Introduction
The Janus kinase/signal transducer and activator of
transcription pathway (JAK/STAT signaling pathway) is an
intracellular signaling pathway possessing interferon-like effects.
The pathway is able to transduce the intracellular signals of
various cytokines [tumor necrosis factor-α (TNF-α), TGF-β and IL]
and thus regulate multiple physiological and pathophysiological
processes, including immune responses, cell proliferation and
differentiation, cell apoptosis, inflammation and cancer (1). The JAK/STAT pathway is also able to
promote gene transcription following activation (2). This signaling pathway may be
activated by several factors that include ischemia, hypoxia,
inflammation and overactive renin-angiotensin processes. Despite
its potential role in the ischemic pathology of cardiac muscle, the
correlation between the JAK/STAT signaling pathway and the onset of
acute myocardial infarction (AMI) has not been elucidated.
Nuclear factor κB (NF-κB) is a nucleoprotein that is
able to specifically bind to the 10-bp nucleotide sequence in the
immunoglobulin κ light chain gene and promote the expression of the
κ gene (3). Following activation,
NF-κB quickly translocates into the nucleus from the cytoplasm and
then binds to the specific κB sites in the inducible gene promoter
sequence. Then, various target gene expression may be induced,
which may lead to cell and tissue damage and the development of
various pathophysiological processes (4–8),
including coronary heart disease and AMI.
In the present study, an AMI rat model was
successfully established and AG490, a specific blocking agent, was
used to inhibit the JAK/STAT signaling pathway. Plasma TNF-α levels
in the AMI rats were measured by ELISA. NF-κB protein expression
was detected in the myocardial cells of the AMI rats using
immunohistochemistry. The role of the JAK/STAT signaling pathway in
the onset of AMI, as well as its effect and significance on NF-κB
expression in the myocardium and TNF-α levels in the plasma of AMI
rats, were investigated.
Materials and methods
Animals
Wistar rats were obtained from the Laboratory Animal
Center of Zhejiang University (Hangzhou, China). All rats were
female and maintained under specific pathogen-free conditions. The
experimental protocol was approved by the Animal Care and Use
Committee of Zhejiang University and was performed in accordance
with the Guide for the Care and Use of Laboratory Animals (NIH
Publication No. 85–23, National Academy Press, WA, USA; revised
1996).
Main reagents
Pepsin, dimethyl sulfoxide (DMSO), TNF-α ELISA kit,
normal goat serum, 3,3′-diaminobenzidine tetrahydrochloride (DAB),
secondary antibody (rabbit anti-goat), streptavidin
biotin-peroxidase complex (SABC), antigen restoration solution,
occlusive solution and rabbit antidigoxin were purchased from
Boster Biological Technology Ltd. (Wuhan, China), AG490 from Sigma
Chemical Co. (St. Louis, MO, USA) and goat anti-rat primary
antibody from Biocompare Co. (South San Francisco, CA, USA).
Establishment of the AMI animal models
and specimen collection
The animals were anesthetized using an
intraperitoneal injection of pentobarbital sodium (40–60 mg/kg).
Following adequate anesthesia, the animals were intubated in a
supine position and ventilated on room air via a small animal
ventilator (HX100E, TME Co., China). A left thoracotomy was
performed at the third intercostal space and the pericardium was
opened. The left coronary artery was ligated permanently beneath
the left atrial appendage with 6–0 sterile silk. The effectiveness
of the ligation was confirmed when the color in the left ventricle
below the ligation site changed from red to white. After the
ligation was completed, the thorax was closed (9–11).
Female SD rats were used. A total of 30 rats were
randomly selected for sham surgery and the rest underwent coronary
artery ligation. Rats that survived the coronary artery ligation
were randomly divided into a further 3 groups. Group A (sham
surgery group, n=30) underwent the surgery without ligation. Plasma
and heart samples were collected at 7, 14 and 28 days after surgery
(groups A1, A2 and A3, respectively, with 10 animals in each
subgroup). Group B (myocardial infarction control group, n=30)
underwent ligation of the left anterior descending coronary artery.
Plasma and heart samples were collected at 7, 14 and 28 days after
surgery (groups B1, B2 and B3, respectively, with 10 animals in
each subgroup). Group C (AG490 treatment + myocardial infarction
group, n=30) also underwent coronary artery ligation. However,
AG490 (5 mg/kg/day) was administered by intraperitoneal injection
at 96 h after ligation. The consecutive treatment lasted until the
27th day. The plasma and heart samples were collected at 7, 14 and
28 days after the surgery (groups C1, C2 and C3, respectively, with
10 animals in each subgroup). Group D (DMSO + myocardial infarction
group, n=30) underwent coronary artery ligation and received 45%
DMSO via intraperitoneal injection at 96 h after ligation. The
consecutive treatment lasted until the 27th day. The plasma and
heart samples were collected at 7, 14 and 28 days after surgery
(groups D1, D2 and D3, respectively, with 10 animals in each
subgroup). Blood (3 ml) was extracted by cardiac puncture and added
to a tube with EDTA at the end of the experiment. The tube was
gently agitated and the blood was centrifuged for 30 min. It was
then stored at −20°C. At the end of the experiment the chest was
also opened and the heart was removed. The heart was fixed in 10%
buffered formalin for 24 h and embedded in paraffin for
immunohistochemical detection.
