Monoamine oxidase (MAO) catalyzes the oxidative
deamination of biogenic and xenobiotic amines and has an important
role in the metabolism of neuroactive and vasoactive amines in the
central nervous system (CNS) and peripheral tissues. The enzyme
preferentially degrades benzylamine and phenylethylamine and
targets a wide variety of specific neurotransmitters involved in
the primary substrates of MAO in the brain, including epinephrine
(EP), norepinephrine (NE), dopamine (DA), serotonin (5-HT), and
β-phenylethylamine (PEA) (1,2). The
unique position of MAO in modulating the function of a diverse
series of specific neurotransmitters in association with various
conditions, including mood disorders (3), anxiety and depression (4,5),
schizophrenia (6), attention
deficit hyperactivity disorder (7–9),
migraine (10), sexual maturation
(11) and neurodegenerative
diseases (12), has attracted
significant attention to the protein as a therapeutic target.
Compelling studies have shown that the involvement
of MAO in AD and neurodegenerative diseases plays an important role
in several key pathophysiological mechanisms (13,14).
MAO-B has been proposed as a biomarker, whereas activated MAO-B
leads to cognitive dysfunction, destroys cholinergic neurons,
causes disorder of the cholinergic system and contributes to the
formation of amyloid plaques.
The present review focused on evidence supporting
the central role that MAO has in AD pathogenesis, including the
formation of amyloid plaques from amyloid β peptide (Aβ) production
and neurofibrillary tangles (NFTs), and cognitive impairment via
the destruction of cholinergic neurons and disorder of the
cholinergic system. Studies reporting that MAO inhibitors improve
cognitive deficits and reverse Aβ pathology by modulating
proteolytic cleavage of amyloid precursor protein (APP) and
decreasing Aβ protein fragments are also discussed. Finally, on the
basis of current advances in the use of MAO inhibitors for the
treatment of AD, MAO inhibitors are discussed as a promising
therapeutic target for AD.
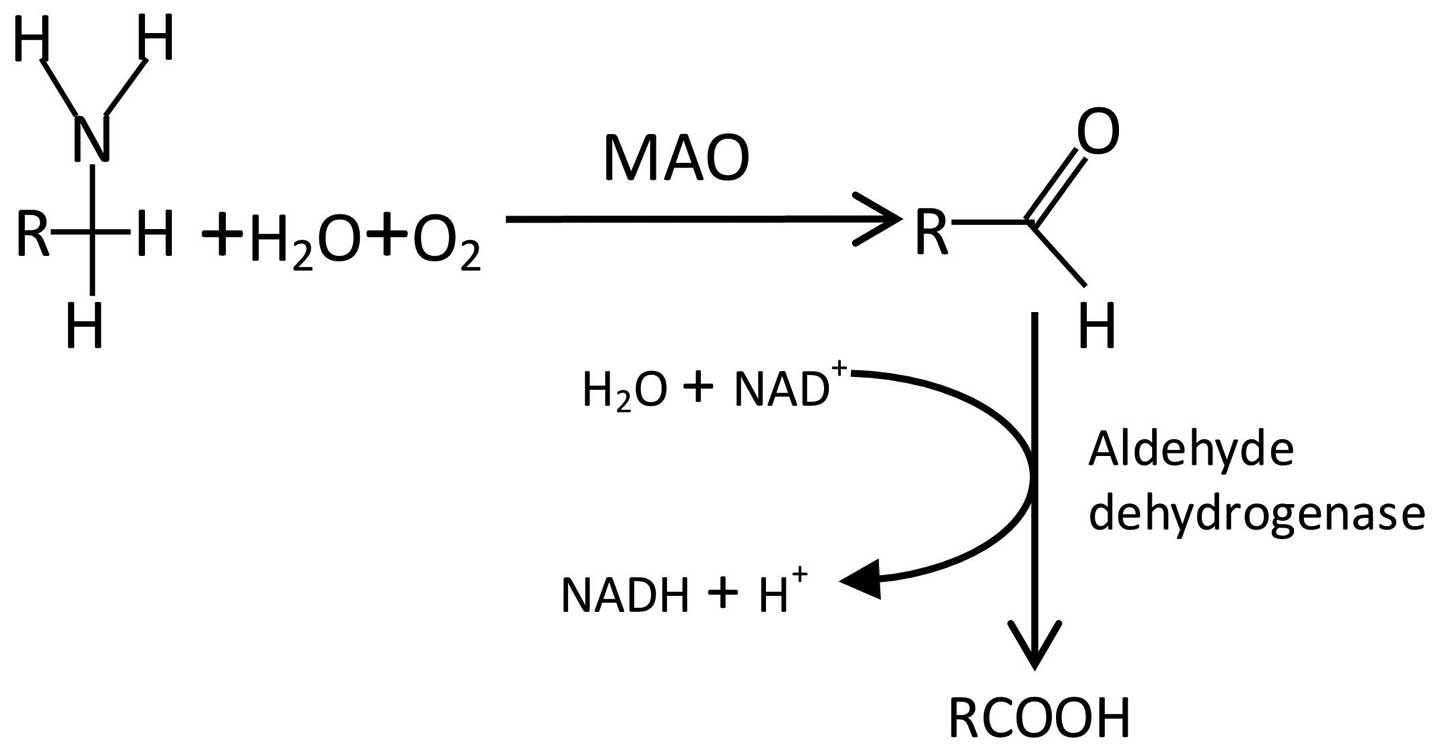
Monoamine oxidase (EC1.4.3.4, a flavin-containing
enzyme) is widely distributed in animal tissue and catalyzes the
oxidative deamination of primary amines by reaction between
dioxygen and R-CH2-NH2 to form R-CHO,
NH3 and H2O2 (Fig. 1). MAO removes an amine group by
catalyzing the oxidative deamination of monoamines, resulting in
the corresponding aldehyde and ammonia. MAO exists as two isozymes
in humans: MAO-A and MAO-B, which are distinct due to different
amino acid sequences, three-dimensional structures, distributions
in organs and tissue, inhibitor sensitivities and substrate
specificity. The two isozymes are found in and outside the CNS.
With regard to the functions of MAO, a wide range of
pathophysiological roles have been suggested, including the
regulation of cardiac function and blood pressure (15), as well as involvement in a number
of psychiatric and neurological disorders, including mood,
depression, schizophrenia, migraine, sexual maturation and
neurodegenerative diseases. The notable role of MAO is in the
regulation of neurotransmitter activity, since the primary
substrates of MAO in the brain are specific neurotransmitters,
including EP, NE, DA, 5-HT and PEA.
An increasing number of studies have demonstrated
the involvement of MAO in neurodegenerative diseases, including
Parkinson’s disease (PD) (16,17),
AD (18,19), Lewy body diseases with dementia
(20) and depression (17,21).
MAO is involved in neurodegeneration via oxidative stress, which
has a central role in neurodegenerative diseases (22). Other mechanisms have been
identified, including neuroinflammation (23), triggering of apoptosis (18,24),
failure of aggregated-protein clearance (25–27)
and glial activation (28) by
MAO.
PD is the second most common age-related
neurodegenerative disease after AD, and is characterized by
progressive loss of dopaminergic neurons in the substantia nigra,
depletion of DA in the striatum, abnormal mitochondrial and
proteasomal functions and accumulation of α-synuclein (29,30).
