than those from
the healthy controls (Fig. 3A and
B). However, the images of the liver sections from the rats
with CCl4-induced fibrosis that were injected with NaHS
had fewer blue pixels (Fig. 3C)
than those from the saline-injected fibrotic rats. The numbers of
blue pixels in the images of the liver sections were measured to
quantify cell numbers. CCl4 significantly increased the
number of blue pixels in the images of the liver sections, but NaHS
significantly reduced the number of blue pixels compared with
saline (Fig. 3D). We further
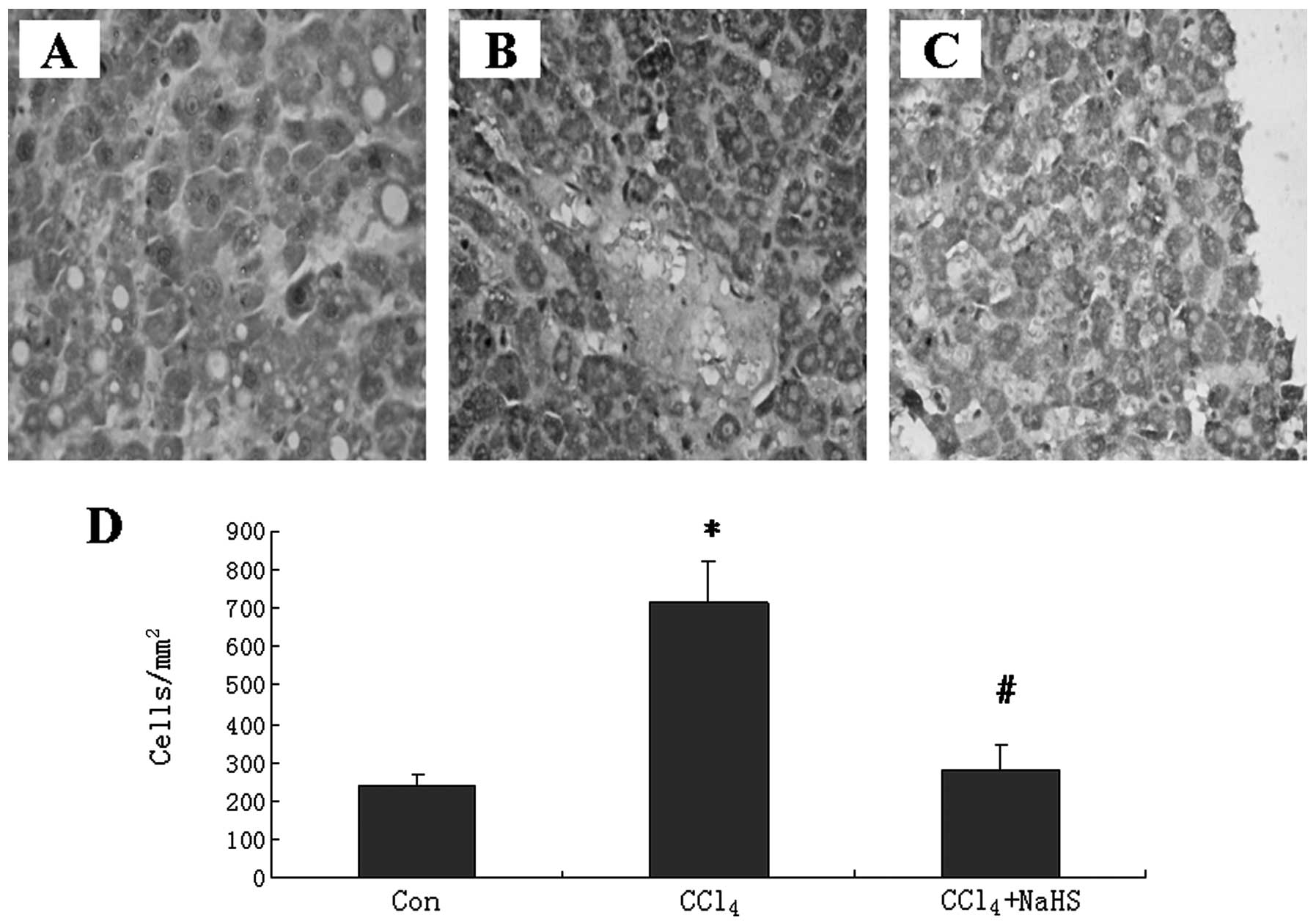
measured collagen I, a marker of fibrosis, by immunohistochemical
analysis. The staining intensity was significantly stronger in the
CCl4 + saline group than in the control group (Fig. 4A and B). However, NaHS treatment
lowered the staining intensity in the CCl4-treated rats
(Fig. 4C). Quantification analysis
demonstrated that the quantities of collagen I+ cells
were significant increased in the CCl4-treated rats
compared with the healthy controls. NaHS treatment decreased the
quantities of collagen I+ cells in the
CCl4-treated rats (Fig.
