Introduction
Obstructive sleep apnea (OSA) is characterized by
intermittent hypoxia/reoxygenation (IHR) as a result of repetitive
episodes of complete or partial obstructions of the upper airway
during sleep. IHR is an independent risk factor for the development
of coronary and cerebral vascular diseases, two common consequences
of atherosclerosis (1–3). The mechanisms by which hypoxic
signaling accelerates the initiation and progression of
atherosclerosis have yet to be fully elucidated.
Nuclear factor-κB (NF-κB) is a transcription factor
that has crucial roles in inflammation, immunity, cell
proliferation and apoptosis (4–6).
Activation of NF-κB is controlled by the inhibitor of κB (I-κB),
which retains NF-κB in the cytoplasm (7). Emerging evidence has revealed that
the activation of NF-κB in the endothelium may contribute to the
pathogenic process of atherosclerosis associated with IHR (8,9).
NF-κB-mediated inflammatory pathways have integrated roles in
classic atherosclerosis induced by a high-cholesterol diet
(9). Patients with OSA have
increased NF-κB activity in circulating neutrophils and monocytes,
and elevated serum levels of NF-κB-dependent gene products
(10–12). Furthermore, using an in
vitro model in cultured cells exposed to repetitive
hypoxia/reoxygenation, previous studies have demonstrated a
selective and dose-dependent activation of NF-κB compared with
adaptive hypoxia-inducible factor-1 (HIF-1)-dependent pathways, and
the p38 mitogen-activated protein kinase (MAPK) signaling pathway
is believed to mediate the activation of NF-κB during IHR (7).
Propofol (2,6-diisopropylphenol) is a potent
intravenous hypnotic agent widely used for the induction and
maintenance of anesthesia. In addition, propofol exhibits
anti-inflammatory properties by decreasing the production of
proinflammatory cytokines, altering the production of nitric oxide
and inhibiting neutrophil function (13). A study reported that propofol
inhibited the activation of p38 MAPK to exert its anti-inflammatory
effect (14). However, it remains
to be elucidated whether propofol inhibits NF-κB and HIF-1 activity
in the vascular endothelial cells during IHR. In the present study,
an in vitro model of human vein endothelial cells was
employed to mimic IHR events and evaluate the effects of propofol
on the IHR-induced activation of NF-κB and HIF-1 and their
molecular mechanisms.
Materials and methods
Cell culture and IHR
The human endothelial cell line EA.hy926, generated
from a fusion of primary human umbilical vein endothelial cells
(HUVECs), was purchased from the Cell Library of Shanghai
Institutes for Biological Sciences (Shanghai, China). The EA.hy926
cells were grown and maintained in Dulbecco’s modified Eagle medium
supplemented with 10% fetal bovine serum (FBS) and kept in a
humidified incubator at 5% CO2. For all experiments, the
cells were grown to 50–70% confluency and starved for 1 day in 1%
FBS medium prior to the different treatments. IHR exposure was
performed in a computer-controlled incubator chamber connected to a
BioSpherix OxyCycler (Biospherix, Redfield, NY, USA), as previously
described (8). The cells were
maintained at 37°C and 5% CO2 in the hypoxic chamber, in
which the O2 levels were shifted between 1% for 10 min
and 21% for 5 min. The cells were exposed to IHR for 64 cycles
based on a previous study in which such cycles of IHR were
determined to sufficiently induce the activation of NF-κB in HUVECs
(8). The cells in the control
group were maintained in normoxic conditions at 21% O2
and 5% CO2.
Propofol treatment
Pure propofol was purchased from Sigma-Aldrich (St.
Louis, MO, USA) to exclude the effect of lipid emulsion. Prepared
propofol was added to the medium at different concentrations of 0,
25, 50 or 100 μM for 30 min prior to IHR exposure, and kept in the
medium throughout the entire experiment. This dose range for
propofol was selected from a previous study and considered as the
range of concentrations that was clinically relevant (15). Cells that were not treated with
either propofol or IHR were used as a control. At the end of the
experiments, a luciferase reporter assay, western blot analysis or
quantitative polymerase chain reaction (qPCR) were performed, as
described below.
Knockdown of p38 MAPK by siRNA
siRNA specifically targeting p38 MAPK and a control
siRNA were purchased from Dharmacon (Lafayette, CO, USA). The cells
were grown to 50% confluence in antibiotic-free medium, and p38
siRNA or control siRNA were transfected using
Lipofectamine® transfection reagent (Invitrogen,
Carlsbad, CA, USA), according to the manufacturer’s instructions.
The cells were incubated at 37°C for 48 h to allow for maximal
knockdown of the target gene and then exposed to the indicated
cycles of IHR. A luciferase reporter assay or western blot analysis
was performed, as described below.
Luciferase reporter assays for NF-κB and
HIF-1 activity
Luciferase reporter assays were conducted, as
described previously (7). Briefly,
the cells were grown to 50–70% confluence on culture dishes and
transiently transfected with NF-κB or HIF-1 luciferase reporter
constructs and then co-transfected with a constitutively active
Renilla luciferase reporter construct (pSV40-Renilla; Promega
Corp., Madison, WI, USA). At 24 h post-transfection, the cells were
lysed in luciferase cell lysis buffer (Promega Corp., Fitchburg,
WI, USA). The luciferase activity was assessed by addition of an
excess of luciferin/adenosine triphosphate (Promega Corp.,
Fitchburg, WI, USA) and luminometry (Berthold Technologies GmbH
& Co. KG, Bad Wildbad, Germany). All the luciferase reporter
values were normalized to the Renilla luciferase activity and
expressed as a fold induction relative to the control group.