It has been suggested that the increased turnover of DA and
dopaminergic neurodegeneration are associated with oxidative stress
derived from an increased production of hydrogen peroxide, which is
formed during the oxidative deamination of DA by MAO (29,31).
A recent study showed that activated MAO induces α-synuclein
aggregation, which may be associated with early Parkinsonism and
dopaminergic neurodegeneration in the substantia nigra in SMAD
family member 3-null mice (32).
The evidence that either inhibition or iron chelation of MAO exerts
neuroprotective effects strongly indicates that MAO is a major
component in the process of PD neurodegeneration under oxidative
stress (17). In PD, dopaminergic
cell death in the substantia nigra is linked to a marked
glutathione decrease and mitochondrial dysfunction (24). MAO in the mitochondrial outer
membrane induces oxidative stress resulting in neuronal
degeneration through the production of hydrogen peroxide by
oxidation of monoamine substrates (18,33,34).
These results suggest that MAO is involved in the neurodegenerative
pathogenesis of PD that manifests as increased oxidative stress and
a major impairment of the glutathione pathway.
Studies have shown that activated MAO in the brains
of patients with AD is a biomarker for AD (35–37).
This was demonstrated by [11C]-L-deprenyl using whole
hemisphere autoradiography (38–40),
epidemiology (41,42), morphology (43), as well as single-photon emission
computed tomography (44). Such
studies demonstrated that: i) MAO activity in platelets was
significantly increased in patients with AD and acted as a marker
of behavioral characteristics in dementia disorders (41,45–48);
ii) there were early and persistent alterations in MAO-A and -B in
the brains of patients with AD (49); iii) activated MAO led to cognitive
dysfunction (50); iv) activated
MAO destroyed cholinergic neurons and caused disorders of the
cholinergic system (51); v)
activated MAO contributed to the formation of amyloid plaques
(13,14) and vi) activated MAO was associated
with the formation of NFTs.
Pharmacological studies have demonstrated that MAO
inhibitors exert neuroprotective effects in patients with AD
(21,52,53)
through the following mechanisms: Improvement of cognitive
impairment (50,54,55);
antioxidant and enhancement of iron chelating activities (56–59);
regulation of APP and Aβ expression processing (56,60),
involving the activation of certain signaling pathways, including
the p42/44 mitogen-activated protein kinase (MAPK) and protein
kinase C (PKC) signaling pathways (61); and inhibition of cholinesterase
(ChE) activity (62–64).
MAO activity has previously been found to be high in
the brain and in platelets in patients with AD, while the activity
of MAO-B in the brain increases with age due to an increased
concentration of MAO-B (35).
Compared with age- and gender-matched controls, MAO activity in
platelets was significantly higher in patients with dementia of the
Alzheimer type, and the MAO-B but not the MAO-A activity was
significantly higher in the hippocampus and cortex of the gyrus
cinguli in the AD group (65). In
the aging controls, MAO-B activity in the brain was positively
correlated with age (35,65).
Studies on the association between platelet MAO-B
activity, clinical features and cerebrospinal fluid (CSF) monoamine
metabolites have demonstrated the importance of MAO-B activity in
platelets as a biological marker of AD (47,66).
An increased activity of this enzyme may constitute a marker for
vulnerability towards behavioral disturbance (47). According to a Mini Mental State
Examination (MMSE) of three groups with 23 patients in the early
(MMSE score of 19–24), 23 patients in the middle (MMSE score of
10–18) and 28 patients in the late (MMSE score of 0–9) phases of
AD, as well as 49 age-matched healthy females, significant
correlations between MMSE scores and MAO-B activity and age were
identified, suggesting that these markers may indicate the severity
and/or clinical progress of AD (42,45).
However, several studies have indicated that MAO
activity in platelets is not associated with the pathogenesis of AD
(67–69). The MMSE indicated that no
correlation was present between platelet MAO-B activity and the
cognitive score in patients with AD (54). No significant differences were
found in the levels of the amine metabolites homovanillic acid and
methoxyhydroxyphenylglycol in the CSF of drug-free patients with AD
as compared with those in a group of controls, showing that AD was
not associated with changes in central catecholamine metabolism and
increased platelet MAO activity (70). In view of MAO being involved in the
metabolic inactivation of several monoaminergic neurotransmitters,
including 5-HT, melatonin, NE and EP (21,70,71),
a complex dysfunction in the MAO system is likely to be present in
AD (48,69). Furthermore, MAO activity may be
used to distinguish patients with AD due to the existence of a
biologically-based behavioral subtype of AD (72). Different results may be obtained
for MAO in AD for different species and in different experimental
settings.
Early and persistent alterations in MAO levels in
the brains of patients with AD have been demonstrated by several
studies that show the role of the hyperoxidation phenomena by MAO
in the mechanism of neuronal cell death in AD. The oxidative
degradation catalyzed by MAO produces free radicals and may thus be
involved in the neurodegenerative process (73,74).
In the CNS, the MAO-A isoform appears to be present mainly in
catecholaminergic neurons, whereas the MAO-B form is primarily
located in the glia and in serotonergic neurons (49). Radioenzymatic assay at brain
autopsy revealed that the changes in MAO-A and -B in the prefrontal
cortex occur in the early stages of AD and remain relatively
constant as the disease progresses (49). It has also been revealed that MAO-A
and -B protein and/or mRNA levels are increased in several brain
areas, including the frontal lobe of the neocortex, as well as the
parietal, occipital, temporal and frontal cortex (37,75–77).
This indicates that the mechanism in MAO enzymes may be
transcriptional or post-transcriptional and may be responsible for
the increase in protein activity as well as important for the
progression of AD.
Although the occurrence of activated MAO-B in the
brains of patients with AD has been evidenced, it appears that
MAO-A has a different appearance in different parts of the brains
of patients with AD. Immunostaining showed that the activity of
MAO-B was significantly increased in the cortical areas and in the
hippocampus in AD, reflecting the underlying cell loss and
substantial gliosis in these areas of the brain (76), while MAO-A was increased in the
hypothalamus and frontal pole (37). Furthermore, MAO-A activity appears
to be lower in the locus ceruleus in patients with AD, and is
accompanied by an ~80% decrease in the number of neurons (78), revealing that activated MAO-A in
neurons is involved in the pathology of this disease as a
predisposing factor. In comparison, increased MAO-A activity
appeared more significant in the glia of patients with AD (79). Thus, the changes in MAO-A levels in
patients with AD appear to have multiple mechanisms.
Numerous studies have shown that monoamine
neurotransmitter systems have a determining role in cognition at
the biomolecular level, including memory (80), orientation (81,82),
attention (83), paranoid thinking
(84), as well as behavior and
emotion (81,85). MAO can disturb the balance of
certain other brain chemical neurotransmitters, including
glutamatergic action, ChE, acetylcholinesterase, 5-HT and NE
(86–91), and, therefore, causes symptoms of
cognitive impairment. MAO-A and -B exhibit different substrate
specificity and inhibitor selectivity. Extensive studies have
revealed that MAO-A preferentially acts on 5-HT and NE (92–94)
and MAO-B acts positively on 2-phenylethylamine and benzylamine
(95,96). NE has a determinant role in
executive function, regulating cognition, motivation and intellect,
which are fundamental in social relationships (97). Activated MAO is a contributing
factor in neuronal NE pathways and highlights the specific role of
NE in the symptoms of disordered executive function (97). Activated MAO is also a detrimental
element for the function of the cholinergic system, which is mostly
associated with memory and emotion (51,86,88).