4D).
Discussion
In the present study, we found that NaHS, a
bioactive compound that releases H2S, suppressed the
proliferation and cell cycle progression and induced the apoptosis
of HSCs. The underlying mechanisms may be related to decreases in
the levels of intracellular ROS, free calcium and collagen I
protein expression. The effects of NaHS were also preliminarily
confirmed by its ability to attenuate liver fibrosis and reduce
collagen I protein expression levels in CCl4-induced
rats.
HSC activation plays a central role in hepatic
fibrosis and is accelerated by a self-amplification effect.
Therefore, the inhibition of HSC activation is the focus of hepatic
fibrosis treatments. HSC-T6 is an immortalized rat liver stellate
cell line showing an activated phenotype of HSCs and a
fibroblast-like morphology, and is often used in the investigation
of hepatic fibrosis (12). In our
study, we found that exogenous H2S suppressed the
proliferation of HSC cells in a dose-dependent manner, which may be
caused by G1 phase cell cycle arrest and enhanced apoptosis in the
NaHS-treated HSCs.
Intracellular ROS and oxidative stress contribute
significantly to the activation of HSC and hepatic fibrosis
(13). The overproduction of ROS
results in oxidative stress, which is a link between chronic liver
injury and hepatic fibrosis (14).
Iron deposition is another characteristic of hepatic fibrosis which
may either directly activate HSC or lead to lipid peroxidation
(15). In the current study, we
applied Fe-NTA to HSC cells to simulate oxidative stress
conditions. The Fe-NTA treatment increased intracellular ROS levels
and this increase was attenuated by NaHS, indicating that an
antioxidation effect may participate in the inhibition of activated
HSC cells. H2S has previously demonstrated a protective
effect through the inhibition of oxidative stress in the rat
gastric mucosal epithelium (16).
The increased contractility of HSCs promotes
fibrosis by regulating sinusoidal blood flow and ECM remodeling
(17), which is mediated by
Ca2+-dependent signaling pathways (18). The inhibition of Ca2+
signaling in HSCs contributes to the attenuation of hepatic
fibrosis (19). Our results
revealed that Fe-NTA increased intracellular Ca2+ levels
in the HSC-T6 cells and that these levels were decreased by NaHS.
Since Fe-NTA induces oxidative stress in HSC-T6 cells, this
indicates that there is a positive correlation between
intracellular ROS and Ca2+ levels in HSCs. This
hypothesis is supported by a previous study in which a retinoic
acid derivative downregulated ROS generation and calcium influx
simultaneously in HSC-T6 cells and also reversed early liver
fibrosis (20).
The increased synthesis and decreased degradation of
the ECM is important in the pathophysiology of liver fibrogenesis,
leading to overproduction and deposition of ECM in the liver. The
ECM is mainly composed of type I and type III collagen, which are
produced primarily by HSCs (21).
In our study, NaHS decreased the intracellular Ca2+
levels in the HSC-T6 cells which indicates it may also affect ECM
remodeling. We further found that NaHS significantly decreased type
I collagen protein in Fe-NTA-induced HSC-T6 cells. Activated and
proliferative HSC is the main cause for the ECM protein deposition
that forms scar tissue during liver fibrogenesis. Our results
demonstrated that NaHS not only inhibits the proliferation and
activation of HSC, but also attenuates ECM remodeling.
