Introduction
Diffuse brain injury (DBI) is a leading cause of
mortality and disability in young individuals. The primary damage
in DBI is thought to result from mechanical forces applied to the
skull and brain at the time of impact, leading to focal or diffuse
brain injury patterns. In comparison, secondary brain damage
following DBI evolves progressively and is characterized by a
complex cascade of biochemical events that cause brain edema and
neuronal death and aggravate neurological dysfunctions, including
learning and memory deficits.
Nicotinamide adenine dinucleotide phosphate (NADPH)
oxidase (NOX) is a multi-subunit enzyme complex localized to the
plasma membrane of cells and is a major source of reactive oxygen
species (ROS). This enzyme is expressed in neurons, astrocytes and
microglia (1–3). There are several subunits of NOX,
termed NOX1–5(1,4).
NOX2 is a catalytic subunit of NOX. A previous study
demonstrated that NOX2 is localized to the cerebral
cortex and hippocampal CA1 subregion (5). Limited knowledge is currently
available on the role of NOX2 in DBI and it remains
unclear whether alterations in NOX2 expression and NOX
activity are associated with secondary damage following DBI in
rats.
Apocynin was first described by Schmiedeberg in 1883
and was isolated from the roots of Apocynum cannabinum
(Canadian hemp). Apocynin has since been utilized in a number of
experimental models as a NOX complex inhibitor (6,7),
however, the mechanism of inhibition of NOX is not well understood
in rats following DBI.
In the present study, alterations in NOX2
protein expression and NOX activity in the CA1 subregion of the
hippocampus following DBI induction were investigated. In addition,
we aimed to determine whether pre-treatment with apocynin results
in the attenuation of brain edema and improves spatial cognitive
functions by modulation of NOX2 protein expression and
NOX activation following DBI induction. The results of the present
study may generate insight into the efficacy of apocynin against
secondary damage following DBI induction, through the inhibition of
NOX2 expression and NOX activity.
Materials and methods
Animals and DBI model
All experimental procedures were performed in
accordance with the guidelines of the Chinese Council on Animal
Protection and were approved by the Hebei Medical University
Committee for the use of animals in research. A total of 140 male
Sprague-Dawley rats (age, 12–16 weeks; weight, 350–375 g; Tangshan,
China) were used in the present study. All animals were housed with
a standard 12 h light/dark cycle and free access to water and food
prior to and following surgery or sham surgery. The rat model of
DBI was created using a modified weight-drop device, as described
previously by Sawauchi et al(8). Briefly, rats were anesthetized with
sodium pentobarbial (Nembutal, 60 mg/kg). Under anaesthesia, a
midline incision was performed to expose the skull the between
bregma and lambda suture lines and a steel disk (diameter, 10 mm;
thickness, 3 mm) was adhered to the skull using dental acrylic.
Animals were placed on a foam mattress underneath a weight-drop
device in which a 450-g weight falls freely through a vertical tube
from 1.5 m onto the steel disk. Sham-operated animals underwent the
same surgical procedure without weight-drop impact. Rats were
housed in individual cages following surgery and placed on heat
pads (37°C) for 24 h to maintain normal body temperature during the
recovery period.
Group and drug administration
Rats were randomly divided into 4 groups: sham, DBI
untreated, DBI treated with vehicle (DMSO) and DBI treated with
apocynin groups. Apocynin (50 mg/kg body mass) (9,10)
and the vehicle were administered by intraperitoneal injection 30
min prior to sham surgery or DBI induction (11).
Western blot analysis of NOX2
protein expression
At 48 and 72 h following DBI induction, rats were
anesthetized and underwent intracardiac perfusion with 0.1 mol/l
phosphate-buffered saline (pH 7.4). Hippocampal CA1 subregions were
rapidly isolated, total proteins were extracted and protein
concentration was determined by the BCA reagent (Solarbio, Beijing,
China) method. Equal amounts (50 μg) of protein were subjected to
10% SDS-PAGE and electrotransferred onto a hydrophobic PVDF
membrane (Roche Diagnostics, Mannheim, Germany). Following
blocking, the membrane was incubated overnight at 4°C with primary
antibodies against NOX2 (1:200) and β-actin (1:200; both
purchased from Santa Cruz Biotechnology; Santa Cruz, CA, USA).