A recent population-based study showed a significant interaction
between MAO-A and catechol-O-methyltransferase genotypes, such that
increased prefrontal catecholamine availability was associated with
an enhanced working memory (80).
Although there are no reports on the direct association between MAO
and the aforementioned neurotransmitter systems in AD, numerous
studies suggest that activated MAO is indirectly involved in the
close association between cognitive impairment and several
neurotransmitter systems in AD (98–100). It is well accepted that oxidative
stress (associated with MAO) contributes to the disturbance of
aforementioned neurotransmitters, including NE and the cholinergic
system, which have a critical role in the cognitive impairment in
AD (100–103). Considering that neuroinflammation
is a key element in cognitive impairment and an intermediate for
oxidative stress (104–107), MAO may act as a proinflammatory
mediator, which causes cognitive impairment in AD. Elevated
monoamine levels in the brain resulting from MAO induce changes in
other neurotransmitter systems and lead to cognitive
impairment.
The amyloid hypothesis was proposed >100 years
before it was demonstrated that the amyloid plaque, a pathological
characteristic of AD, is induced by the generation and deposition
of Aβ (108). Aβ is the root
cause of the pathogenesis of AD, and various mechanisms of neuronal
degeneration have been proposed, including the formation of free
radicals triggered by Aβ, the interaction between oxidative stress
and the production of Aβ, the association between inflammatory
processes and Aβ (109,110), as well as genetic factors and
apoptosis associated with the generation of Aβ.
Studies on the pathogenesis of AD have revealed that
oxidative damage is present in AD, which is the progressive
neurodegenerative disease of ageing. Increased oxidative stress in
patients with AD contributes to Aβ generation and the formation of
amyloid plaques. It has been established that MAO, a marker of
oxidative stress, is linked to the production of reactive oxygen
species and other molecules that cause oxidative stress, which
results in neuronal damage and neurodegeneration, including AD,
indicating that excessive MAO activity contributes to
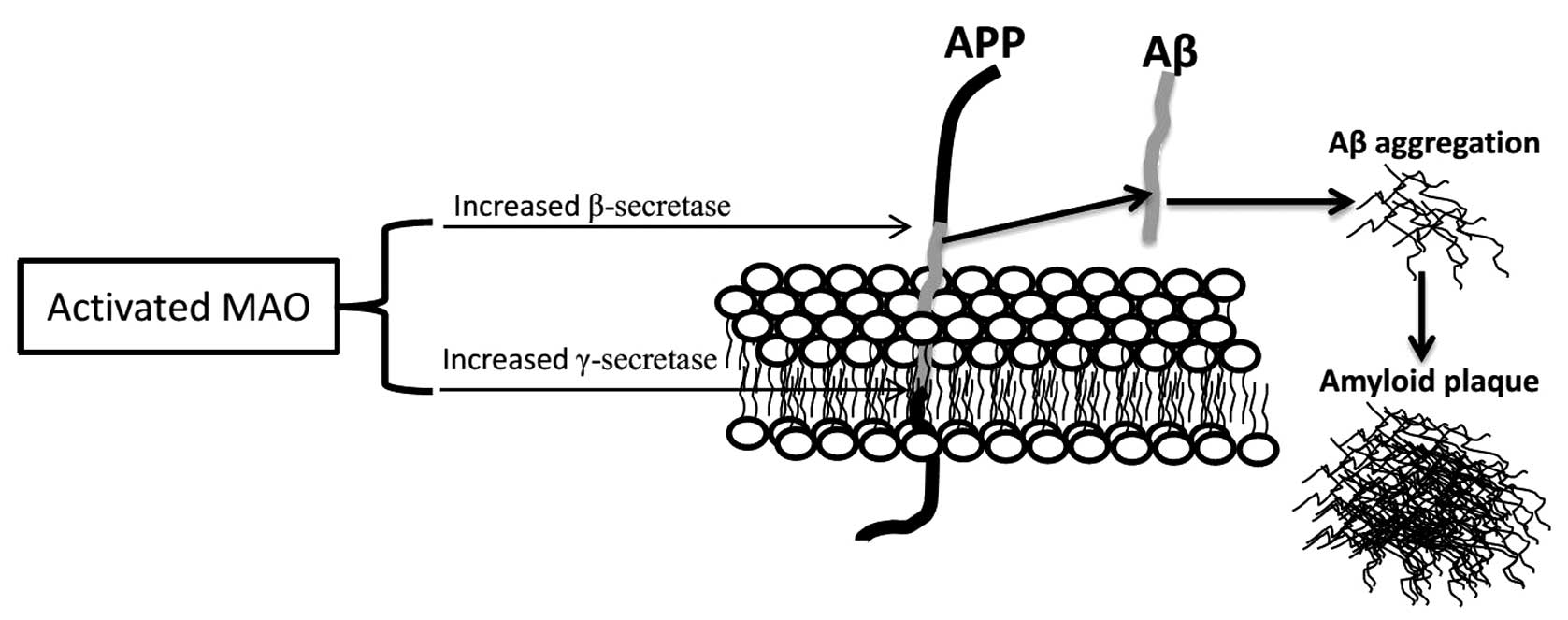
neurodegeneration in AD (62,111–113). Molecular biology studies have
shown the critical role of Aβ generation through the modulation of
APP processing by MAO (60,61,114,115)
(Fig. 2).
Aβ generation is the result of two sequential
cleavages of APP by β-secretase (β-site APP cleavage enzyme, BACE)
and γ-secretase. Extracellular cleavage by β-secretase is the first
step of Aβ production, between the Met671 and Asp672 residues of
APP, which results in a soluble extracellular fragment and a cell
membrane-bound fragment (C99). C99 is then further cleaved by
γ-secretase within the hydrophobic transmembrane domain at either
Val711 or Ile713, finally releasing Aβ and the intracellular domain
of APP. The non-amyloidogenic pathway, in which APP is sequentially
cleaved by α- and γ-secretase, may prevent the production of Aβ
(116,117). Several studies have reported that
propargylamine-containing compounds, including ladostigil and M30,
irreversible and selective MAO-B inhibitors, act as modulators of
the proteolytic cleavage of APP via activation of the p42/44MAPK
and PKC signaling pathways (61,118–120). It was also demonstrated that M30
effectively inhibited Aβ accumulation and tau phosphorylation in
APP/presenilin 1 mice, where it markedly downregulated the levels
of phosphorylated cyclin-dependent kinase 5 and increased PKC and
glycogen synthase kinase-3β phosphorylation (121). Furthermore, deprenyl, an
irreversible MAO-B inhibitor, was able to increase processing of
APP through the non-amyloidogenic pathway via MAPK and
PKC-dependent signaling pathways, and increase α-secretase activity
in a dose-dependent manner in vitro through the involvement
of protein trafficking (122). In
Aβ-injected mice it was found that co-administration of donepezil
with selegiline significantly alleviated cognitive impairment,
indicating a synergistic cognition-improving effect by MAO
inhibitors (123).