To further confirm the antifibrotic effect of NaHS
in vivo, we established a hepatic fibrosis model by the
intraperitoneal injection of CCl4 to rats.
CCl4 has been widely used as a chemical agent to induce
hepatic fibrosis, and it is metabolized into trichloromethyl
radicals which lead to increased lipid peroxidation, depletion of
GSH and necrosis of hepatocytes (22). The present study demonstrated that
the administration of NaHS attenuated CCl4-induced
hepatic fibrosis, as evidenced by the reduction in the number of
HSCs and decreased expression of collagen I protein in liver
tissues. This is in accordance with previous reports on the
cytoprotective effects of NaHS against hepatotoxicity, liver
cirrhosis and portal hypertension in rats (23). This suggests that NaHS is able to
inhibit HSC proliferation and ECM synthesis in vivo and thus
shows therapeutic promise for hepatic fibrogenesis. It has been
reported that in rats with CCl4-induced hepatic
fibrosis, CCl4 downregulated the expression of CSE, the
major enzyme for H2S production in the liver, and that
NaHS did not change either hepatic CSE expression or
H2S-producing activity (23). In our study, it appears that the
injected NaHS released H2S in the body and directly
mediated its antifibrotic effect through the HSCs. It remains to be
further investigated whether the in vivo function of NaHS is
mediated by inhibition of proliferation, cell cycle arrest,
induction of apoptosis and decreased intracellular ROS and
Ca2+ levels in the HSCs of rats with hepatic
fibrosis.
In conclusion, the present study demonstrates that
NaHS suppresses the proliferation of HSC-T6 cells in a
dose-dependent manner and induces G1 cell cycle arrest and
apoptosis in Fe-NTA-induced HSC cells. It also attenuates
CCl4-induced hepatic fibrosis and ECM expression. These
findings suggest that exogenous H2S has promise in the
development of new therapeutic strategies for hepatic fibrosis.
References
|
1
|
Lanthier N, Horsmans Y and Leclercq IA:
The metabolic syndrome: how it may influence hepatic stellate cell
activation and hepatic fibrosis. Curr Opin Clin Nutr Metab Care.
12:404–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tarrats N, Moles A, Morales A, García-Ruiz
C, Fernández-Checa JC and Marí M: Critical role of tumor necrosis
factor receptor 1, but not 2, in hepatic stellate cell
proliferation, extracellular matrix remodeling, and liver
fibrogenesis. Hepatology. 54:319–327. 2011. View Article : Google Scholar
|
|
3
|
Li JT, Liao ZX, Ping J, Xu D and Wang H:
Molecular mechanism of hepatic stellate cell activation and
antifibrotic therapeutic strategies. J Gastroenterol. 43:419–428.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Łowicka E and Bełtowski J: Hydrogen
sulfide (H2S) - the third gas of interest for
pharmacologists. Pharmacol Rep. 59:4–24. 2007.
|
|
5
|
Gao Y, Yao X, Zhang Y, Li W, Kang K, Sun L
and Sun X: The protective role of hydrogen sulfide in myocardial
ischemia-reperfusion-induced injury in diabetic rats. Int J
Cardiol. 152:177–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ahmad FU, Sattar MA, Rathore HA, Abdullah
MH, Tan S, Abdullah NA and Johns EJ: Exogenous hydrogen sulfide
(H2S) reduces blood pressure and prevents the
progression of diabetic nephropathy in spontaneously hypertensive
rats. Ren Fail. 34:203–210. 2012.PubMed/NCBI
|
|
7
|
Zhang LM, Jiang CX and Liu DW: Hydrogen
sulfide attenuates neuronal injury induced by vascular dementia via
inhibiting apoptosis in rats. Neurochem Res. 34:1984–1992. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fiorucci S, Distrutti E, Cirino G and
Wallace JL: The emerging roles of hydrogen sulfide in the
gastrointestinal tract and liver. Gastroenterology. 131:259–271.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kabil O, Vitvitsky V, Xie P and Banerjee
R: The quantitative significance of the transsulfuration enzymes
for H2S production in murine tissues. Antioxid Redox
Signal. 15:363–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Siebert N, Cantré D, Eipel C and Vollmar
B: H2S contributes to the hepatic arterial buffer
response and mediates vasorelaxation of the hepatic artery via
activation of K(ATP) channels. Am J Physiol Gastrointest Liver
Physiol. 295:G1266–G1273. 2008.