Measurement of infarct size
After the rat heart was harvested, the left
ventricle was separated from the heart and weighed. It was sliced
into 2–3-mm sections parallel to the atrioventricular groove. The
sections were then incubated in 1% triphenyltetrazolium chloride
(TTC) solution prepared in a pH 7.4 phosphate buffer for 30 min at
37°C. The slices were then incubated in the stain for 20 min at
37°C with constant agitation. In viable myocardium, TTC was
converted by dehydrogenase enzymes to formazan, a red pigment that
stained the tissue dark red. Non-viable infarcted myocardium that
did not take up the TTC stain remained pale in color. The pale
necrotic tissue was separated from the stained portions and weighed
on an electronic balance. The weight ratio of the infarct size was
calculated as the weight of the infarction zone divided by the
weight of the heart × 100.
ELISA test of plasma TNF-α (9)
The coated antibody for TNF-α was added to the ELISA
plate and stored at 4°C for 48 h. The plate was then rinsed three
times. The diluted sample solution and the standard solution were
added and incubated at 37°C for 1 h. The HRP-conjugated anti-TNF-α
solution was added and incubated at 37°C for 1 h. The plate was
washed three times and the prepared ABTS
[2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid)] chromogenic
substrate reagent was added at 37°C for 25 min. The optical density
(OD) value was measured by a microplate reader set at 450 nm.
Immunohistochemistry of NF-κB protein
(9)
The immunostaining procedure was performed on rat
myocardium sections embedded in paraffin. The sections were
deparaffinized in xylene and rapidly rehydrated using graded
alcohols. Excess liquid was removed, and the sections were washed
in phosphate-buffered solution (PBS; pH 7.4) with 0.05% Tween-20
(Sigma Chemical Co.). To reduce non-specific binding, normal goat
serum (1% in PBS) was applied to the slides for 30 min at 37°C and
then incubated with monoclonal goat anti-rat primary antibody on
consecutive sections. Following rinsing with PBS-T, the sections
were incubated with specific secondary antibodies (rabbit
anti-goat) for 1 h and then incubated with the biotinylated
tyramide and streptavidin-peroxidase complexes. The immunoreaction
was visualized using 0.015% H2O2 in DAB/TBS
for 10 min at room temperature. In order to evaluate the extent of
non-specific binding in the immunohistochemical experiments,
control sections were incubated in the absence of the primary
antibody.
The slides were examined under a microscope
(Olympus, Tokyo, Japan) at ×400 magnification. Eight areas per
slide and six non-successive slides per sample were counted for the
NF-κB-positive stained cells. The number of positive cells was
expressed as a percentage (%) according to this formula: Percentage
of NF-κB-positive stained cells (%) = number of NF-κB-positive
cardiomyocytes/total number of cardiomyocytes × 100.
Detection of left ventricular mass index
(LVMI)
The body weight, left ventricular weight and heart
weight in each group were measured at 28 days after surgery. The
rats were sacrificed rapidly after their body weights were
measured. The heart was removed quickly and placed into cold normal
saline to clear the residual blood from the heart chamber. The
heart was then dried with filter paper and weighed. The left
ventricle was then separated along the auriculoventricular ring and
the interventricular groove. The left ventricle was weighed and the
LVMI was calculated. LVMI = left ventricular weight/body weight
(mg/g).
Statistical analysis
Statistical analyses were performed to evaluate the
differences between the experimental and control groups. The
differences between groups were evaluated by using the one-way
analysis of variance and the Student’s t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
JAK/STAT signaling pathway and the change
of myocardial infarct area
The infarction area in the AG490 treatment group
(group C) was significantly less than that of the myocardial
infarction control group (group B; P<0.05; Table I). There was no significant
difference between the DMSO treatment group (group D) and group
B.
 | Table IPercentage change in the myocardial
infarct area (%). |
Table I
Percentage change in the myocardial
infarct area (%).