Substantial basic biomolecular and clinical studies
suggest that neuroinflammation, with overexpression of cytokines
and inflammatory mediators, is centrally involved in the
pathogenesis of AD (124–126). One notable feature of the
pathophysiology of the brains of patients with AD is that oxidative
stress can induce an active, self-perpetuating cycle of chronic
neuroinflammation, which further promotes oxidative stress and
contributes to irreversible neuronal dysfunction and cell death
(127). The interaction between
oxidative stress and neuroinflammation is an important contributing
factor to Aβ generation. Furthermore, oxidative stress and
neuroinflammation are critical in the pathogenic cascade of
neurodegeneration in AD, suggesting that therapeutic efforts aimed
at these two mechanisms may be beneficial. It has been evidenced
that several MAO inhibitors restrain the production of Aβ by
inhibiting neuroinflammation, such as the inhibition of nuclear
factor κB activity, the downregulation of the expression of
interleukin 1β and tumor necrosis factor α, and the limitation of
glial activation (38,128,129).
NFTs are well known as a primary pathological marker
of AD. It has been indicated that oxidative stress not only is a
major factor in the early stages of AD, but also contributes to the
formation of NFTs via the aggregation of hyperphosphorylated tau
protein. Activated MAO, a trigger for oxidative stress, produces
reactive oxygen species in mitochondria and benefits the
pathogenesis of neurodegeneration (130). Theoretically, it is possible that
activated MAO is associated with the formation of NFTs (131). However, it remains to be
elucidated whether activated MAO directly leads to the aggregation
of hyperphosphorylated tau protein and the formation of NFTs since,
to the best of our knowledge, it has not been reported.
An increasing number of molecular biology and
pharmacology studies have shown the neuroprotective effects of MAO
inhibitors on the prevention and treatment of AD (21,52,53)
(Table I) (15,56,57,61,62,112,114,131–144). The main neuroprotective
mechanisms of MAO inhibitors in AD include the following: i)
Improvement of cognitive impairment (50,54,55),
where MAO inhibitors correct chemical imbalances in the brain; ii)
antioxidant activities and enhancement of iron-chelating activities
(56–59), where chelators can modulate Aβ
accumulation, protect against tau hyperphosphorylation and block
metal-associated oxidative stress, thereby holding considerable
promise as effective anti-AD drugs (145,146); iii) regulation of APP and Aβ
expression processing (56,60),
for example ladostigil (TV3326), a selective MAO-B inhibitor, which
regulates APP translation and processing (114); iv) the selective MAO inhibitors
selegiline and rasagiline have been proven to possess
neuroprotective activities in cell cultures and animal models of
neurodegenerative diseases through the activation of certain
signaling pathways, including p42/44 MAPK and PKC (61); v) inhibition of ChE activity by the
MAO inhibitor rasagiline (62–64),
with MAO inhibitors also affecting other chemicals throughout the
body and acting by correcting chemical imbalances in the brain.
Laboratory and clinical studies have evidenced that
the MAO inhibitors are a potential therapeutic approach for the
treatment of AD. Certain novel pyrazole derivatives as dual MAO
inhibitors and anti-inflammatory analgesics (148) are also a novel therapeutic
approach in AD. Thus, selective MAO inhibitors may be a promising
alternative for AD therapy. An enhanced understanding of MAO
inhibitors may result in improved treatment of AD in the future
(149,150).
Activated MAO-B in the brains of patients with AD is
a biomarker for AD. Studies have shown that activated MAO
contributes to cognitive dysfunction, destroys cholinergic neurons,
causes disorder of the cholinergic system and leads to the
formation of amyloid plaques and NFTs.
Numerous studies support the involvement of
activated MAO in AD. Thus, drugs used to inhibit activated MAO,
including rasagiline, ladostigil and selegiline, may provide
neuroprotection against AD by improving cognitive impairment,
modulating the proteolytic cleavage of APP and decreasing levels of
Aβ protein fragments that accumulate in the brain. Although
clinical trials involving the MAO-deactivating drugs have been
largely conducted, numerous questions remain to be answered
regarding the clinical trials of the drugs. As MAO is well known
for its effects on neurotransmitter substance and MAO inhibitors
are infamous for their numerous drug interactions, these drugs may
produce a number of unwanted side effects. It remains to be
elucidated whether MAO inhibitors have severe long-term effects via
the inhibition of chemicals that break down 5-HT, NE and DA, which
may lead to intolerably high levels of any of these neurochemicals.
MAO inhibitors may be particularly harmful when taken with certain
foods, beverages and medicines. Future clinical studies on MAO
inhibitors for the treatment of AD are required, and these may
provide further insights into the mechanism of action of
antioxidants as therapeutic agents for AD.
The present study was supported by a grant from the
Provincial Nature Science Foundation of Anhui (1308085MH158) to Dr
Zhiyou Cai.
|
1
|
Said UZ, Saada HN, Abd-Alla MS, Elsayed ME
and Amin AM: Hesperidin attenuates brain biochemical changes of
irradiated rats. Int J Radiat Biol. 88:613–618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bodkin JA, Cohen BM, Salomon MS, Cannon
SE, Zornberg GL and Cole JO: Treatment of negative symptoms in
schizophrenia and schizoaffective disorder by selegiline
augmentation of antipsychotic medication. A pilot study examining
the role of dopamine. J Nerv Ment Dis. 184:295–301. 1996.
View Article : Google Scholar
|
|
3
|
Dunleavy DL: Mood and sleep changes with
monoamine-oxidase inhibitors. Proc R Soc Med. 66:9511973.PubMed/NCBI
|
|
4
|
Shabbir F, Patel A, Mattison C, Bose S,
Krishnamohan R, Sweeney E, Sandhu S, Nel W, Rais A, Sandhu R, Ngu N
and Sharma S: Effect of diet on serotonergic neurotransmission in
depression. Neurochem Int. 62:324–329. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Merikangas KR and Merikangas JR:
Combination monoamine oxidase inhibitor and beta-blocker treatment
of migraine, with anxiety and depression. Biol Psychiatry.
38:603–610. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Samson JA, Gurrera RJ, Nisenson L and
Schildkraut JJ: Platelet monoamine oxidase activity and deficit
syndrome schizophrenia. Psychiatry Res. 56:25–31. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lawson DC, Turic D, Langley K, Pay HM,
Govan CF, Norton N, Hamshere ML, Owen MJ, O’Donovan MC and Thapar
A: Association analysis of monoamine oxidase A and attention
deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr
Genet. 116B:84–89. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wargelius HL, Malmberg K, Larsson JO and
Oreland L: Associations of MAOA-VNTR or 5HTT-LPR alleles with
attention-deficit hyperactivity disorder symptoms are moderated by
platelet monoamine oxidase B activity. Psychiatr Genet. 22:42–45.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nedic G, Pivac N, Hercigonja DK,
Jovancevic M, Curkovic KD and Muck-Seler D: Platelet monoamine
oxidase activity in children with attention-deficit/hyperactivity
disorder. Psychiatry Res. 175:252–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marziniak M, Mössner R, Benninghoff J,
Syagailo YV, Lesch KP and Sommer C: Association analysis of the
functional monoamine oxidase A gene promotor polymorphism in
migraine. J Neural Transm. 111:603–609. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moreno ML, Villanúa MA and Esquifino AI:
Serum prolactin and luteinizing hormone levels and the activities
of hypothalamic monoamine oxidase A and B and
phenylethanolamine-N-methyl transferase are changed during sexual
maturation in male rats treated neonatally with melatonin. J Pineal
Res. 13:167–1731. 1992. View Article : Google Scholar
|
|
12
|
Youdim MB, Fridkin M and Zheng H: Novel
bifunctional drugs targeting monoamine oxidase inhibition and iron
chelation as an approach to neuroprotection in Parkinson’s disease
and other neurodegenerative diseases. J Neural Transm.