Western blot analysis
Cell homogenates were separated by 10% SDS-PAGE and
transferred to a polyvinylidene fluoride membrane. The membrane was
blocked in Tris-buffered saline Tween-20 (TBST) with 5% skimmed
milk and incubated overnight with primary rabbit polyclonal
anti-I-κBα, mouse monoclonal anti-phosphorylated I-κBα (1:200;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), rabbit
monoclonal anti-p38 MAPK, rabbit monoclonal anti-phosphorylated p38
MAPK (1:1,000 and 1:500, respectively; Cell Signaling Technology,
Inc., Danvers, MA, USA) and mouse monoclonal anti-β-actin (1:200;
Santa Cruz Biotechnology, Inc.) antibodies. Next, the membrane was
incubated for 1 h with secondary antibodies diluted with TBST. The
signals of the detected proteins were visualised by an enhanced
chemiluminescence (ECL) reaction system (Millipore, Billerica, MA,
USA) and quantified by ImageJ software. (National Institutes of
Health, Bethesda, MD, USA)
qPCR
Total RNA was prepared from the cells that remained
in the wells by extraction with TRIzol® reagent
(Invitrogen, Carlsbad, CA, USA) and purification using an RNeasy
Mini kit (Qiagen, Valencia, CA, USA), according to the
manufacturer’s instructions. RNA with an A260/280 ratio between 1.8
and 2.0 was used for reverse transcription using the qScript cDNA
kit (Quanta BioSciences, Gaithersburg, MD, USA). qPCR was performed
in the ABI prism 7000 Sequence Detection System (Applied
Biosystems, Carlsbad, CA, USA) using SYBR® Green
(SuperArray Bioscience, Valencia, CA, US). The primer sequences
were as follows: Tumor necrosis factor α (TNF-α) forward,
5′-CGAGTGACAAGCCTGTAGC-3′ and reverse, 5′-GGTGTG GGTGAGGAGCACAT-3′;
interleukin-1β (IL-1β) forward, 5′-AAACAGATGAAGTGCTCCTTCCAGG-3′ and
reverse, 5′-TGGAGAACACCACTTGTTGCTCCA-3′; interleukin-6 (IL-6)
forward, 5′-AAATGCCAGCCTGCTGACGAAC-3′ and reverse,
5′-AACAACAATCTGAGGTGCCCATGCTAC-3′; and GAPDH forward,
5′-TGGGCTACACTGAGCACCAG-3′ and reverse, 5′-GGGTGTCGCTGTTGAAGTCA-3′.
The values were normalized to GAPDH and the final concentration of
mRNA was calculated using the formula x = 2−ΔΔCt, where
x is the fold difference relative to the control.
Statistical analysis
Values are expressed as the mean ± standard errpr
from at least three independent experiments. Each treatment was
performed in the triplicate culture wells. Statistically
significant values were tested by one-way analysis of variance
followed by Bonferroni’s post-hoc test for multiple comparisons.
P<0.05 was used to indicate a statistically significant
difference.
Results
Effects of propofol on IHR-induced NF-κB
and HIF-1 activity
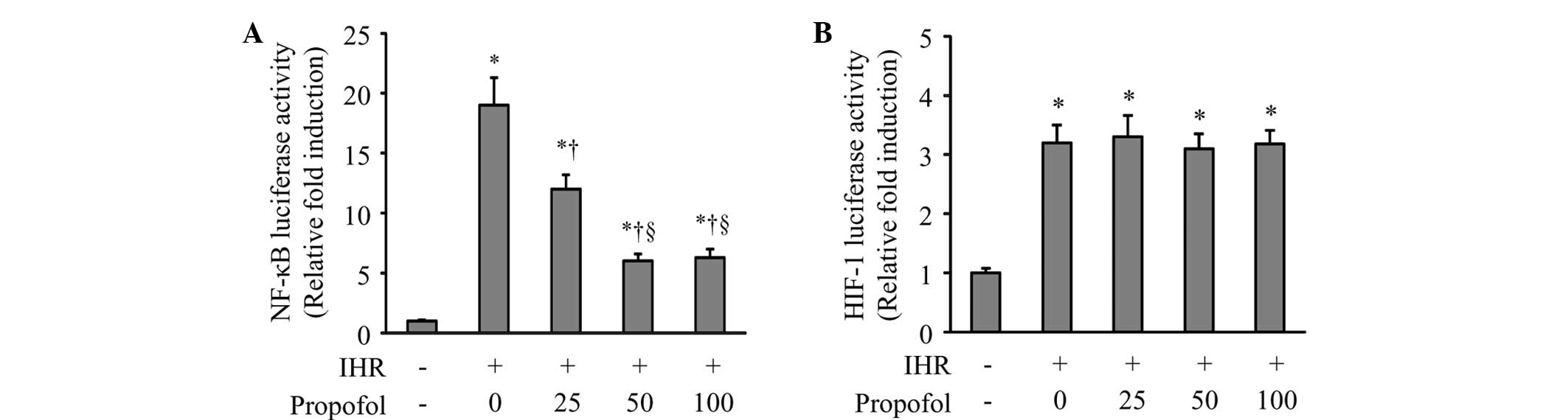
Using luciferase reporter assays, the NF-κB and
HIF-1 signaling pathway activity in HUVECs was measured in response
to propofol treatment. As shown in Fig. 1, 64 cycles of IHR significantly
induced the activation of NF-κB and HIF-1 compared with the control
group. Propofol at 25 and 50 μM dose-dependently inhibited the
NF-κB activity compared with IHR alone. High-dose propofol (100 μM)
did not further reduce NF-κB activity (Fig. 1A). By contrast, neither a high nor
a low dose of propofol affected the IHR-induced HIF-1 activity
(Fig. 1B).