| Group | 7 days | 14 days | 28 days |
|---|
| A | 0 | 0 | 0 |
| B | 23.65±5.33 | 20.79±7.08 | 21.46±5.81 |
| C | 16.25±5.21a | 15.87±3.86a | 15.58±5.03a |
| D | 22.90±5.07b | 21.26±5.71b | 22.03±4.98b |
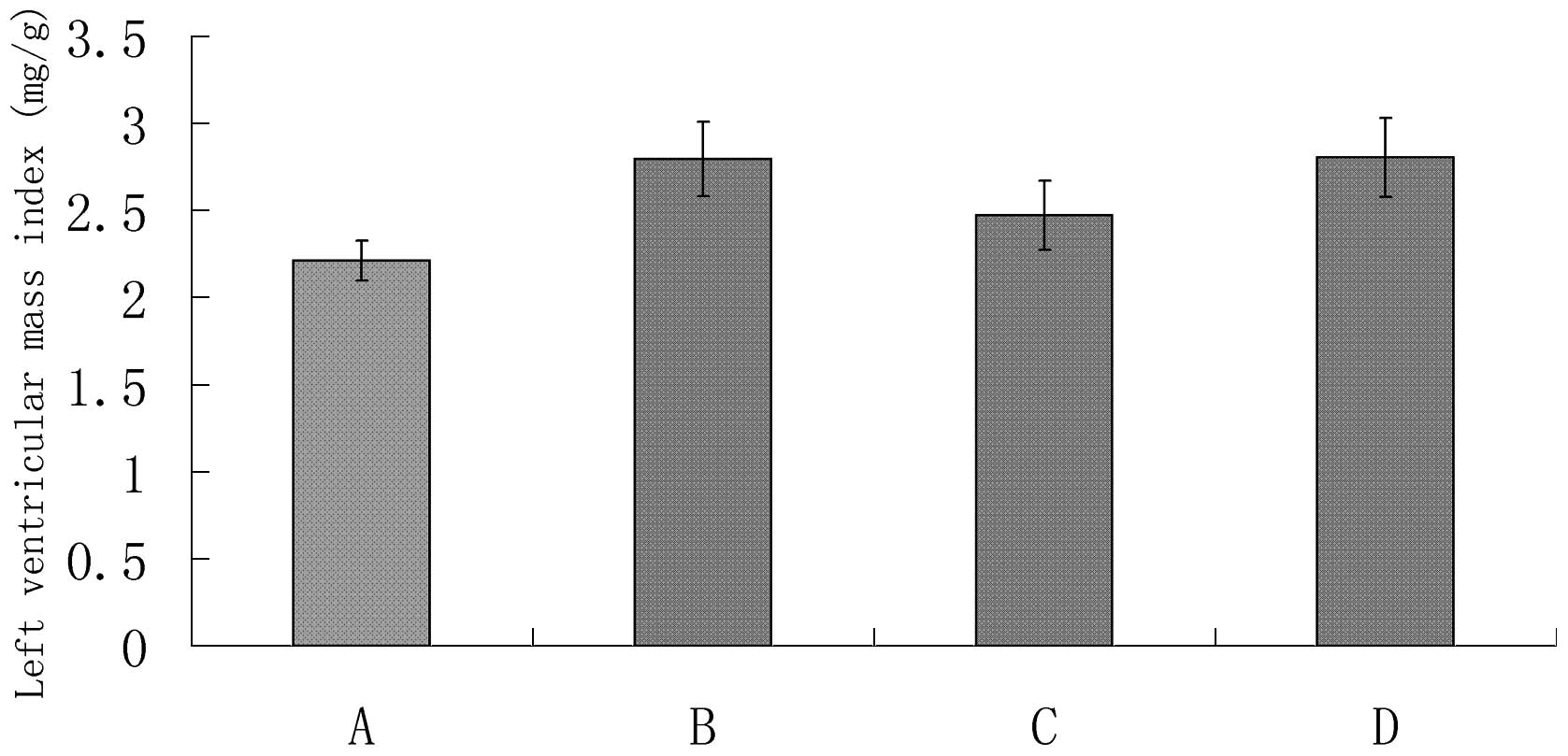
Results of LVMI
The LVMIin the sham surgery group (group A) was
significantly lower than that of group B (P<0.05; Table II and Fig. 1). The LVMI in the AG490 treatment
group (group C) was significantly lower than that of the myocardial
infarction control group (group B; P<0.05). There was no
significant difference in the LVMI between the DMSO treatment group
(group D) and group B (P>0.05).
| Table IILeft ventricular mass index of AMI
rats. |
Table II
Left ventricular mass index of AMI
rats.
| Group | Weight (g) | Left ventricular
weight (mg) | LVMI (mg/g) |
|---|
| A | 324.6±7.60 | 718.2±37.08 | 2.213±0.115 |