111:1455–1471. 2004.PubMed/NCBI
|
|
13
|
Huang L, Lu C, Sun Y, Mao F, Luo Z, Su T,
Jiang H, Shan W and Li X: Multitarget-directed benzylideneindanone
derivatives: anti-β-amyloid (Aβ) aggregation, antioxidant, metal
chelation, and monoamine oxidase B (MAO-B) inhibition properties
against Alzheimer’s disease. J Med Chem. 55:8483–8492.
2012.PubMed/NCBI
|
|
14
|
Zheng H, Fridkin M and Youdim MB: From
antioxidant chelators to site-activated multi-target chelators
targeting hypoxia inducing factor, beta-amyloid,
acetylcholinesterase and monoamine oxidase A/B. Mini Rev Med Chem.
12:364–370. 2012. View Article : Google Scholar
|
|
15
|
Gal S, Abassi ZA and Youdim MB: Limited
potentiation of blood pressure in response to oral tyramine by the
anti-Parkinson brain selective multifunctional monoamine oxidase-AB
inhibitor, M30. Neurotox Res. 18:143–150. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Youdim MB and Lavie L: Selective MAO-A and
B inhibitors, radical scavengers and nitric oxide synthase
inhibitors in Parkinson’s disease. Life Sci. 55:2077–2082.
1994.PubMed/NCBI
|
|
17
|
Youdim MB and Bakhle YS: Monoamine
oxidase: isoforms and inhibitors in Parkinson’s disease and
depressive illness. Br J Pharmacol. 147(Suppl 1): S287–S296.
2006.
|
|
18
|
Naoi M, Maruyama W, Akao Y, Yi H and
Yamaoka Y: Involvement of type A monoamine oxidase in
neurodegeneration: regulation of mitochondrial signaling leading to
cell death or neuroprotection. J Neural Transm Suppl. 67–77. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zheng H, Youdim MB and Fridkin M:
Site-activated chelators targeting acetylcholinesterase and
monoamine oxidase for Alzheimer’s therapy. ACS Chem Biol.
5:603–610. 2010.
|
|
20
|
Cummings JL: Lewy body diseases with
dementia: pathophysiology and treatment. Brain Cogn. 28:266–280.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Drozak J and Kozłowski M: Monoamine
oxidase as a target for drug action. Postepy Hig Med Dosw (Online).
60:498–515. 2006.(In Polish).
|
|
22
|
Siddiqui A, Mallajosyula JK, Rane A and
Andersen JK: Ability to delay neuropathological events associated
with astrocytic MAO-B increase in a Parkinsonian mouse model:
implications for early intervention on disease progression.
Neurobiol Dis. 43:527–532. 2011. View Article : Google Scholar
|
|
23
|
Bielecka AM, Paul-Samojedny M and
Obuchowicz E: Moclobemide exerts anti-inflammatory effect in
lipopolysaccharide-activated primary mixed glial cell culture.
Naunyn Schmiedebergs Arch Pharmacol. 382:409–417. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Merad-Boudia M, Nicole A, Santiard-Baron
D, Saillé C and Ceballos-Picot I: Mitochondrial impairment as an
early event in the process of apoptosis induced by glutathione
depletion in neuronal cells: relevance to Parkinson’s disease.
Biochem Pharmacol. 56:645–655. 1998.PubMed/NCBI
|
|
25
|
Hüll M, Berger M and Heneka M:
Disease-modifying therapies in Alzheimer’s disease: how far have we
come? Drugs. 66:2075–2093. 2006.
|
|
26
|
Rodríguez S, Ito T, He XJ, Uchida K and
Nakayama H: Resistance of the golden hamster to
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-neurotoxicity
is not only related with low levels of cerebral monoamine
oxidase-B. Exp Toxicol Pathol. 65:127–133. 2013.PubMed/NCBI
|
|
27
|
Konradi C, Riederer P, Jellinger K and
Denney R: Cellular action of MAO inhibitors. J Neural Transm Suppl.
25:15–25. 1987.
|
|
28
|
Weinstock M, Luques L, Poltyrev T, Bejar C
and Shoham S: Ladostigil prevents age-related glial activation and
spatial memory deficits in rats. Neurobiol Aging. 32:1069–1078.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Spina MB and Cohen G: Dopamine turnover
and glutathione oxidation: implications for Parkinson disease. Proc
Natl Acad Sci USA. 86:1398–1400. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Farooqui T and Farooqui AA: Lipid-mediated
oxidative stress and inflammation in the pathogenesis of
Parkinson’s disease. Parkinsons Dis. 2011:2474672011.PubMed/NCBI
|
|
31
|
Loeffler DA, DeMaggio AJ, Juneau PL,
Havaich MK and LeWitt PA: Effects of enhanced striatal dopamine
turnover in vivo on glutathione oxidation. Clin Neuropharmacol.
17:370–379. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tapia-González S, Giráldez-Pérez RM,
Cuartero MI, Casarejos MJ, Mena MÁ, Wang XF and Sánchez-Capelo A:
Dopamine and α-synuclein dysfunction in Smad3 null mice. Mol
Neurodegener. 6:722011.
|
|
33
|
Oberpichler-Schwenk H: Rasagiline. A new
monoamine oxidase b inhibitor for Parkinson treatment. Med
Monatsschr Pharm. 28:224–227. 2005.(In German).
|
|
34
|
Chen JJ and Wilkinson JR: The monoamine
oxidase type B inhibitor rasagiline in the treatment of Parkinson
disease: is tyramine a challenge? J Clin Pharmacol. 52:620–628.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oreland L and Gottfries CG: Brain and
brain monoamine oxidase in aging and in dementia of Alzheimer’s
type. Prog Neuropsychopharmacol Biol Psychiatry. 10:533–540.
1986.
|
|
36
|
Sherif F, Gottfries CG, Alafuzoff I and
Oreland L: Brain gamma-aminobutyrate aminotransferase (GABA-T) and
monoamine oxidase (MAO) in patients with Alzheimer’s disease. J
Neural Transm Park Dis Dement Sect. 4:227–240. 1992.PubMed/NCBI
|
|
37
|
Sparks DL, Woeltz VM and Markesbery WR:
Alterations in brain monoamine oxidase activity in aging,
Alzheimer’s disease, and Pick’s disease. Arch Neurol. 48:718–721.
1991.
|
|
38
|
Gulyás B, Pavlova E, Kása P, Gulya K,
Bakota L, Várszegi S, Keller E, Horváth MC, Nag S, Hermecz I,
Magyar K and Halldin C: Activated MAO-B in the brain of Alzheimer
patients, demonstrated by [11C]-L-deprenyl using whole hemisphere
autoradiography. Neurochem Int. 58:60–68. 2011.PubMed/NCBI
|
|
39
|
Hirvonen J, Kailajärvi M, Haltia T,
Koskimies S, Någren K, Virsu P, Oikonen V, Sipilä H, Ruokoniemi P,
Virtanen K, Scheinin M and Rinne JO: Assessment of MAO-B occupancy
in the brain with PET and [11C]-L-deprenyl-D2: a dose-finding study
with a novel MAO-B inhibitor, EVT 301. Clin Pharmacol Ther.