Following incubation with a titrated secondary antibody (1:2,000;
Cell Signaling Technology, Inc., Danvers, MA, USA), the immunoblot
on the membrane was visualized by development with an enhanced
chemiluminescence detection system and densitometric signals were
quantified using an imaging program. Immunoreactive bands were
normalized to intensity of corresponding bands for β-actin. Results
were analyzed using the National Institutes of Health Image 1.41
software (Bethesda, MD, USA).
NOX activity assay
NOX activity was determined by a colorimetric method
(12,13) based on changes in NADPH consumption
monitored by the decrease in absorbance at λ=340 nm in the presence
of DPI. Hippocampal CA1 tissue samples collected at 48 and 72 h
following DBI induction were homogenized in Krebs-Ringer phosphate
buffer at pH 7.4. Homogenates were centrifuged at 1,000 × g for 10
min at 4°C and the pellets were discarded. Supernatants were spun
at 13,000 × g in an ultracentrifuge for 20 min at 4°C and membrane
fractions were separated. Enzyme assays were performed in a final
volume of 1 ml containing 50 mM Krebs-Ringer phosphate buffer (pH
7.0), 1 mM EGTA, 150 mM sucrose, 0.5 mM lucigenin, 0.1 mM NADPH
solution and 50 μg membrane fractions. Photoemissions, expressed in
terms of relative light units (RLU), were recorded every 1 min
continuously for 5 min by a standard luminometer. All values were
standardized to the amount of protein and were calculated as
RLU/μg/minute.
Evaluation of brain edema
Brain edema was evaluated by analysis of brain water
content with the wet-dry weight method as described previously
(14). Following this, animals
were sacrificed by decapitation under anesthesia at 48 and 72 h
following DBI induction or sham surgery. Brains were separated and
weighed immediately to obtain wet weight and dried in a desiccating
oven for 24 h at 100°C. Dry tissues were weighed again. The
percentage of water in the tissues was calculated according to the
formula: % brain water = [(wet weight - dry weight)/wet weight]
×100.
Assessment of the spatial learning
ability using the Morris water maze
Spatial learning ability was assessed using a Morris
water maze as described previously (15). The maze consists of a black
circular pool (diameter, 180 cm; height, 45 cm) filled with water
(depth, 30 cm) at 26°C and virtually divided into 4 equivalent
quadrants: north (N), west (W), south (S) and east (E). A 2-cm
submerged escape platform (diameter, 12 cm; height, 28 cm; made
opaque with paint) was placed in the center of one of the
quadrants, equidistant from the sidewall and the center of the
pool. Rats were trained to find the platform prior to DBI or sham
surgery. For each trial, the rat was randomly placed into a
quadrant start point (N, S, E or W) facing the wall of the pool and
allowed a maximum of 60 sec to escape to a platform, rats which
failed to escape within 90 sec were placed on the platform for a
maximum of 20 sec and returned to the cage for a new trial
(intertrial interval 20 sec). Maze performance was recorded by a
video camera suspended above the maze and interfaced with a video
tracking system (HVS Imaging, Hampton, UK). The average escape
latency of a total of five trials was calculated. Tests were
conducted 72 h following DBI induction.
Statistical analysis
Data are expressed as the means ± standard error.