| B | 303.4±9.32 | 847.0±57.48 | 2.795±0.214a |
| C | 310.8±8.84 | 757.6±54.62 | 2.473±0.198b |
| D | 303.0±13.91 | 848.6±57.03 | 2.806±0.227c |
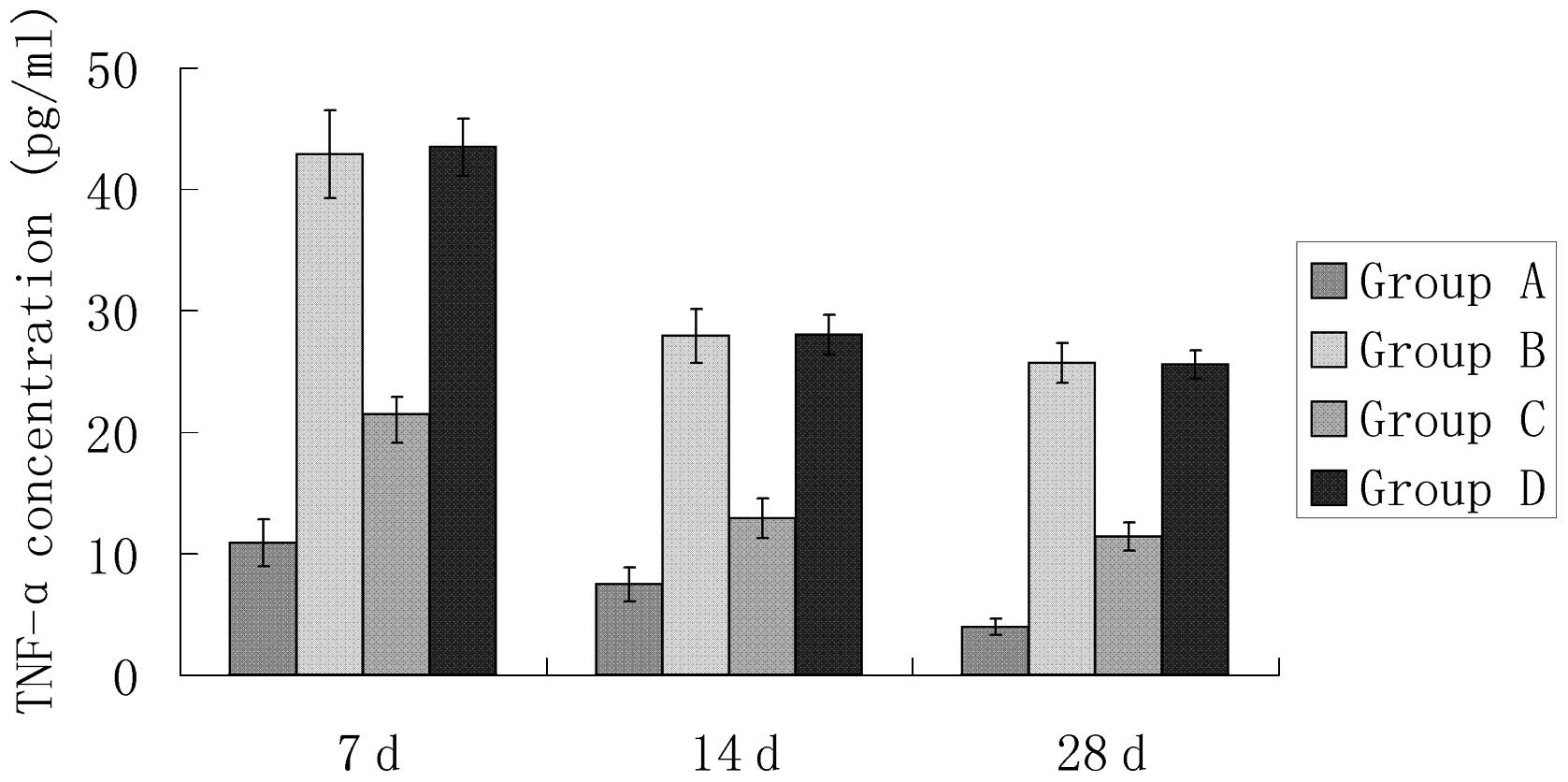
Plasma TNF-α in AMI rats
The TNF-α concentration was calculated according to
the OD value of the sample. The TNF-α plasma concentration in the
myocardial infarction control group (group B) was significantly
greater than that of the sham surgery group (group A; P<0.05)
and the AG490 treatment group (group C; P<0.05; Table III and Fig. 2). No significant difference was
detected in TNF-α between the DMSO treatment group (group D) and
the myocardial infarction control group (group B).
| Table IIIPlasma TNF-α concentration in AMI
rats (pg/ml). |
Table III
Plasma TNF-α concentration in AMI
rats (pg/ml).
| Group | 7 days | 14 days | 28 days |