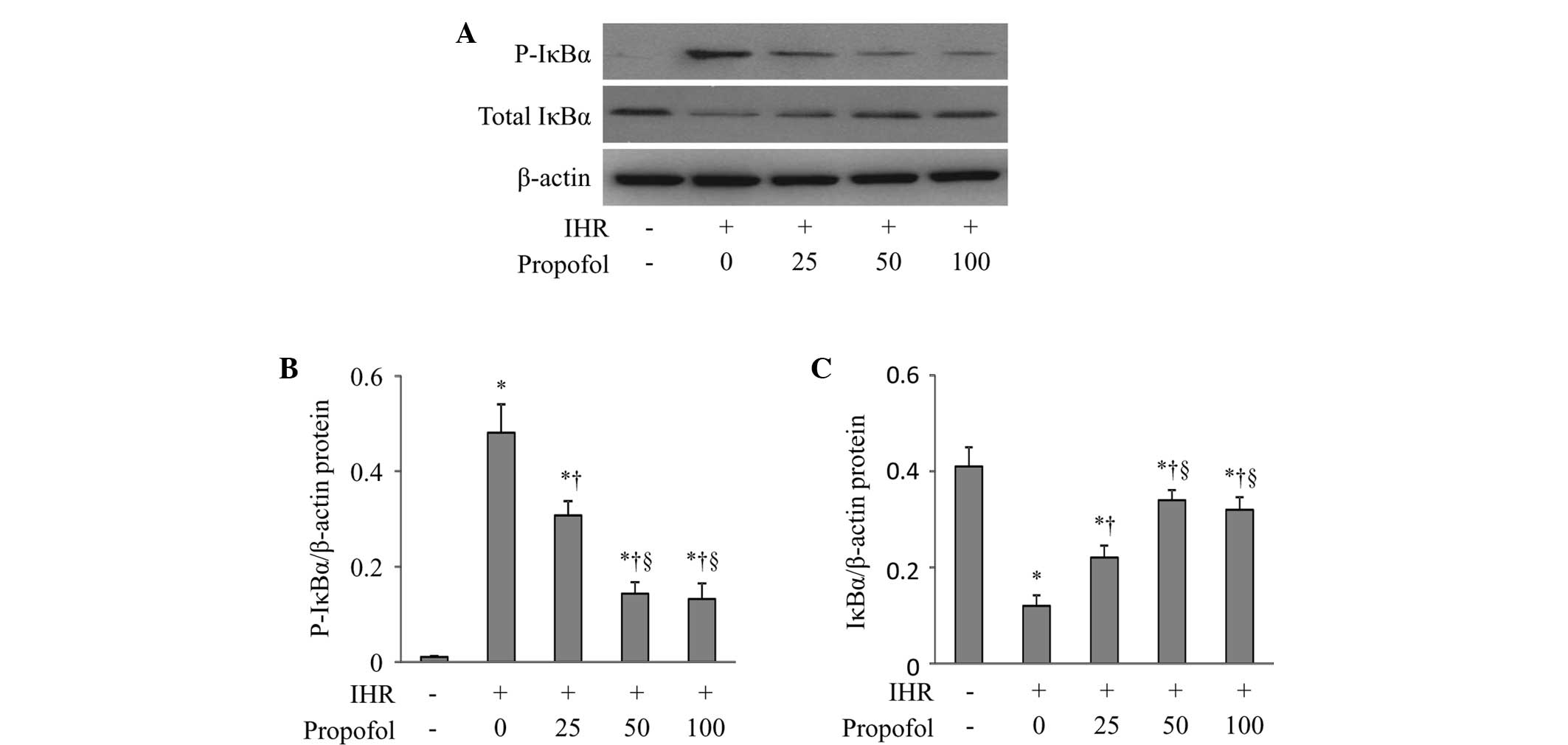
To examine whether propofol inhibited the activation
of NF-κB through regulating its inhibitor, I-κB, in the cytoplasm,
the protein levels of total and phosphorylated I-κBα were assessed
(Fig. 2). IHR alone caused a
significant increase in I-κBα phosphorylation (Fig. 2B) and a decrease in the total I-κBα
levels (Fig. 2C), and these
changes were prevented by propofol in a dose-dependent manner.