Statistical analysis was performed using ANOVA and followed by the
Student-Newman-Keuls post-hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
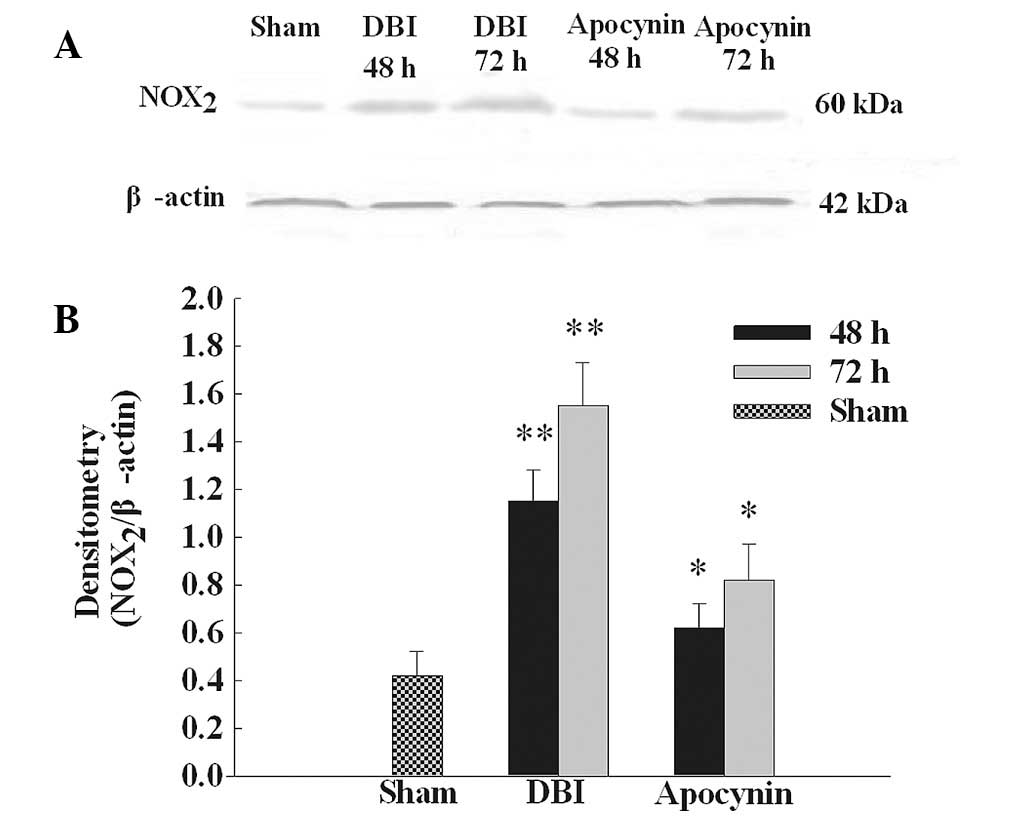
Apocynin treatment reduces upregulation
of NOX2 protein expression
NOX2 protein expression was analyzed by
western blot analysis (Fig. 1A).
NOX2 protein expression was identified at low levels in
the CA1 region of the hippocampus in the sham group. Levels were
markedly increased at 48 and 72 h following DBI induction. As
demonstrated in Fig. 1B,
NOX2 protein band intensity was quantified and results
demonstrated that apocynin pre-treatment significantly inhibited
the upregulation of NOX2 protein levels compared with
the DBI groups.
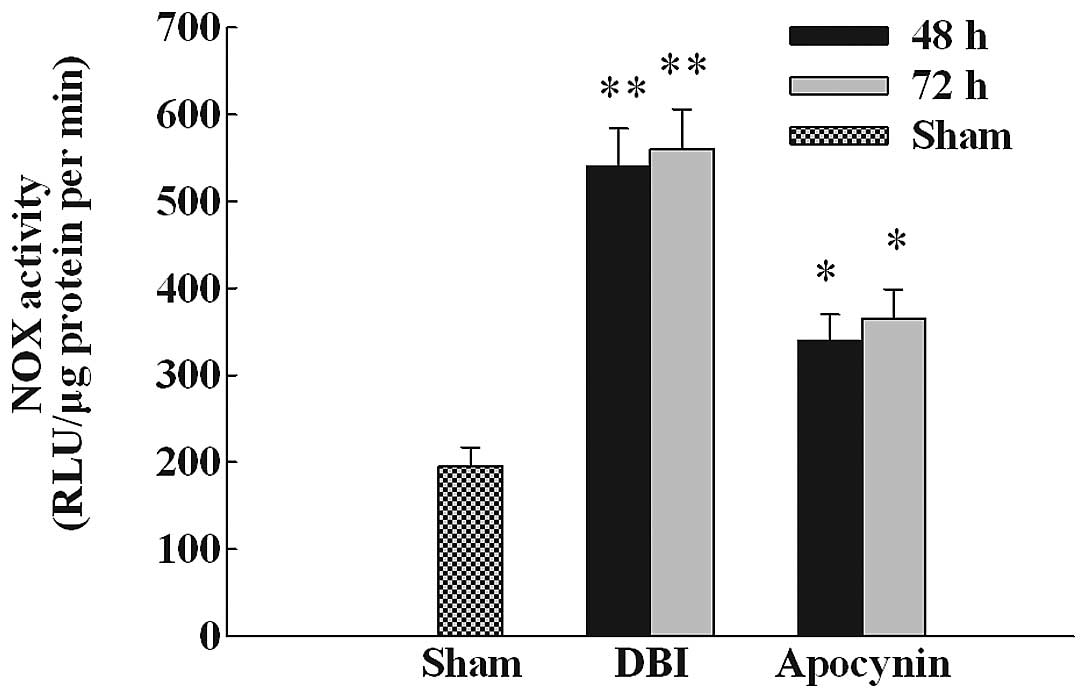
Apocynin treatment depresses NOX
activity
Since apocynin was found to inhibit the increased
protein expression of NOX2 in the CA1 subregion of the
hippocampus following DBI induction, we then performed a
colorimetric assay to determine whether apocynin treatment reduces
NOX activity. As demonstrated in Fig.