|---|
| A | 10.88±1.94 | 7.48±1.41 | 3.98±0.68 |
| B | 42.91±3.62a | 27.93±2.21a | 25.71±1.64a |
| C | 21.50±1.39b | 12.91±1.65b | 11.43±1.16b |
| D | 43.48±2.36c | 28.03±1.63c | 25.56±1.19c |
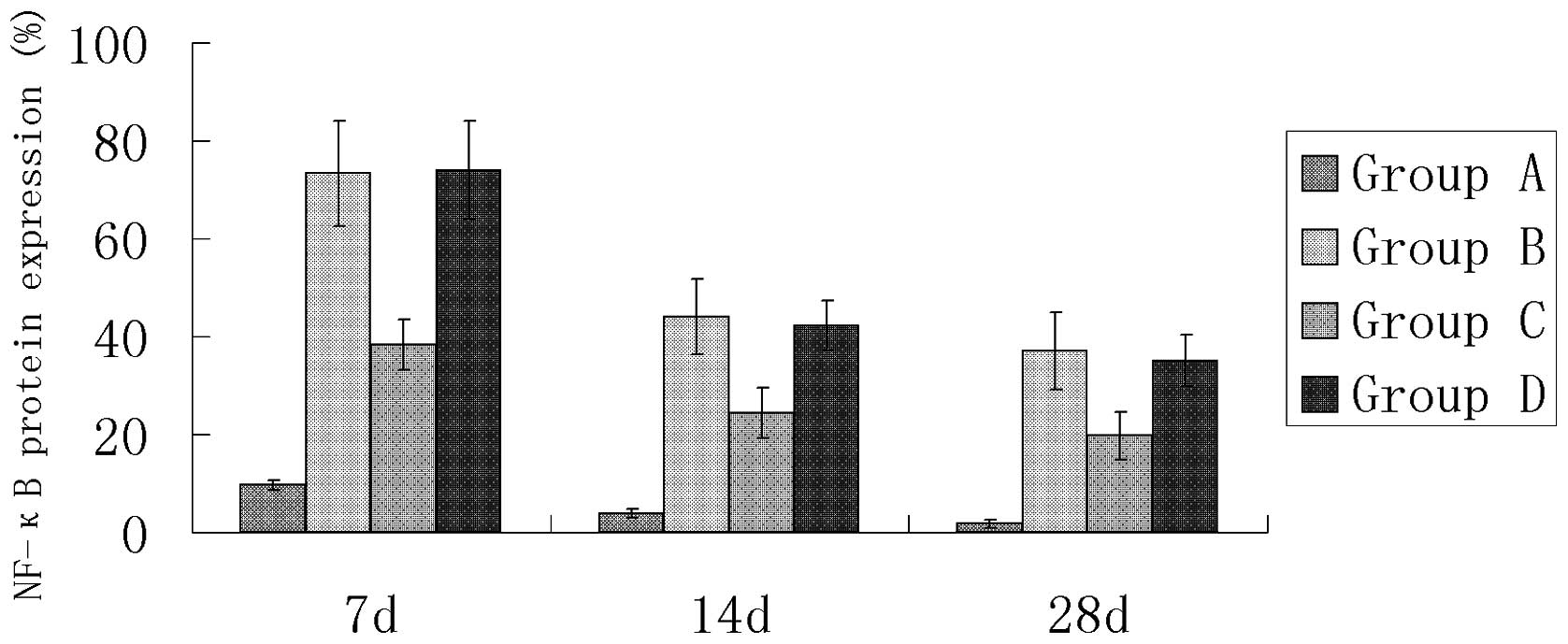
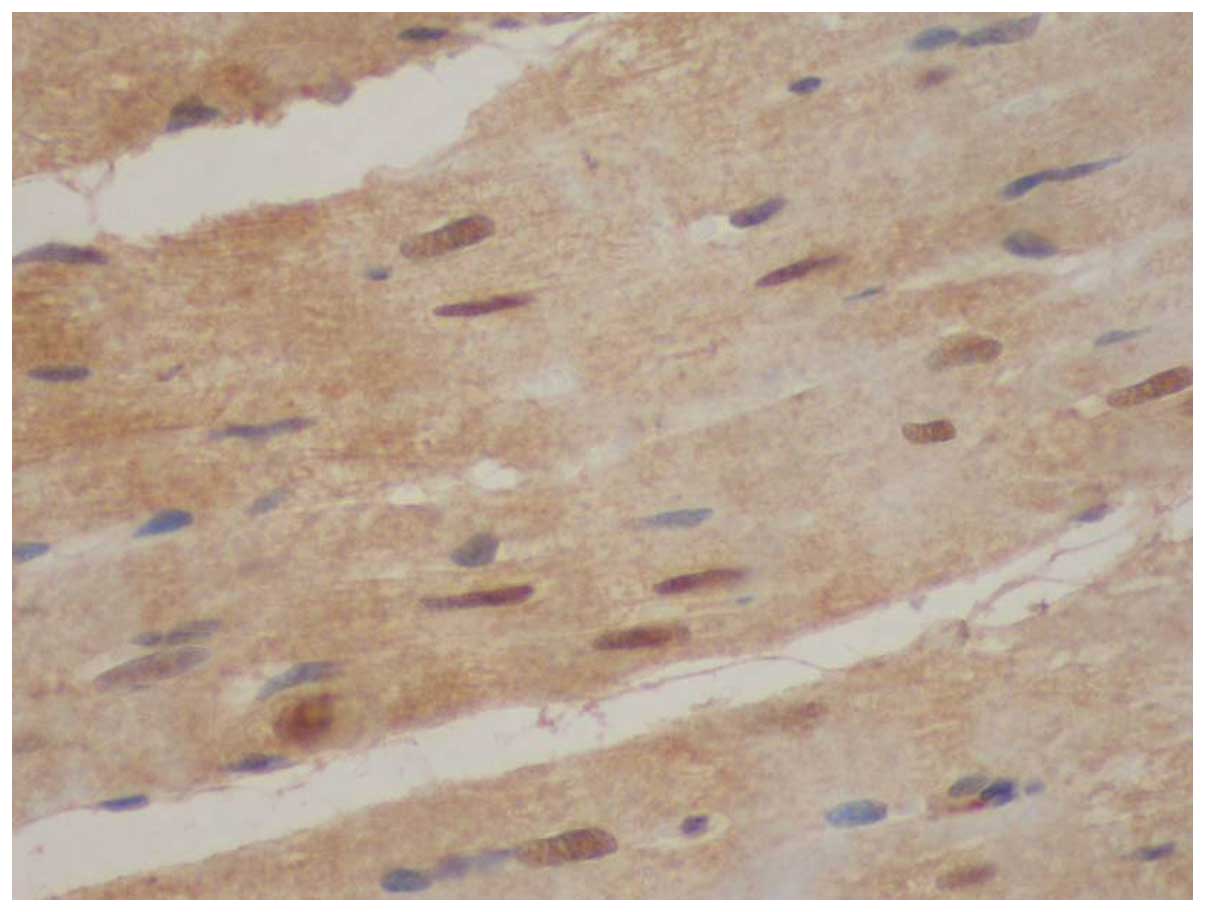
Immunohistochemical detection of NF-κB
protein expression
In the sham surgery group (group A), only a small
number of cardiomyocytes positive for NF-κB protein expression were
observed (Table IV and Figs. 3 and 4). The number of cardiomyocytes positive
for NF-κB protein expression was significantly greater in groups B,
C and D compared with group A (P<0.05). There was no significant
difference between groups B and D, but the number of cardiomyocytes
positive for NF-κB protein expression was significantly less in the
AG490 treatment group (group C; Fig.