85:506–512. 2009.PubMed/NCBI
|
|
40
|
Jossan SS, Gillberg PG, Karlsson I,
Gottfries CG and Oreland L: Visualization of brain monoamine
oxidase B (MAO-B) in dementia of Alzheimer’s type by means of large
cryosection autoradiography: a pilot study. J Neural Transm Suppl.
32:61–65. 1990.
|
|
41
|
Fischer P, Götz ME, Ellinger B, Streifler
M, Riederer P and Danielczyk W: Platelet monoamine oxidase B
activity and vitamin B12 in dementia. Biol Psychiatry. 35:772–774.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Muck-Seler D, Presecki P, Mimica N,
Mustapic M, Pivac N, Babic A, Nedic G and Folnegovic-Smalc V:
Platelet serotonin concentration and monoamine oxidase type B
activity in female patients in early, middle and late phase of
Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry.
33:1226–1231. 2009.PubMed/NCBI
|
|
43
|
Riederer P and Jellinger K: Morphological
and biochemical changes in the aging brain: pathophysiological and
possible therapeutic consequences. Exp Brain Res. (Suppl 5):
158–166. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Battistin L, Rigo A, Bracco F, Dam M and
Pizzolato G: Metabolic aspects of aging brain and related
disorders. Gerontology. 33:253–258. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mimica N, Mück-Seler D, Pivac N, Mustapić
M, Dezeljin M, Stipcević T, Presecki P, Radonić E and
Folnegović-Smalc V: Platelet serotonin and monoamine oxidase in
Alzheimer’s disease with psychotic features. Coll Antropol.
32(Suppl 1): 119–122. 2008.
|
|
46
|
Götz ME, Fischer P, Gsell W, Riederer P,
Streifler M, Simanyi M, Müller F and Danielczyk W: Platelet
monoamine oxidase B activity in dementia. A 4-year follow-up.
Dement Geriatr Cogn Disord. 9:74–77. 1998.PubMed/NCBI
|
|
47
|
Parnetti L, Reboldi GP, Santucci C,
Santucci A, Gaiti A, Brunetti M, Cecchetti R and Senin U: Platelet
MAO-B activity as a marker of behavioural characteristics in
dementia disorders. Aging (Milano). 6:201–207. 1994.PubMed/NCBI
|
|
48
|
Bonuccelli U, Piccini P, Marazziti D,
Cassano GB and Muratorio A: Increased platelet 3H-imipramine
binding and monoamine oxidase B activity in Alzheimer’s disease. J
Neural Transm Park Dis Dement Sect. 2:139–147. 1990.
|
|
49
|
Kennedy BP, Ziegler MG, Alford M, Hansen
LA, Thal LJ and Masliah E: Early and persistent alterations in
prefrontal cortex MAO A and B in Alzheimer’s disease. J Neural
Transm. 110:789–801. 2003.PubMed/NCBI
|
|
50
|
Delumeau JC, Bentué-Ferrer D, Gandon JM,
Amrein R, Belliard S and Allain H: Monoamine oxidase inhibitors,
cognitive functions and neurodegenerative diseases. J Neural Transm
Suppl. 41:259–266. 1994.PubMed/NCBI
|
|
51
|
Grailhe R, Cardona A, Even N, Seif I,
Changeux JP and Cloëz-Tayarani I: Regional changes in the
cholinergic system in mice lacking monoamine oxidase A. Brain Res
Bull. 78:283–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Riederer P, Danielczyk W and Grünblatt E:
Monoamine oxidase-B inhibition in Alzheimer’s disease.
Neurotoxicology. 25:271–277. 2004.
|
|
53
|
Thomas T: Monoamine oxidase-B inhibitors
in the treatment of Alzheimer’s disease. Neurobiol Aging.
21:343–348. 2000.
|
|
54
|
Soto J, Ulibarri I, Jauregui JV,
Ballesteros J and Meana JJ: Dissociation between I2-imidazoline
receptors and MAO-B activity in platelets of patients with
Alzheimer’s type dementia. J Psychiatr Res. 33:251–257.
1999.PubMed/NCBI
|
|
55
|
Finali G, Piccirilli M, Oliani C and
Piccinin GL: L-deprenyl therapy improves verbal memory in amnesic
Alzheimer patients. Clin Neuropharmacol. 14:523–536. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Weinreb O, Mandel S, Bar-Am O and Amit T:
Iron-chelating backbone coupled with monoamine oxidase inhibitory
moiety as novel pluripotential therapeutic agents for Alzheimer’s
disease: a tribute to Moussa Youdim. J Neural Transm. 118:479–492.
2011.PubMed/NCBI
|
|
57
|
Guay DR: Rasagiline (TVP-1012): a new
selective monoamine oxidase inhibitor for Parkinson’s disease. Am J
Geriatr Pharmacother. 4:330–346. 2006.
|
|
58
|
Grünblatt E, Schlösser R, Fischer P,
Fischer MO, Li J, Koutsilieri E, Wichart I, Sterba N, Rujescu D,
Möller HJ, Adamcyk W, Dittrich B, Müller F, Oberegger K, Gatterer
G, Jellinger KJ, Mostafaie N, Jungwirth S, Huber K, Tragl KH,
Danielczyk W and Riederer P: Oxidative stress related markers in
the ‘VITA’ and the centenarian projects. Neurobiol Aging.
26:429–438. 2005.
|
|
59
|
Wu RM, Mohanakumar KP, Murphy DL and
Chiueh CC: Antioxidant mechanism and protection of nigral neurons
against MPP+ toxicity by deprenyl (selegiline). Ann NY
Acad Sci. 738:214–221. 1994.PubMed/NCBI
|
|
60
|
Youdim MB, Amit T, Bar-Am O, Weinreb O and
Yogev-Falach M: Implications of co-morbidity for etiology and
treatment of neurodegenerative diseases with multifunctional
neuroprotective-neurorescue drugs; ladostigil. Neurotox Res.
10:181–192. 2006. View Article : Google Scholar
|
|
61
|
Bar-Am O, Amit T, Weinreb O, Youdim MB and
Mandel S: Propargylamine containing compounds as modulators of
proteolytic cleavage of amyloid-beta protein precursor: involvement
of MAPK and PKC activation. J Alzheimers Dis. 21:361–371.
2010.PubMed/NCBI
|
|
62
|
Weinreb O, Mandel S, Bar-Am O,
Yogev-Falach M, Avramovich-Tirosh Y, Amit T and Youdim MB:
Multifunctional neuroprotective derivatives of rasagiline as
anti-Alzheimer’s disease drugs. Neurotherapeutics. 6:163–174.
2009.PubMed/NCBI
|
|
63
|
Weinstock M and Groner E: Rational design
of a drug for Alzheimer’s disease with cholinesterase inhibitory
and neuroprotective activity. Chem Biol Interact. 175:216–221.
2008.
|
|
64
|
Youdim MB, Fridkin M and Zheng H:
Bifunctional drug derivatives of MAO-B inhibitor rasagiline and
iron chelator VK-28 as a more effective approach to treatment of
brain ageing and ageing neurodegenerative diseases. Mech Ageing
Dev. 126:317–326. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Adolfsson R, Gottfries CG, Oreland L,
Wiberg A and Winblad B: Increased activity of brain and platelet
monoamine oxidase in dementia of Alzheimer type. Life Sci.