|
|
11
|
Ebrahimkhani MR, Mani AR and Moore K:
Hydrogen sulphide and the hyperdynamic circulation in cirrhosis: a
hypothesis. Gut. 54:1668–1671. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vogel S, Piantedosi R, Frank J, Lalazar A,
Rockey DC, Friedman SL and Blaner WS: An immortalized rat liver
stellate cell line (HSC-T6): a new cell model for the study of
retinoid metabolism in vitro. J Lipid Res. 41:882–893.
2000.PubMed/NCBI
|
|
13
|
Ming-Ju H, Yih-Shou H, Tzy-Yen C and
Hui-Ling C: Hepatitis C virus E2 protein induce reactive oxygen
species (ROS)-related fibrogenesis in the HSC-T6 hepatic stellate
cell line. J Cell Biochem. 112:233–243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ghatak S, Biswas A, Dhali GK, Chowdhury A,
Boyer JL and Santra A: Oxidative stress and hepatic stellate cell
activation are key events in arsenic induced liver fibrosis in
mice. Toxicol Appl Pharmacol. 251:59–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo L, Enzan H, Hayashi Y, Miyazaki E, Jin
Y, Toi M, Kuroda N and Hiroi M: Increased iron deposition in rat
liver fibrosis induced by a high-dose injection of
dimethylnitrosamine. Exp Mol Pathol. 81:255–261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yonezawa D, Sekiguchi F, Miyamoto M,
Taniguchi E, Honjo M, Masuko T, Nishikawa H and Kawabata A: A
protective role of hydrogen sulfide against oxidative stress in rat
gastric mucosal epithelium. Toxicology. 241:11–18. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Soon RK Jr and Yee HF Jr: Stellate cell
contraction: role, regulation, and potential therapeutic target.
Clin Liver Dis. 12:791–803. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iizuka M, Murata T, Hori M and Ozaki H:
Increased contractility of hepatic stellate cells in cirrhosis is
mediated by enhanced Ca2+-dependent and
Ca2+-sensitization pathways. Am J Physiol Gastrointest
Liver Physiol. 300:G1010–G1021. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim SY, Cho BH and Kim UH: CD38-mediated
Ca2+ signaling contributes to angiotensin II-induced
activation of hepatic stellate cells: attenuation of hepatic
fibrosis by CD38 ablation. J Biol Chem. 285:576–582.
2010.PubMed/NCBI
|
|
20
|
Yang KL, Chang WT, Chuang CC, Hung KC and
Li EI: Antagonizing TGF-beta induced liver fibrosis by a retinoic
acid derivative through regulation of ROS and calcium influx.
Biochem Biophys Res Commun. 365:484–489. 2008. View Article : Google Scholar
|
|
21
|
Wells RG: Cellular sources of
extracellular matrix in hepatic fibrosis. Clin Liver Dis.
12:759–768. 2008.PubMed/NCBI
|
|
22
|
Weber LW, Boll M and Stampfl A:
Hepatotoxicity and mechanism of action of haloalkanes: carbon
tetrachloride as a toxicological model. Crit Rev Toxicol.
33:105–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tan G, Pan S, Li J, Dong X, Kang K, Zhao
M, Jiang X, Kanwar JR, Qiao H, Jiang H and Sun X: Hydrogen sulfide
attenuates carbon tetrachloride-induced hepatotoxicity, liver
cirrhosis and portal hypertension in rats. PLoS One. 6:e259432011.
View Article : Google Scholar : PubMed/NCBI
|