6) compared with the myocardial infarction control group (group
B; Fig. 5; P<0.05).
| Table IVNF-κB protein expression in the AMI
rats (%). |
Table IV
NF-κB protein expression in the AMI
rats (%).
| Group | 7 days | 14 days | 28 days |
|---|
| A | 9.66±1.06 | 3.82±0.91 | 1.78±0.88 |
| B | 73.33±10.74a | 43.96±7.70a | 37.08±7.90a |
| C | 38.41±5.12b | 24.42±5.12b | 19.73±4.80b |
| D | 74.03±10.06c | 42.13±5.12c | 35.07±5.28c |
Discussion
A variety of cell signaling pathways play
significant roles in the pathological changes that occur during and
following myocardial infarction. These pathways may be activated by
various factors that include inflammation and the overactive
renin-angiotensin system. These pathways are able to protect the
myocardium or cause further damage via downstream cytokines. The
JAK/STAT signaling pathway plays a critical role in the signal
transduction of cytokines, which are directly responsible for
transferring the stimulation signals to the nucleus and promoting
gene transcription. This pathway is widely involved in cell stress
responses, apoptosis, inflammation and other biological processes,
making it a key player in the occurrence and development of
numerous cardiovascular diseases (12).
There are four kinase members in the JAK family,
JAK1, JAK2, JAK3 and Tyk2. AG490 is a specific JAK tyrosine
phosphorylation inhibitor, capable of effectively blocking the
transduction of downstream signaling and inhibiting the activation
of STAT (13). STAT is the
downstream substrate of JAK and is a transcription factor in the
cytoplasm that is able to bind to the specific DNA sequence of the
regulatory region in the target gene. The JAK/STAT signaling
pathway in the myocardium may be activated by various factors,
including IL-6 (14), granulocyte
colony stimulating factor, hypoxia and inflammation. Marked
myocardial hypertrophy has been shown to develop in transgenic mice
with STAT overexpression in the cardiac muscle. Thus, the
overexpression of STAT and the activation of the JAK/STAT signaling
pathway by the mechanical tension of the ventricular wall after
myocardial infarction is a significant mechanism of myocardial
hypertrophy following myocardial infarction.
Activation of the JAK/STAT signaling pathway and
expression of the downstream substrate STAT is dependent on time
passed since infarction, the region of myocardial infarction and
the type of cells. Usually, ischemic pretreatment activates STAT1
and STAT3, ischemia-reperfusion activates STAT1, STAT5a and STAT6
and permanent myocardial ischemia activates STAT3 (12,15,16).
In addition, the JAK/STAT signaling pathway is closely correlated
with myocardial hypertrophy after AMI and cardiac remodeling
following myocardial infarction. The JAK/STAT signaling pathway is
activated in AMI and has been revealed as playing a key role in
cytoprotective signaling (15). In
the present study, the infarct area was significantly reduced in
the AG490 treatment group compared with that in the infarction
control group; however, there was no significant difference in the
DMSO treatment group. The infarct area likely decreased after
blocking the JAK/STAT signaling pathway using AG490, suggesting
that the JAK/STAT signaling pathway was correlated with the area of
myocardial infarction and was involved in its onset.
LVMI is an important indicator used to evaluate left
ventricular remodeling and left ventricular function. Our results
revealed that the LVMI in the myocardial infarction group was
significantly higher than that of the non-infarction groups. This
finding may be attributed to left ventricular remodeling, left
ventricular hypertrophy, growth promotion of catecholamine
activated by infarction, proliferation via the renin-angiotensin
system, proliferation and migration of the smooth muscle cells and
collagen deposition caused by certain cytokines. These events
contribute to the increased heart weight, which was particularly
evident in the left ventricular region. The LVMI in the AG490
treatment group was significantly lower than in that the myocardial
infarction control group, indicating that blocking the JAK/STAT
signaling pathway inhibited cardiac hypertrophy and collagen matrix
deposition, thereby inhibiting left ventricular remodeling.
Collectively, these findings suggest that the JAK/STAT signaling
pathway may be activated and involved in left ventricular
remodeling after myocardial infarction.
NF-κB is a specific DNA binding protein that is
found throughout eukaryotic cells and is responsible for
multi-directional transcription regulation. A variety of
stimulatory signals inside and outside cells activate NF-κB and
stimulate the gene expression of a variety of active substances,
including corresponding cytokines, adhesion molecules and
immune-related receptors. The nucleoprotein is involved in the
growth and differentiation of cells, apoptosis, inflammation and
tumorigenesis (17,18). NF-κB was detected in the nuclear
extracts of mature B lymphocytes for the first time by Sen and
Baltimore (19) in 1986. It was
revealed to specifically bind with the enhancer sequence in the
immunoglobulin κ light chain gene and promote its expression. It is
known that there are more than 150 genes that may be regulated by
NF-κB (20). Previous studies
(21–24) have shown that the NF-κB activity in
coronary atherosclerotic plaques increased, particularly in
patients with unstable angina; the NF-κB activity in peripheral
blood leukocytes in patients with unstable angina was higher than
that in patients with stable angina. In addition, the NF-κB
activity in cardiomyocytes in the region of the myocardial
infarction was higher than that in the areas that were absent of
any infarction. In STAT1 mutant fibroblasts, STAT1 and NF-κB
synergistically promoted pro-inflammatory cytokine transcription,
indicating that the JAK/STAT signaling pathway promoted NF-κB
activation and then induced or enhanced the inflammatory responses.