2, a marked elevation of NOX activity in the CA1 region was
observed at 48 and 72 h in the DBI untreated group following DBI
induction compared with the sham group. Apocynin treatment
significantly attenuated NOX activity in the treated group compared
with the DBI untreated or the DBI group treated with the vehicle;
however, NOX activity levels remained higher than the sham
group.
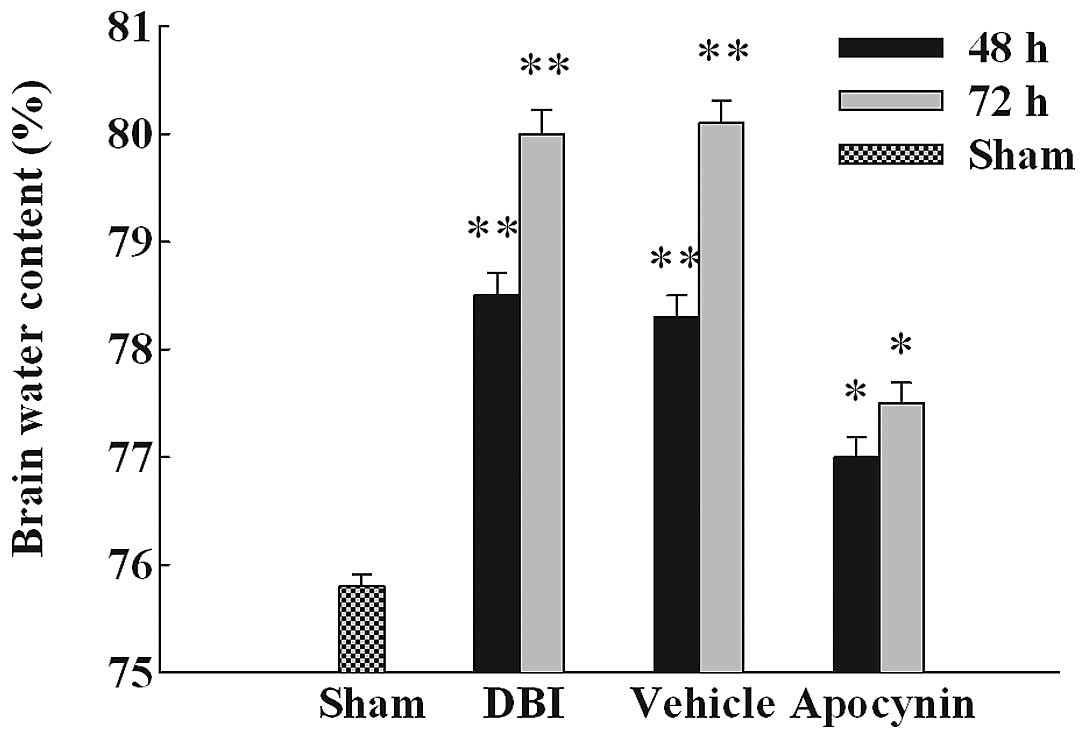
Apocynin treatment attenuates brain
edema
The wet-dry method was used to evaluate brain edema.
In order to examine whether brain edema is associated with the
upregulation of NOX2 and NOX activation, we used
apocynin, a specific inhibitor of NOX, as pre-treatment prior to
DBI. As demonstrated in Fig. 3,
DBI induced a significant increase in brain edema at 48 and 72 h in
the DBI group compared with sham control. Pre-treatment with
apocynin significantly reduced brain edema following DBI
induction.