Effects of propofol on IHR-induced
proinflammatory cytokines
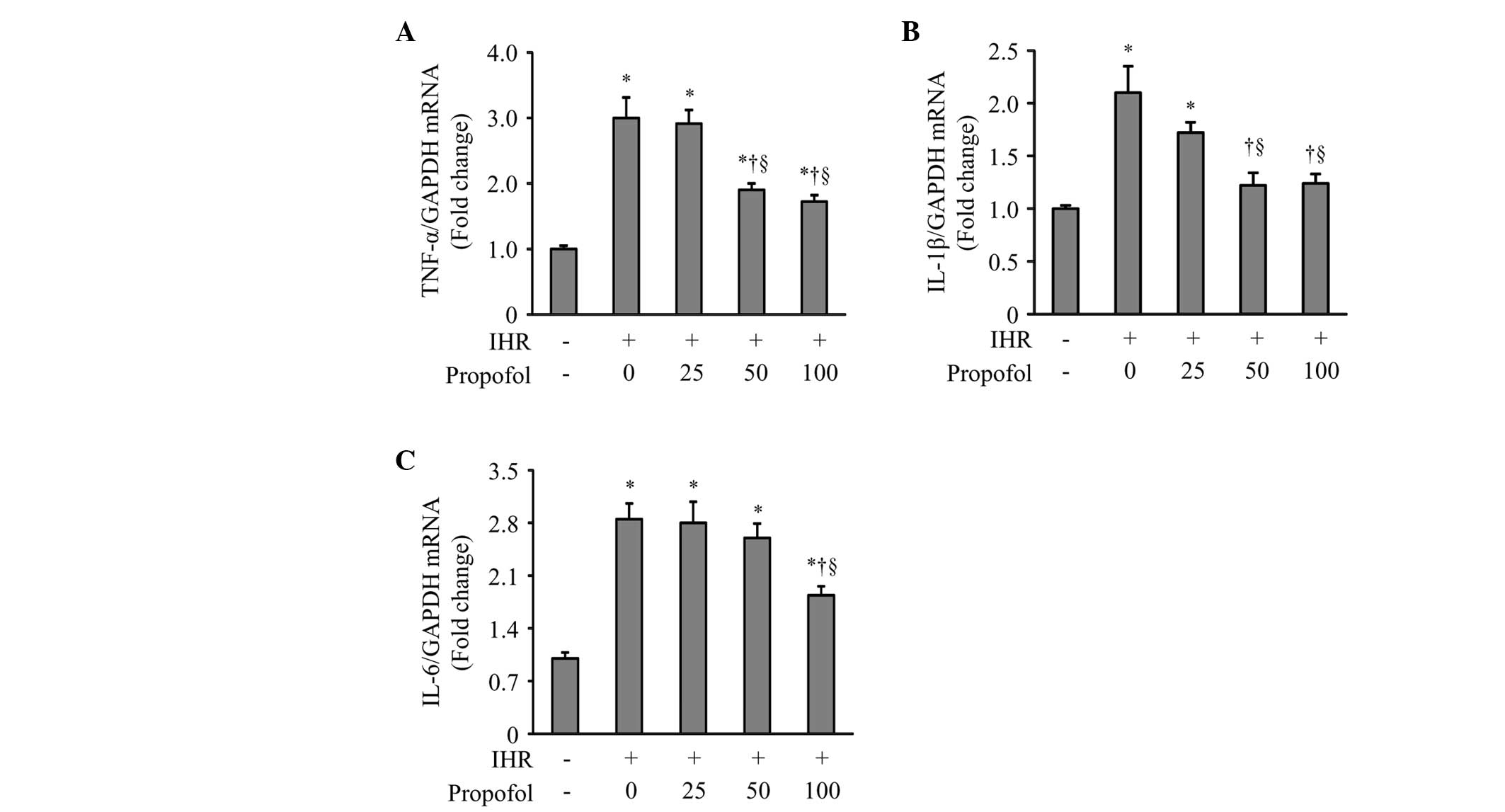
Due to NF-κB being a key component in the regulation
of inflammation, the present study examined whether propofol
treatment reduces IHR-induced expression of proinflammatory
cytokines in HUVECs. qPCR demonstrated that the mRNA expression of
the proinflammatory cytokines TNF-α, IL-1β and IL-6, were markedly
enhanced by IHR compared with the control group (Fig. 3). Propofol at 50 or 100 μM
significantly inhibited the increase of mRNA expression of TNF-α,
IL-1β and IL-6 in the HUVECs exposed to IHR.
Effects of propofol on p38 MAPK
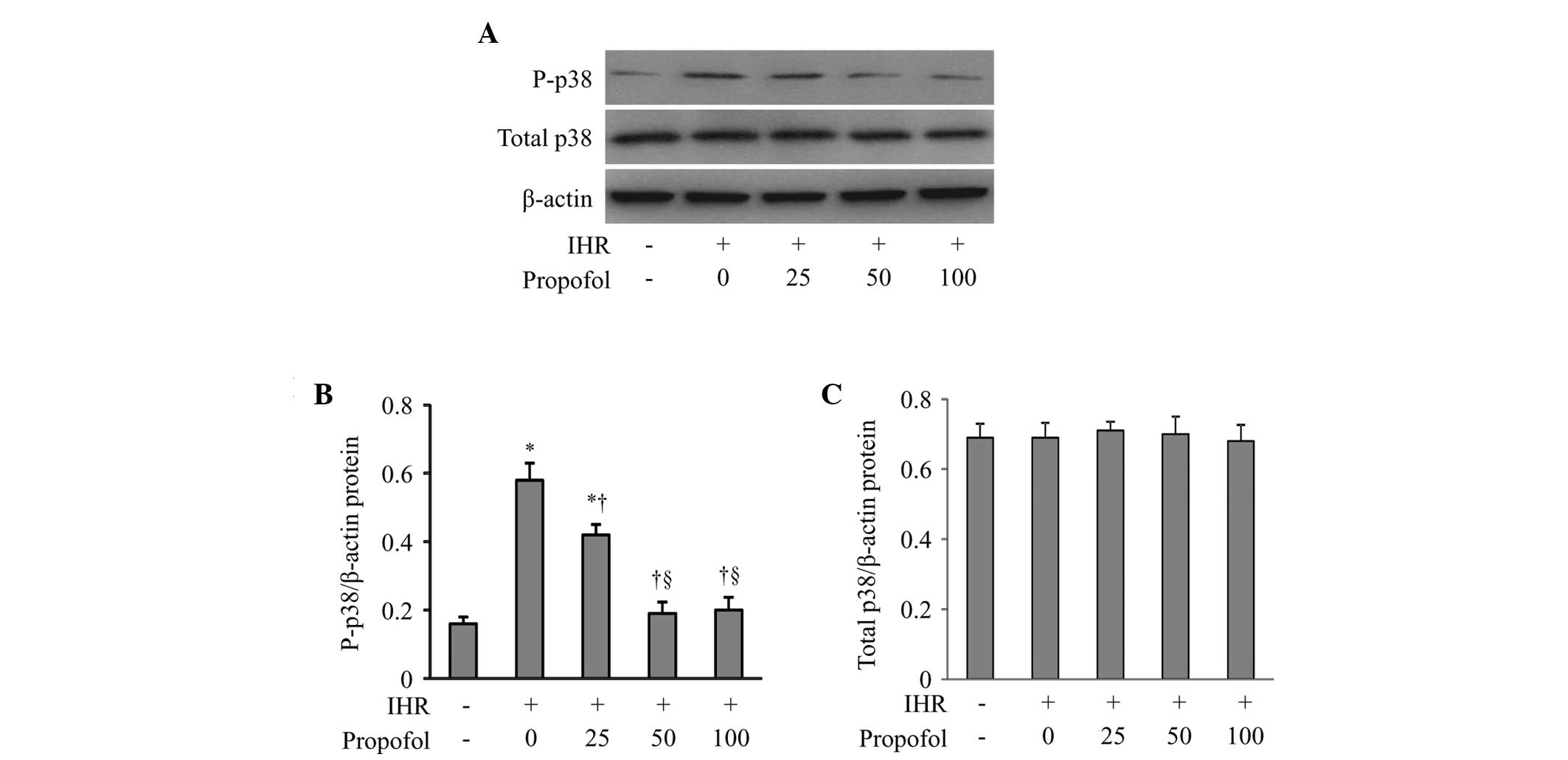
It was hypothesized that propofol attenuates the
IHR-induced activation of NF-κB and its downstream proinflammatory
cytokines by inhibiting p38 MAPK signaling. To test this
hypothesis, the protein levels of total and phosphorylated p38 MAPK