NF-κB is closely associated with the occurrence and development of
atherosclerosis, coronary heart disease, myocardial infarction,
myocardial hypertrophy and congestive heart failure after
myocardial infarction. In the present study, a greater number of
cardiomyocytes positive for NF-κB protein expression were observed
in the myocardial infarction control group compared with the
non-infarction group. The number of positive cells was
significantly reduced with the treatment of AG490, indicating that
NF-κB expression was enhanced by AMI, and that blocking the
JAK/STAT signaling pathway significantly reduced NF-κB expression.
This finding suggests that a correlation exists between NF-κB
expression and the activity of the JAK/STAT signaling pathway.
Therefore, activation of the JAK/STAT signaling pathway may affect
NF-κB expression and NF-κB may be involved in the occurrence and
development of myocardial infarction.
In 1975, Carswell et al(25) found an active tumor necrosis factor
that induced tumor cell necrosis, but caused no damage to the
normal tissues and cells. Tumor necrosis factor may be divided into
three subtypes: TNF-α, -β and -γ. TNF-α is mainly secreted by
macrophages, although lymphocytes, monocytes, smooth muscle cells,
fibroblasts and vascular endothelial cells are also able to produce
and release TNF-α under certain conditions (25–30).
It was identified that TNF-α levels increased in AMI and may be
involved in the onset of myocardial infarction. Ridker et
al(31) found that the risk of
coronary event recurrence was greater in patients with higher
plasma TNF-α levels after AMI. These authors also found that the
frequency of recurrence of coronary events was positively
correlated with the plasma TNF-α level. Animal experiments revealed
that after ligation of the left anterior descending artery, TNF-α
gene expression was promoted, which may be associated with vascular
and ventricular remodeling after AMI (32). TNF-α is able to induce
extracellular matrix changes, collagen matrix remodeling and
promote the hypertrophy of cardiomyocytes by the destruction of
collagen and an increase in the amount of denatured collagen fibers
in myocardium. TNF-α is also able to promote myocardial cell
apoptosis, which may result in fewer myocardial cells and more
fibrous tissues. Our results showed that the plasma TNF-α
concentration was increased in the AMI group and was significantly
decreased following the inhibition of the JAK/STAT signaling
pathway by AG490. The results indicated that the TNF-α level was
correlated with and dependent on the JAK/STAT signaling pathway.
This finding may be attributed to the binding sites of STAT in the
mRNA promoter of TNF-α. The inhibition of STAT activity by AG490
may reduce TNF-α mRNA transcription (33). Several studies have shown that
following myocardial infarction, TNF-α concentration in the
infarction area was increased. However, increases in TNF-α
concentration were also noted in the normal myocardium of
non-infarct areas. Thus, AG490 may inhibit the inflammatory
responses after myocardial infarction, improve the cardiac
hypertrophy, reduce fibrosis and attenuate the renin-angiotensin
system response. This may ultimately reduce myocardial remodeling
following AMI. These findings suggest that the JAK/STAT signaling
pathway is able to affect TNF-α concentration and that the latter
may be involved in the development of myocardial infarction.
References
|
1
|
Igaz P, Toth S and Falus A: Biological and
clinical significance of the JAK-STAT pathway; lessons from
knockout mice. Inflamm Res. 50:435–441. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kisseleva T, Bhattacharya S, Braunstein J
and Schindler CW: Signaling through the JAK/STAT pathway, recent
advances and future challenges. Gene. 285:1–24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sen R and Baltimore D: Multiple nuclear
factors interact with the immunoglobulin enhancer sequences. Cell.
46:705–716. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yeh CH, Chen TP, Wu YC, Lin YM and Jing
Lin P: Inhibition of NFkappaB activation with curcumin attenuates
plasma inflammatory cytokines surge and cardiomyocytic apoptosis
following cardiac ischemia/reperfusion. J Surg Res. 125:109–116.
2005. View Article : Google Scholar
|
|
5
|
Altavilla D, Deodato B, Campo GM, et al:
IRFI 042, a novel dual vitamin E-like antioxidant, inhibits
activation of nuclear factor-kappaB and reduces the inflammatory
response in myocardial ischemia-reperfusion injury. Cardiovasc Res.
47:515–528. 2000. View Article : Google Scholar
|
|
6
|
Christman JW, Lancaster LH and Blackwell
TS: Nuclear factor kappa B: a pivotal role in the systemic
inflammatory response syndrome and new target for therapy.
Intensive Care Med. 24:1131–1138. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Jiang H, Yang J, Chen SS and Xu L:
Down-regulation of CREB-binding protein expression blocks
thrombin-mediated endothelial activation by inhibiting acetylation
of NF-kappaB. Int J Cardiol. 154:147–152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fan Y, Wang J, Wei L, He B, Wang C and
Wang B: Iron deficiency activates pro-inflammatory signaling in
macrophages and foam cells via the p38 MAPK-NF-kappaB pathway. Int
J Cardiol. 152:49–55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang S, He B, Goldstein S, Ge J, Wang Z
and Ruiz G: Changes in adiponectin expression in acute myocardial
infarction rats and the significance of bisoprolol intervention.