27:1029–1034. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bongioanni P, Gemignani F, Boccardi B,
Borgna M and Rossi B: Platelet monoamine oxidase molecular activity
in demented patients. Ital J Neurol Sci. 18:151–156. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ahlskog JE, Uitti RJ, Tyce GM, O’Brien JF,
Petersen RC and Kokmen E: Plasma catechols and monoamine oxidase
metabolites in untreated Parkinson’s and Alzheimer’s diseases. J
Neurol Sci. 136:162–168. 1996.PubMed/NCBI
|
|
68
|
Konings CH, Scheltens P, Kuiper MA and
Wolters EC: No evidence for abnormalities in kinetics of platelet
monoamine oxidase in Alzheimer’s disease. Clin Chim Acta.
240:99–102. 1995.PubMed/NCBI
|
|
69
|
Winblad B, Gottfries CG, Oreland L and
Wiberg A: Monoamine oxidase in platelets and brains of
non-psychiatric and non-neurological geriatric patients. Med Biol.
57:129–132. 1979.PubMed/NCBI
|
|
70
|
Mann JJ, Stanley M, Neophytides A, de Leon
MJ, Ferris SH and Gershon S: Central amine metabolism in
Alzheimer’s disease: in vivo relationship to cognitive deficit.
Neurobiol Aging. 2:57–60. 1981.
|
|
71
|
da Silva VB, de Andrade P, Kawano DF,
Morais PA, de Almeida JR, Carvalho I, Taft CA and da Silva CH: In
silico design and search for acetylcholinesterase inhibitors in
Alzheimer’s disease with a suitable pharmacokinetic profile and low
toxicity. Future Med Chem. 3:947–960. 2011.
|
|
72
|
Schneider LS, Severson JA, Chui HC,
Pollock VE, Sloane RB and Fredrickson ER: Platelet tritiated
imipramine binding and MAO activity in Alzheimer’s disease patients
with agitation and delusions. Psychiatry Res. 25:311–322.
1988.PubMed/NCBI
|
|
73
|
Rodríguez MJ, Saura J, Billett EE, Finch
CC and Mahy N: Cellular localization of monoamine oxidase A and B
in human tissues outside of the central nervous system. Cell Tissue
Res. 304:215–220. 2001.PubMed/NCBI
|
|
74
|
Sivasubramaniam SD, Finch CC, Rodriguez
MJ, Mahy N and Billett EE: A comparative study of the expression of
monoamine oxidase-A and -B mRNA and protein in non-CNS human
tissues. Cell Tissue Res. 313:291–300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Palmer AM and DeKosky ST: Monoamine
neurons in aging and Alzheimer’s disease. J Neural Transm Gen Sect.
91:135–159. 1993.
|
|
76
|
Reinikainen KJ, Paljärvi L, Halonen T,
Malminen O, Kosma VM, Laakso M and Riekkinen PJ: Dopaminergic
system and monoamine oxidase-B activity in Alzheimer’s disease.
Neurobiol Aging. 9:245–252. 1988.
|
|
77
|
Emilsson L, Saetre P, Balciuniene J,
Castensson A, Cairns N and Jazin EE: Increased monoamine oxidase
messenger RNA expression levels in frontal cortex of Alzheimer’s
disease patients. Neurosci Lett. 326:56–60. 2002.PubMed/NCBI
|
|
78
|
Burke WJ, Li SW, Schmitt CA, Xia P, Chung
HD and Gillespie KN: Accumulation of
3,4-dihydroxyphenylglycolaldehyde, the neurotoxic monoamine oxidase
A metabolite of norepinephrine, in locus ceruleus cell bodies in
Alzheimer’s disease: mechanism of neuron death. Brain Res.
816:633–637. 1999.PubMed/NCBI
|
|
79
|
Chan-Palay V, Höchli M, Savaskan E and
Hungerecker G: Calbindin D-28k and monoamine oxidase A
immunoreactive neurons in the nucleus basalis of Meynert in senile
dementia of the Alzheimer type and Parkinson’s disease. Dementia.
4:1–15. 1993.PubMed/NCBI
|
|
80
|
Barnett JH, Xu K, Heron J, Goldman D and
Jones PB: Cognitive effects of genetic variation in monoamine
neurotransmitter systems: a population-based study of COMT, MAOA,
and 5HTTLPR. Am J Med Genet B Neuropsychiatr Genet. 156:158–167.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Klinteberg B, Levander SE, Oreland L,
Asberg M and Schalling D: Neuropsychological correlates of platelet
monoamine oxidase (MAO) activity in female and male subjects. Biol
Psychol. 24:237–252. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Heimberg RG, Liebowitz MR, Hope DA,
Schneier FR, Holt CS, Welkowitz LA, Juster HR, Campeas R, Bruch MA,
Cloitre M, Fallon B and Klein DF: Cognitive behavioral group
therapy vs phenelzine therapy for social phobia: 12-week outcome.
Arch Gen Psychiatry. 55:1133–1141. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Goldstein DM and Goldberg RL: Monoamine
oxidase inhibitor-induced speech blockage. J Clin Psychiatry.
47:6041986.PubMed/NCBI
|
|
84
|
Delcker A and Gaertner HJ: Tolerability
and antidepressive effect of brofaromine, a short-acting reversible
MAO inhibitor - an open study. Eur Neuropsychopharmacol. 1:177–180.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Danilova RA, Moskvityna TA, Obukhova MF,
Belopolskaya MV and Ashmarin IP: Pargyline conjugate-induced
long-term activation of monoamine oxidase as an immunological model
for depression. Neurochem Res. 24:1147–1151. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Giacobini G, Marchisio PC, Giacobini E and
Koslow SH: Developmental changes of cholinesterases and monoamine
oxidase in chick embryo spinal and sympathetic ganglia. J
Neurochem. 17:1177–1185. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Van der Schyf CJ, Gal S, Geldenhuys WJ and
Youdim MB: Multifunctional neuroprotective drugs targeting
monoamine oxidase inhibition, iron chelation, adenosine receptors,
and cholinergic and glutamatergic action for neurodegenerative
diseases. Expert Opin Investig Drugs. 15:873–886. 2006.
|
|
88
|
Ikemoto K, Kitahama K, Maeda T, Jouvet M
and Nagatsu I: Cholinergic neurons with monoamine oxidase type B
(MAOB)-activity in the laterodorsal tegmental nucleus of the mouse.
Neurosci Lett. 271:53–56. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Nakamura S, Akiguchi I and Kimura J: A
subpopulation of mouse striatal cholinergic neurons show monoamine
oxidase activity. Neurosci Lett. 161:141–144. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Panagiotidis G, Stenström A and Lundquist
I: Effects of adrenergic and cholinergic stimulation on islet
monoamine oxidase activity and insulin secretion in the mouse. Eur
J Pharmacol. 233:285–290. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Pintar JE, Breakefield XO and Patterson
PH: Differences in monoamine oxidase activity between cultured
noradrenergic and cholinergic sympathetic neurons. Dev Biol.
120:305–308. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Garrick NA and Murphy DL: Monoamine
oxidase type A: differences in selectivity towards l-norepinephrine
compared to serotonin. Biochem Pharmacol. 31:4061–4066. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Spector S: Monoamine oxidase in control of
brain serotonin and norepinephrine content. Ann NY Acad Sci.