were determined by western blot analysis (Fig. 4). Compared with the control group,
IHR increased the protein levels of phosphorylated p38 MAPK
(Fig. 4B). Propofol at 25 and 50
μM dose-dependently reduced the protein levels of phosphorylated
p38 MAPK in the HUVECs exposed to IHR, and high-dose propofol (100
μM) had no further effect on the phosphorylation of p38 MAPK. By
contrast, the protein levels of total p38 MAPK were not affected by
either IHR or propofol among the groups (Fig. 4C).
Effects of p38 siRNA on IHR-induced NF-κB
and HIF-1 activity
To further confirm that propofol selectively reduces
the NF-κB activity through inhibition of p38 MAPK signaling, the
effects of p38 MAPK knockdown with siRNA were examined on the
IHR-induced NF-κB and HIF-1 activity. Compared with the control
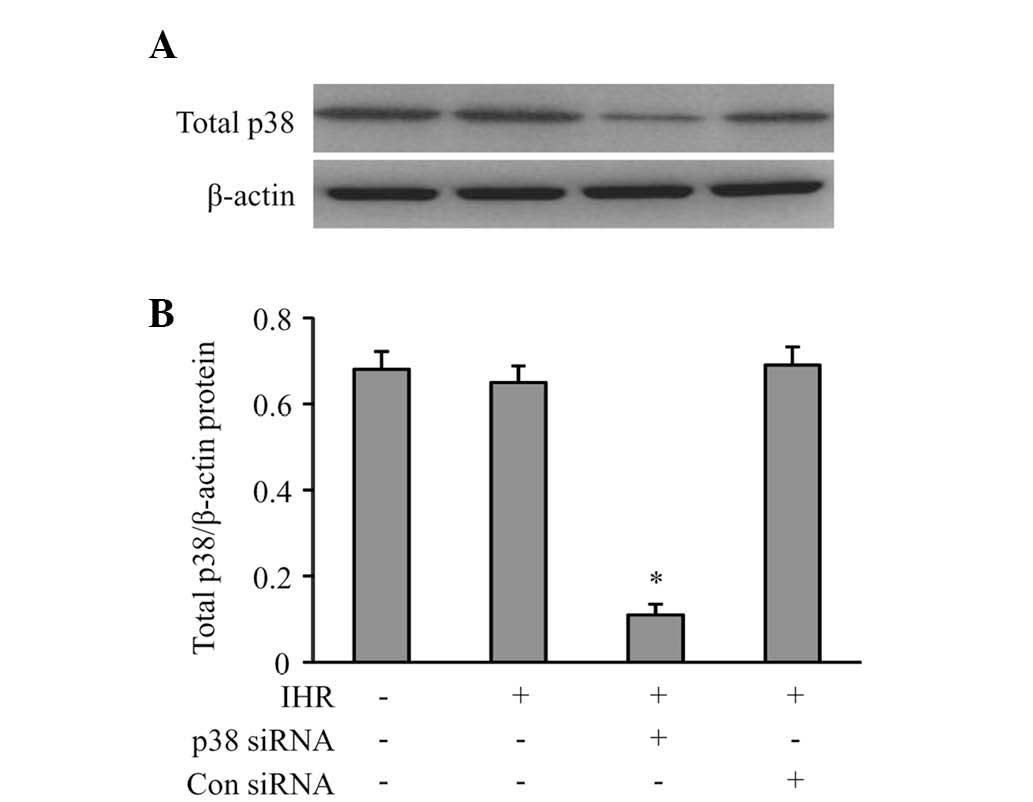
siRNA, p38 siRNA effectively produced a knockdown of p38 MAPK by
84% in the HUVECs (Fig. 5), which
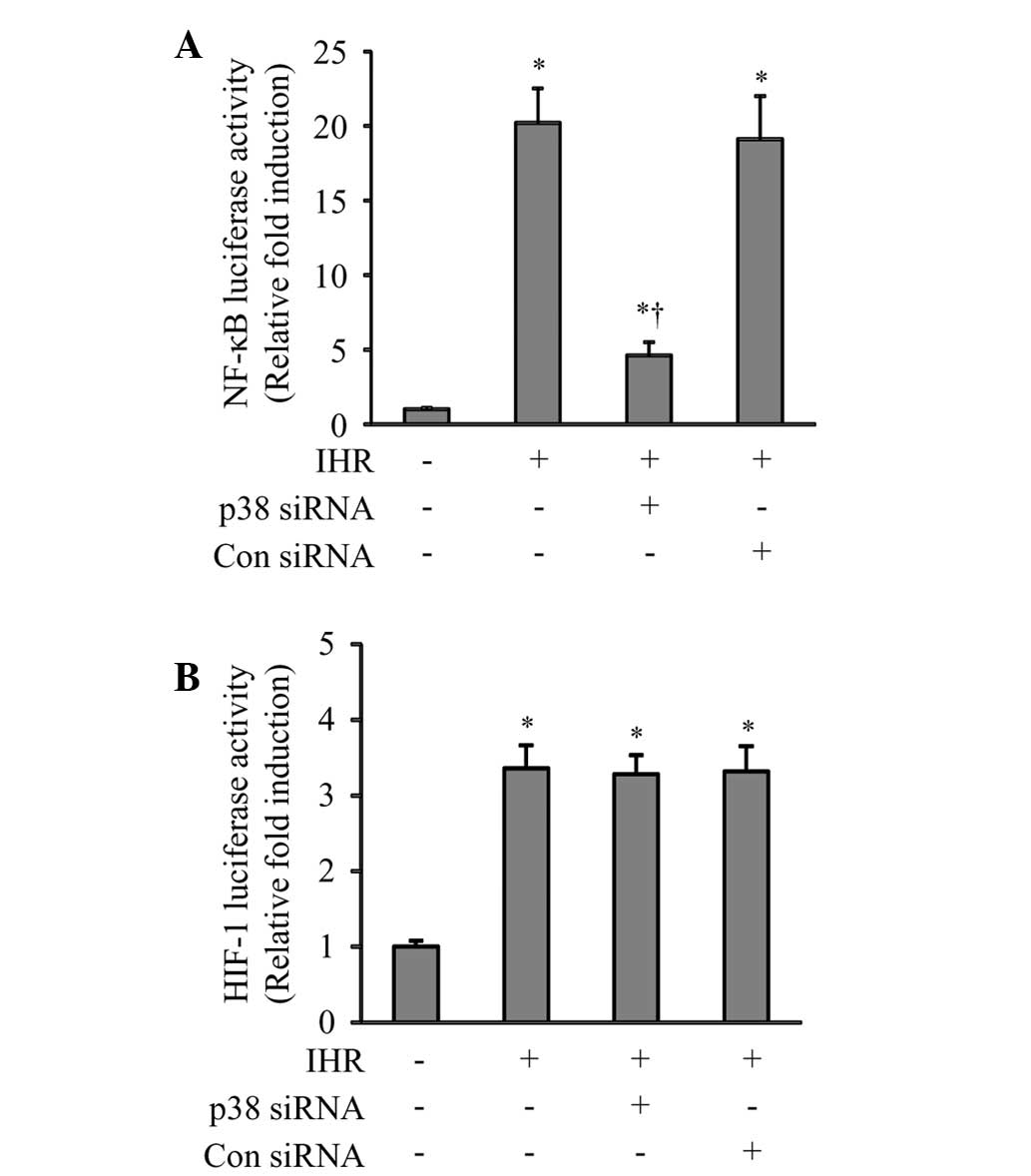
was accompanied by a significant reduction in the IHR-induced
activation of NF-κB (Fig. 6A), but
not HIF-1 (Fig. 6B).
Discussion
The novel findings of the present study are
summarized as follows: i) Propofol selectively inhibited the
activation of NF-κB, but not HIF-1, in the HUVECs during IHR; ii)
the reduced activation of NF-κB by propofol was accompanied by
decreases in the levels of proinflammatory cytokines; and iii) the
inhibitory effect of propofol on IHR-induced NF-κB activity was
likely be based on the suppression of the p38 MAPK signaling
pathway.
IHR is a hallmark feature of OSA and is associated
with atherosclerosis, which is a chronic inflammatory disease
involving a plethora of cell types and multiple pathological
processes (16,17). Atherogenesis is triggered by
vascular endothelial dysfunction that is characterized by a
pro-inflammatory state of the endothelium (18,19),
leading to endothelial apoptosis (19,20).