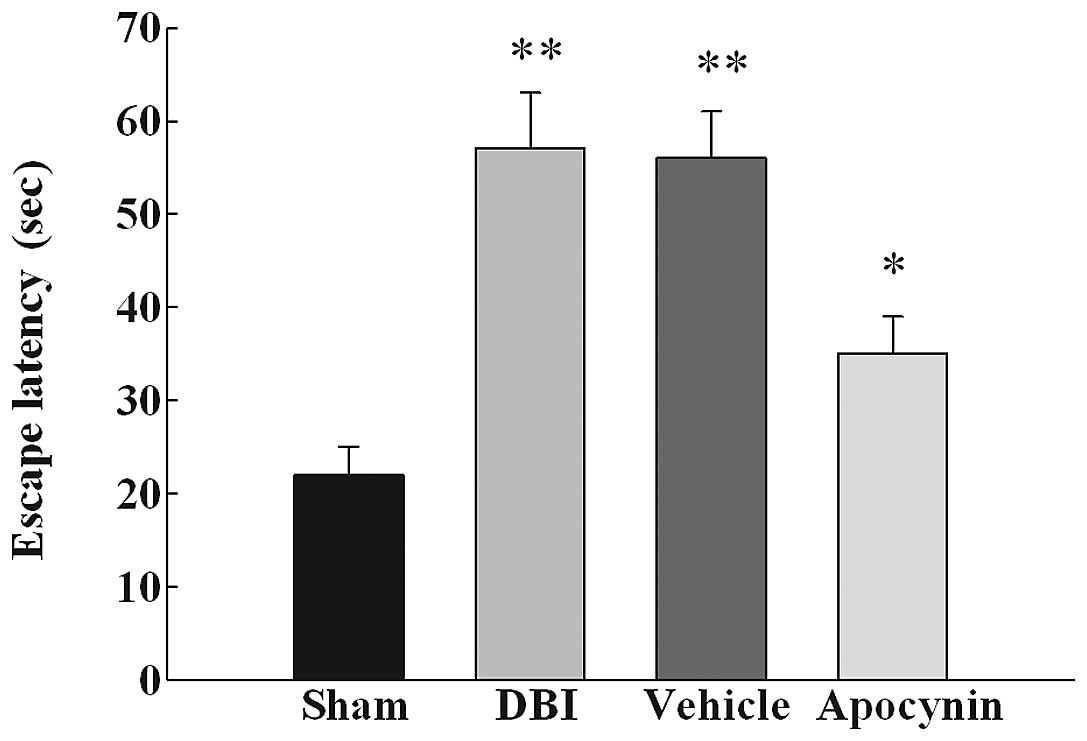
Apocynin treatment improves learning and
memory ability
Since apocynin treatment was found to inhibit NOX
activation and attenuate brain edema, we then examined whether
apocynin treatment can improve spatial learning function using a
Morris water maze 72 h following DBI induction or sham surgery. As
demonstrated in Fig. 4, DBI caused
a significant deficit in spacial learning at 72 h following DBI
induction in the DBI groups compared with the sham group. Apocynin
treatment reduced the escape latency in the treated group compared
with DBI untreated group or the DBI group treated with the
vehicle.
Discussion
Secondary damage following primary DBI leads to
brain edema and neuronal cell death and aggravates neurological
disfunctions. Effective management is imperative for promoting
anatomical and functional recovery. NOX is a major complex that
produces reactive ROS and has previously been associated with
secondary damage, leading to secondary brain injury, following
brain ischemia reperfusion (16)
and cerebral ischemia stroke (17,18).
However, the role of NOX in the aggravation of brain damage
following DBI in the rat model remains poorly understood.
In the present study, we confirmed that
NOX2 protein expression and NOX activity were enhanced
at 48 and 72 h following DBI induction. These observations were
accompanied by an increase in brain water content and profound
neurological dysfunction. Treatment with apocynin not only reduced
upregulated NOX2 protein expression and NOX activity,
but markedly attenuated brain edema and spatial learning deficits,
as determined by Morris water maze performance. These results
indicate that following DBI, NOX activation is pivotal in the
additional aggravation of secondary brain damage.
Elucidation of the molecular mechanisms of brain
edema is an important area of investigation, with the ultimate aim
to develop new therapeutic interventions for the prevention of
edema formation following traumatic brain injury. NOX2,
a membrane catalytic subunit of NOX, has been demonstrated to be
upregulated in the ischemic period (19). Previous studies using
NOX2 mutant knockout mice or pre-treatment with the
specific NOX2 inhibitor, gp91ds-tat, have demonstrated
significantly attenuated neuronal damage and edema following
traumatic brain injury (20–22).