Can J Physiol Pharmacol. 89:109–115. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang S, He B, Ge J, Zhai C, Liu X and Liu
P: Characterization of chemical composition of Agaricus
brasiliensis polysaccharides and its effect on myocardial SOD
activity, MDA and caspase-3 level in ischemia-reperfusion rats. Int
J Biol Macromol. 46:363–366. 2010.PubMed/NCBI
|
|
11
|
Zhang S, He B, Ge J, et al: Extraction,
chemical analysis of Angelica sinensis polysaccharides and
antioxidant activity of the polysaccharides in ischemia-reperfusion
rats. Int J Biol Macromol. 47:546–550. 2010.PubMed/NCBI
|
|
12
|
Booz GW, Day JN and Baker KM: Interplay
between the cardiac renin angiotensin system and JAK-STAT
signaling: role in cardiac hypertrophy, ischemia/reperfusion
dysfunction, and heart failure. J Mol Cell Cardiol. 34:1443–1453.
2002. View Article : Google Scholar
|
|
13
|
Strutz F and Muller GA: The role of
tubulo-interstitial processes in progression of primary renal
diseases. Nephr Dial Transplant. 9:10–20. 1994. View Article : Google Scholar
|
|
14
|
Boengler K, Hilfiker-Kleiner D, Drexler H,
Heusch G and Schulz R: The myocardial JAK/STAT pathway: from
protection to failure. Pharmacol Ther. 120:172–185. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Negoro S, Kunisada K, Tone E, et al:
Activation of JAK/STAT pathway transduces cytoprotective signal in
rat acute myocardial infarction. Cardiovasc Res. 47:797–805. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mascareno E, El-Shafei M, Maulik N, et al:
JAK/STAT signaling is associated with cardiac dysfunction during
ischemia and reperfusion. Circulation. 104:325–329. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heyninck K, Kreike MM and Beyaert R:
Structure-function analysis of the A20-binding inhibitor of
NF-kappa B activation, ABIN-1. FEBS Lett. 536:135–140. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nair A, Venkatraman M, Maliekal TT, Nair B
and Karunagaran D: NF-kappaB is constitutively activated in
high-grade squamous intraepithelial lesions and squamous cell
carcinomas of the human uterine cervix. Oncogene. 22:50–58. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sen R and Baltimore D: Inducibility of
kappa immunoglobulin enhancer-binding protein Nf-kappa B by a
posttranslational mechanism. Cell. 47:921–928. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rodriguez-Porcel M, Lerman LO, Holmes DR
Jr, Richardson D, Napoli C and Lerman A: Chronic antioxidant
supplementation attenuates nuclear factor-kappa B activation and
preserves endothelial function in hypercholesterolemic pigs.
Cardiovasc Res. 53:1010–1018. 2002. View Article : Google Scholar
|
|
22
|
Dichtl W, Nilsson L, Goncalves I, et al:
Very low-density lipoprotein activates nuclear factor-kappaB in
endothelial cells. Circ Res. 84:1085–1094. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ritchie ME: Nuclear factor-kappaB is
selectively and markedly activated in humans with unstable angina
pectoris. Circulation. 98:1707–1713. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kawano S, Kubota T, Monden Y, et al:
Blockade of NF-kappaB improves cardiac function and survival after
myocardial infarction. Am J Physiol Heart Circ Physiol.
291:H1337–H1344. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carswell EA, Old LJ, Kassel RL, Green S,
Fiore N and Williamson B: An endotoxin-induced serum factor that
causes necrosis of tumors. Proc Natl Acad Sci U S A. 72:3666–3670.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shaw T, Nixon JS and Bottomley KM:
Metalloproteinase inhibitors: new opportunities for the treatment
of rheumatoid arthritis and osteoarthritis. Expert Opin Investig
Drugs. 9:1469–1478. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang NL, Chiang SH, Hsueh CH, Liang YJ,
Chen YJ and Lai LP: Metformin inhibits TNF-alpha-induced IkappaB
kinase phosphorylation, IkappaB-alpha degradation and IL-6
production in endothelial cells through PI3K-dependent AMPK
phosphorylation. Int J Cardiol. 134:169–175. 2009. View Article : Google Scholar
|
|
28
|
Cao YL, Wang YX, Wang DF, Meng X and Zhang
J: Correlation between omental TNF-alpha protein and plasma PAI-1
in obesity subjects. Int J Cardiol. 128:399–405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Conraads VM, Denollet J, De Clerck LS,
Stevens WJ, Bridts C and Vrints CJ: Type D personality is
associated with increased levels of tumour necrosis factor
(TNF)-alpha and TNF-alpha receptors in chronic heart failure. Int J
Cardiol. 113:34–38. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sharma R and Anker SD: Cytokines,
apoptosis and cachexia: the potential for TNF antagonism. Int J
Cardiol. 85:161–171. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ridker PM, Rifai N, Pfeffer M, Sacks F,
Lepage S and Braunwald E: Elevation of tumor necrosis factor-alpha
and increased risk of recurrent coronary events after myocardial
infarction. Circulation. 101:2149–2153. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Irwin MW, Mak S, Mann DL, et al: Tissue
expression and immunolocalization of tumor necrosis factor-alpha in
postinfarction dysfunctional myocardium. Circulation. 99:1492–1498.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Held TK, Weihua X, Yuan L, Kalvakolanu DV
and Cross AS: Gamma interferon augments macrophage activation by
lipopolysaccharide by two distinct mechanisms, at the signal
transduction level and via an autocrine mechanism involving tumor
necrosis factor alpha and interleukin-1. Infect Immun. 67:206–212.
1999.
|