NF-κB is a well-known redox-sensitive transcription factor involved
in numerous pathological conditions, including inflammatory
processes and cell apoptosis. In resting cells, NF-κB is
predominantly localized in the cytoplasm in a complex with I-κB,
which undergoes phosphorylation, ubiquitination and degradation
upon stimulation, leading to the translocation of NF-κB into the
nucleus followed by transcription of a battery of genes (21). Increasing evidence suggests that
IHR may induce the activation of NF-κB and the release of
proinflammatory cytokines. In addition to NF-κB, HIF-1, a
transcription factor that is essential for regulating oxygen
homeostasis, also regulates the expression of target genes,
including proinflammatory cytokines (22). HIF-1 is a heterodimer composed of
an oxygen-regulated HIF-1α subunit and a constitutively expressed
HIF-1β subunit. Under hypoxic conditions, HIF-1α accumulates,
translocates into the nucleus and determines the activity of HIF-1,
which promotes the production of inflammatory cytokines. HIF-1
activity has been shown to be regulated through the NF-κB pathway
(22). In the present study, 64
cycles of IHR significantly induced activation of NF-κB and HIF-1
in the HUVECs, accompanied by increased mRNA expression of TNF-α,
IL-1β and IL-6. These results were consistent with previous
findings in vivo and in vitro, demonstrating that
NF-κB and HIF-1 are activated or produced in response to IHR and
contribute to the expression of proinflammatory cytokines (11,23).
Thus, inhibition of the proinflammatory cytokines by suppressing
NF-κB or HIF-1 activity in the vascular endothelium may be crucial
to prevent atherosclerosis in patients with OSA.
The anti-inflammatory effects of propofol, an
intravenous general anesthetic agent, have attracted attention in
the studies of multiple diseases associated with inflammation. An
in vitro study has demonstrated that propofol
post-conditioning inhibits the activation of NF-κB induced by
hypoxia/reoxygenation and protects cardiomyocytes against apoptosis
(15). Other studies reported that
propofol suppresses the activation of HIF-1 induced by
lipopolysaccharides or hypoxia in macrophages (24), alveolar epithelial cells (25) and lung epithelial cells (22). However, the effects of propofol on
NF-κB and HIF-1 activity in vascular endothelial cells subjected to
IHR have not been previously assessed. In the present study, using
HUVECs, propofol was demonstrated to dose-dependently inhibit
IHR-induced NF-κB activity by suppressing the phosphorylation of
I-κBa. However, propofol did not change the HIF-1 activity. In
addition, the reduced NF-κB activity caused by propofol was
accompanied by decreased mRNA expression of proinflammatory
cytokines. These observations indicate that the anti-inflammatory
effects of propofol in HUVECs during IHR are mainly due to the
inhibition of the NF-κB pathway, which may have a dominant role
over the HIF-1 pathway in regulating proinflammatory cytokines in
the vascular endothelial cells. A previous study revealed that
inhibition of the NF-κB pathway resulted in reduced HIF-1 activity
in mouse embryonic stem cells, indicating that HIF-1 is downstream
of the NF-κB pathway (26).
Furthermore, propofol, at a similar dose range to that used in the
present study, was reported to inhibit NF-κB and HIF-1 activity
induced by lipopolysaccharides in lung epithelial cells (22). The discrepancy between the current
data and previous findings may be due to the different cell types
or methods used for studying the activation of NF-κB and HIF-1.
p38 MAPK is critical for the production of
NF-κB-dependent proinflammatory cytokines (27,28).
A previous study demonstrated that IHR activated NF-κB via
activation of the p38 MAPK signaling pathway (7). The hypothesis that propofol may
reduce IHR-induced NF-κB activity in HUVECs by suppressing p38 MAPK
activity was tested. The data of the present study revealed that
the phosphorylation of p38 MAPK was dose-dependently inhibited by
propofol in a similar manner to the reduction in NF-κB activity.
Furthermore, the knockdown of p38 MAPK with p38 siRNA led to a
significant reduction in IHR-induced NF-κB activity. Together,
these results indicate that the reduced NF-κB activity caused by
propofol acts through the inhibition of the p38 MAPK signaling
pathway in HUVECs exposed to IHR. Indeed, propofol has been
identified to attenuate the lipopolysaccharide-induced production
of proinflammatory cytokines and monocyte chemotactic protein-1 by
inhibiting the phosphorylation of p38 MAPK in human THP-1 cells
(14).
In conclusion, the present study demonstrated that
propofol attenuates the IHR-induced activation of NF-κB, but not
HIF-1, in vascular endothelial cells, and that these beneficial
effects are possibly based on the inhibition of the p38 MAPK
signaling pathway. Propofol may have the potential to prevent
atherosclerosis in patients with OSA by inhibiting NF-κB-mediated
inflammation in the vascular endothelium.
Acknowledgements
The present study was supported by the Shandong
Provincial Natural Science Foundation of China (no.
ZR2010HM120).
References
|
1
|
Yaggi HK, Concato J, Kernan WN, Lichtman
JH, Brass LM and Mohsenin V: Obstructive sleep apnea as a risk
factor for stroke and death. N Engl J Med. 353:2034–2041. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Quan SF and Gersh BJ; National Center on
Sleep Disorders Research; National Heart, Lung, and Blood
Institute. Cardiovascular consequences of sleep-disordered
breathing: past, present and future: report of a workshop from the
National Center on Sleep Disorders Research and the National Heart,
Lung, and Blood Institute. Circulation. 109:951–957. 2004.