Dohi et al(20) and Lo
et al(23) found that the
inhibition of NOX activity improved neurological functions
following surgically-induced brain injury or traumatic brain injury
using transgenic mice lacking the NOX2 subunit of NOX.
These studies all indicate that the beneficial neuroprotective
effects of apocynin are specifically due to the inhibition of
NOX2 expression. Consistent with these previous studies,
the results of the present study revealed that treatment with
apocynin reduced enhanced NOX2 expression and attenuated
brain edema at 48 and 72 h following DBI induction. In addition,
cognitive impairment was improved by apocynin treatment, which may
be mediated, in part, by the downregulation of NOX2
following DBI.
NOX activity has previously been demonstrated to
increase during brain injury following experimental ischemia
(24). The activation of NOX is
mediated by the translocation of cytosolic subunits to the cell
membrane and fusion with NOX2. The active complex
transfers electrons to oxygen, producing superoxide anions, a
precursor of ROS (1,25). The neuroprotective effects of
apocynin have been hypothesized to be achieved by the
downregulation of NOX2 expression and NOX activity.
These events may depress the reduction of ROS (26), attenuating the permeability of the
blood-brain barrier and further reducing brain edema following DBI.
Alternative mechanisms of NOX activity associated with brain
damage, including inflammation (27) and neuronal death (28) i.e., apoptosis (29) and autophagy cell death (30) should be investigated further in
order to evaluate the detailed role of NOX in secondary brain
damage following DBI.
In conclusion, the results of the present study
demonstrate that the upregulation of NOX2 expression and
NOX activation are involved in secondary brain damage following
DBI. Pre-treatment with apocynin attenuates brain edema and
improves spatial learning ability. This neuroprotection is
associated with the blockage of NOX activity. In addition, the
present study indicates that targeting NOX with specific NOX
inhibitors may have clinical efficacy in DBI.
Acknowledgements
The present study was supported by a grant from the
Natural Science Foundation of Hebei Province (no. H2012401071).
Abbreviations:
|
DMSO
|
dimethyl sulphoxide
|
|
DBI
|
diffuse brain injury
|
|
NOX
|
nicotinamide adenine dinucleotide
phosphate oxidase
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Infanger DW, Sharma RV and Davisson RL:
NADPH oxidases of the brain: distribution, regulation and function.
Antioxid Redox Signal. 8:1583–1596. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim MJ, Shin KS, Chung YB, Jung KW, Cha CI
and Shin DH: Immunohistochemical study of p47Phox and gp91Phox
distributions in rat brain. Brain Res. 1040:178–186. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sorce S and Krause KH: NOX enzymes in the
central nervous system: from signaling to disease. Antioxid Redox
Signal. 11:2481–2504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Serrano F, Kolluri NS, Wientjes FB, Card
JP and Klann E: NADPH oxidase immunoreactivity in the mouse brain.
Brain Res. 24:193–198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lafeber FP, Beukelman CJ, van den Worm E,
et al: Apocynin, a plant-derived, cartilage-saving drug, might be
useful in the treatment of rheumatoid arthritis. Rheumatology
(Oxford). 38:1088–1093. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang Y, Chan MM, andrews MC, et al:
Apocynin but not allopurinol prevents and reverses
adrenocorticotropic hormone-induced hypertension in the rat. Am J
Hypertens. 18:910–916. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sawauchi S, Marmarou A, Beaumont A, Tomita
Y and Fukui S: A new rat model of diffuse brain injury associated
with acute subdural hematoma: assessment of varying hematoma
volume, insult severity and the presence of hypoxemia. J
Neurotrauma. 20:613–622. 2003.
|
|
9
|
Kahles T, Luedike P, Endres M, et al:
NADPH oxidase plays a central role in blood-brain barrier damage in
experimental stroke. Stroke. 38:3000–3006. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang LL, Ye K, Yang XF and Zheng JS:
Apocynin attenuates cerebral infarction after transient focal
ischaemia in rats. J Int Med Res. 35:517–522. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Connell BJ, Saleh MC, Khan BV and Saleh
TM: Apocynin may limit total cell death following cerebral ischemia
and reperfusion by enhancing apoptosis. Food Chem Toxicol.
49:3063–3069. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Murillo MM, Carmona-Cuenca I, Del Castillo
G, et al: Activation of NADPH oxidase by transforming growth
factor-beta in hepatocytes mediates up-regulation of epidermal
growth factor receptor ligands through a nuclear
factor-kappaB-dependent mechanism. Biochem J. 405:251–259. 2007.