View Article : Google Scholar
|
|
3
|
Fang G, Song D, Ye X, Mao SZ, Liu G and
Liu SF: Chronic intermittent hypoxia exposure induces
atherosclerosis in ApoE knockout mice: role of NF-κB p50. Am J
Pathol. 181:1530–1539. 2012.PubMed/NCBI
|
|
4
|
Lan L, Tao J, Chen A, et al:
Electroacupuncture exerts anti-inflammatory effects in cerebral
ischemia-reperfusion injured rats via suppression of the TLR4/NF-κB
pathway. Int J Mol Med. 31:75–80. 2013.PubMed/NCBI
|
|
5
|
Tang Z, Jiang L, Peng J, et al: PCSK9
siRNA suppresses the inflammatory response induced by oxLDL through
inhibition of NF-κB activation in THP-1-derived macrophages. Int J
Mol Med. 30:931–938. 2012.PubMed/NCBI
|
|
6
|
Abe J: Role of PKCs and NF-kappaB
activation in myocardial inflammation: enemy or ally? J Mol Cell
Cardiol. 43:404–408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ryan S, McNicholas WT and Taylor CT: A
critical role for p38 map kinase in NF-kappaB signaling during
intermittent hypoxia/reoxygenation. Biochem Biophys Res Commun.
355:728–733. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han Q, Yeung SC, Ip MS and Mak JC:
Intermittent hypoxia-induced NF-κB and HO-1 regulation in human
endothelial EA.hy926 cells. Cell Biochem Biophys. 66:431–441.
2013.
|
|
9
|
Song D, Fang G, Mao SZ, et al: Chronic
intermittent hypoxia induces atherosclerosis by NF-κB-dependent
mechanisms. Biochim Biophys Acta. 1822:1650–1659. 2012.
|
|
10
|
Htoo AK, Greenberg H, Tongia S, et al:
Activation of nuclear factor kappaB in obstructive sleep apnea: a
pathway leading to systemic inflammation. Sleep Breath. 10:43–50.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ryan S, Taylor CT and McNicholas WT:
Selective activation of inflammatory pathways by intermittent
hypoxia in obstructive sleep apnea syndrome. Circulation.
112:2660–2667. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryan S, Taylor CT and McNicholas WT:
Systemic inflammation: a key factor in the pathogenesis of
cardiovascular complications in obstructive sleep apnoea syndrome?
Thorax. 64:631–636. 2009.
|
|
13
|
Marik PE: Propofol: an immunomodulating
agent. Pharmacotherapy. 25:28S–33S. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang J, Chen X, Tu W, et al: Propofol
inhibits the activation of p38 through up-regulating the expression
of annexin A1 to exert its anti-inflammation effect. PLoS One.
6:e278902011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li H, Tan J, Zou Z, Huang CG and Shi XY:
Propofol post-conditioning protects against cardiomyocyte apoptosis
in hypoxia/reoxygenation injury by suppressing nuclear factor-kappa
B translocation via extracellular signal-regulated kinase
mitogen-activated protein kinase pathway. Eur J Anaesthesiol.
28:525–534. 2011. View Article : Google Scholar
|
|
16
|
Drager LF, Polotsky VY and Lorenzi-Filho
G: Obstructive sleep apnea: an emerging risk factor for
atherosclerosis. Chest. 140:534–542. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arnaud C, Dematteis M, Pepin JL, Baguet JP
and Lévy P: Obstructive sleep apnea, immuno-inflammation, and
atherosclerosis. Semin Immunopathol. 31:113–125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Endemann DH and Schiffrin EL: Endothelial
dysfunction. J Am Soc Nephrol. 15:1983–1992. 2004. View Article : Google Scholar
|
|
19
|
Kutuk O and Basaga H: Bcl-2 protein
family: implications in vascular apoptosis and atherosclerosis.
Apoptosis. 11:1661–1675. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martinet W and Kockx MM: Apoptosis in
atherosclerosis: focus on oxidized lipids and inflammation. Curr
Opin Lipidol. 12:535–541. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thurberg BL and Collins T: The nuclear
factor-kappa B/inhibitor of kappa B autoregulatory system and
atherosclerosis. Curr Opin Lipidol. 9:387–396. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yeh CH, Cho W, So EC, et al: Propofol
inhibits lipopolysaccharide-induced lung epithelial cell injury by
reducing hypoxia-inducible factor-1alpha expression. Br J Anaesth.
106:590–599. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li S, Qian XH, Zhou W, et al:
Time-dependent inflammatory factor production and NFκB activation
in a rodent model of intermittent hypoxia. Swiss Med Wkly.
141:w133092011.
|
|
24
|
Tanaka T, Takabuchi S, Nishi K, et al: The
intravenous anesthetic propofol inhibits lipopolysaccharide-induced
hypoxia-inducible factor 1 activation and suppresses the glucose
metabolism in macrophages. J Anesth. 24:54–60. 2010. View Article : Google Scholar
|
|
25
|
He XY, Shi XY, Yuan HB, Xu HT, Li YK and
Zou Z: Propofol attenuates hypoxia-induced apoptosis in alveolar
epithelial type II cells through down-regulating hypoxia-inducible
factor-1α. Injury. 43:279–283. 2012.PubMed/NCBI
|
|
26
|
Lee SH, Lee YJ and Han HJ: Effect of
arachidonic acid on hypoxia-induced IL-6 production in mouse ES
cells: Involvement of MAPKs, NF-kappaB, and HIF-1alpha. J Cell
Physiol. 222:574–585. 2010.PubMed/NCBI
|
|
27
|
Kumar S, Boehm J and Lee JC: p38 MAP
kinases: key signalling molecules as therapeutic targets for
inflammatory diseases. Nat Rev Drug Discov. 2:717–726. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karin M: Inflammation-activated protein
kinases as targets for drug development. Proc Am Thorac Soc.
2:386–390. 2005. View Article : Google Scholar : PubMed/NCBI
|