View Article : Google Scholar
|
|
13
|
Zhang QG, Raz L, Wang R, et al: Estrogen
attenuates ischemic oxidative damage via an estrogen receptor
alpha-mediated inhibition of NADPH oxidase activation. J Neurosci.
29:13823–13836. 2009. View Article : Google Scholar
|
|
14
|
Tang J, Liu J, Zhou C, et al: Mmp-9
deficiency enhances collagenase-induced intracerebral hemorrhage
and brain injury in mutant mice. J Cereb Blood Flow Metab.
24:1133–1145. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hui-guo L, Kui L, Yan-ning Z and Yong-jian
X: Apocynin attenuate spatial learning deficits and oxidative
responses to intermittent hypoxia. Sleep Med. 11:205–212. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Woodfin A, Hu DE, Sarker M, Kurokawa T and
Fraser P: Acute NADPH oxidase activation potentiates
cerebrovascular permeability response to bradykinin in
ischemia-reperfusion. Free Radic Biol Med. 50:518–524. 2011.
View Article : Google Scholar
|
|
17
|
Yoshioka H, Niizuma K, Katsu M, et al:
NADPH oxidase mediates striatal neuronal injury after transient
global cerebral ischemia. J Cereb Blood Flow Metab. 31:868–880.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kelly KA, Li X, Tan Z, VanGilder RL, Rosen
CL and Huber JD: NOX2 inhibition with apocynin worsens stroke
outcome in aged rats. Brain Res. 1292:165–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hur J, Lee P, Kim MJ, Kim Y and Cho YW:
Ischemia-activated microglia induces neuronal injury via activation
of gp91phox NADPH oxidase. Biochem Biophys Res Commun.
391:1526–1530. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dohi K, Ohtaki H, Nakamachi T, et al:
Gp91phox (NOX2) in classically activated microglia exacerbates
traumatic brain injury. J Neuroinflammation. 7:412010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang QG, Laird MD, Han D, et al: Critical
role of NADPH oxidase in neuronal oxidative damage and microglia
activation following traumatic brain injury. PLoS One.
7:e345042012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jackman KA, Miller AA, De Silva TM, Crack
PJ, Drummond GR and Sobey CG: Reduction of cerebral infarct volume
by apocynin requires pretreatment and is absent in Nox2-deficient
mice. Br J Pharmacol. 156:680–688. 2009.PubMed/NCBI
|
|
23
|
Lo W, Bravo T, Jadhav V, Titova E, Zhang
JH and Tang J: NADPH oxidase inhibition improves neurological
outcomes in surgically-induced brain injury. Neurosci Lett.
414:228–232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vallet P, Charnay Y, Steger K, et al:
Neuronal expression of the NADPH oxidase NOX4 and its regulation in
mouse experimental brain ischemia. Neuroscience. 132:233–238. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cairns B, Kim JY, Tang XN and Yenari MA:
NOX inhibitors as a therapeutic strategy for stroke and
neurodegenerative disease. Curr Drug Targets. 13:199–206. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zia MT, Csiszar A, Labinskyy N, et al:
Oxidative-nitrosative stress in a rabbit pup model of germinal
matrix hemorrhage: role of NAD(P)H oxidase. Stroke. 40:2191–2198.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mander PK, Jekabsone A and Brown GC:
Microglia proliferation is regulated by hydrogen peroxide from
NADPH oxidase. J Immunol. 176:1046–1052. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jana A and Pahan K: Fibrillar amyloid-beta
peptides kill human primary neurons via NADPH oxidase-mediated
activation of neutral sphingomyelinase. Implications for
Alzheimer’s disease. J Biol Chem. 279:51451–51459. 2004.PubMed/NCBI
|
|
29
|
Bobba A, Atlante A, Petragallo VA and
Marra E: Different sources of reactive oxygen species contribute to
low potassium-induced apoptosis in cerebellar granule cells. Int J
Mol Med. 21:737–745. 2008.PubMed/NCBI
|
|
30
|
Luo CL, Li BX, Li QQ, et al: Autophagy is
involved in traumatic brain injury-induced cell death and
contributes to functional outcome deficits in mice. Neuroscience.
184:54–63. 2011. View Article : Google Scholar : PubMed/NCBI
|