107:856–864. 1963. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kumagae Y, Matsui Y and Iwata N:
Deamination of norepinephrine, dopamine, and serotonin by type A
monoamine oxidase in discrete regions of the rat brain and
inhibition by RS-8359. Jpn J Pharmacol. 55:121–128. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Baron M, Perumal AS, Levitt M and Cannova
G: Platelet monoamine oxidase in schizophrenia with
beta-phenylethylamine and benzylamine as substrates. Biol
Psychiatry. 17:479–483. 1982.PubMed/NCBI
|
|
96
|
Lewinsohn R, Glover V and Sandler M:
Beta-phenylethylamine and benzylamine as substrates for human
monoamine oxidase A: A source of some anomalies? Biochem Pharmacol.
29:777–781. 1980.PubMed/NCBI
|
|
97
|
Moret C and Briley M: The importance of
norepinephrine in depression. Neuropsychiatr Dis Treat. 7(Suppl 1):
9–13. 2011.PubMed/NCBI
|
|
98
|
Levitan MN, Chagas MH, Crippa JA, Manfro
GG, Hetem LA, Andrada NC, Salum GA, Isolan L, Ferrari MC and Nardi
AE; Brazilian Medical Association. Guidelines of the Brazilian
Medical Association for the treatment of social anxiety disorder.
Rev Bras Psiquiatr. 33:292–302. 2011.(In Portugese).
|
|
99
|
Schneier FR: Pharmacotherapy of social
anxiety disorder. Expert Opin Pharmacother. 12:615–625. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Engelborghs S and De Deyn PP: The
neurochemistry of Alzheimer’s disease. Acta Neurol Belg. 97:67–84.
1997.
|
|
101
|
Ishrat T, Parveen K, Khan MM, Khuwaja G,
Khan MB, Yousuf S, Ahmad A, Shrivastav P and Islam F: Selenium
prevents cognitive decline and oxidative damage in rat model of
streptozotocin-induced experimental dementia of Alzheimer’s type.
Brain Res. 1281:117–127. 2009.PubMed/NCBI
|
|
102
|
Schaeffer EL and Gattaz WF: Cholinergic
and glutamatergic alterations beginning at the early stages of
Alzheimer disease: participation of the phospholipase A2 enzyme.
Psychopharmacology (Berl). 198:1–27. 2008. View Article : Google Scholar
|
|
103
|
Tran MH, Yamada K and Nabeshima T: Amyloid
beta-peptide induces cholinergic dysfunction and cognitive
deficits: a minireview. Peptides. 23:1271–1283. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Dhull DK, Jindal A, Dhull RK, Aggarwal S,
Bhateja D and Padi SS: Neuroprotective effect of cyclooxygenase
inhibitors in ICV-STZ induced sporadic Alzheimer’s disease in rats.
J Mol Neurosci. 46:223–235. 2012.PubMed/NCBI
|
|
105
|
McNaull BB, Todd S, McGuinness B and
Passmore AP: Inflammation and anti-inflammatory strategies for
Alzheimer’s disease - a mini-review. Gerontology. 56:3–14.
2010.
|
|
106
|
Cai ZY, Yan Y and Chen R: Minocycline
reduces astrocytic reactivation and neuroinflammation in the
hippocampus of a vascular cognitive impairment rat model. Neurosci
Bull. 26:28–36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Agostinho P, Cunha RA and Oliveira C:
Neuroinflammation, oxidative stress and the pathogenesis of
Alzheimer’s disease. Curr Pharm Des. 16:2766–2778. 2010.
|
|
108
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer’s disease: progress and problems on the
road to therapeutics. Science. 297:353–356. 2002.
|
|
109
|
Song W, Zhou LJ, Zheng SX and Zhu XZ:
Amyloid-beta 25–35 peptide induces expression of monoamine oxidase
B in cultured rat astrocytes. Acta Pharmacol Sin. 21:557–563.
2000.
|
|
110
|
Carter SF, Schöll M, Almkvist O, Wall A,
Engler H, Långström B and Nordberg A: Evidence for astrocytosis in
prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a
multitracer PET paradigm combining 11C-Pittsburgh compound B and
18F-FDG. J Nucl Med. 53:37–46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Hu MK, Liao YF, Chen JF, Wang BJ, Tung YT,
Lin HC and Lee KP: New 1,2,3,4-tetrahydroisoquinoline derivatives
as modulators of proteolytic cleavage of amyloid precursor
proteins. Bioorg Med Chem. 16:1957–1965. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Avramovich-Tirosh Y, Amit T, Bar-Am O,
Zheng H, Fridkin M and Youdim MB: Therapeutic targets and potential
of the novel brain- permeable multifunctional iron
chelator-monoamine oxidase inhibitor drug, M-30, for the treatment
of Alzheimer’s disease. J Neurochem. 100:490–502. 2007.PubMed/NCBI
|
|
113
|
Ono K, Hasegawa K, Naiki H and Yamada M:
Anti-Parkinsonian agents have anti-amyloidogenic activity for
Alzheimer’s beta-amyloid fibrils in vitro. Neurochem Int.
48:275–285. 2006.PubMed/NCBI
|
|
114
|
Yogev-Falach M, Bar-Am O, Amit T, Weinreb
O and Youdim MB: A multifunctional, neuroprotective drug,
ladostigil (TV3326), regulates holo-APP translation and processing.
FASEB J. 20:2177–2179. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Weinreb O, Amit T, Bar-Am O, Sagi Y,
Mandel S and Youdim MB: Involvement of multiple survival signal
transduction pathways in the neuroprotective, neurorescue and APP
processing activity of rasagiline and its propargyl moiety. J
Neural Transm Suppl. 457–465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhiyou C, Yong Y, Shanquan S, Jun Z,
Liangguo H, Ling Y and Jieying L: Upregulation of BACE1 and
beta-amyloid protein mediated by chronic cerebral hypoperfusion
contributes to cognitive impairment and pathogenesis of Alzheimer’s
disease. Neurochem Res. 34:1226–1235. 2009.PubMed/NCBI
|
|
117
|
Liu H, Li Z, Qiu D, Gu Q, Lei Q and Mao L:
The inhibitory effects of different curcuminoids on β-amyloid
protein, β-amyloid precursor protein and β-site amyloid precursor
protein cleaving enzyme 1 in swAPP HEK293 cells. Neurosci Lett.
485:83–88. 2010.
|
|
118
|
Yogev-Falach M, Amit T, Bar-Am O,
Weinstock M and Youdim MB: Involvement of MAP kinase in the
regulation of amyloid precursor protein processing by novel
cholinesterase inhibitors derived from rasagiline. FASEB J.
16:1674–1676. 2002.PubMed/NCBI
|
|
119
|
Youdim MB, Bar Am O, Yogev-Falach M,
Weinreb O, Maruyama W, Naoi M and Amit T: Rasagiline:
neurodegeneration, neuroprotection, and mitochondrial permeability
transition. J Neurosci Res. 79:172–179. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Youdim MB, Amit T, Bar-Am O, Weinstock M
and Yogev-Falach M: Amyloid processing and signal transduction
properties of antiparkinson-antialzheimer neuroprotective drugs
rasagiline and TV3326. Ann NY Acad Sci. 993:378–393. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Kupershmidt L, Amit T, Bar-Am O, Youdim MB
and Weinreb O: The novel multi-target iron chelating-radical
scavenging compound M30 possesses beneficial effects on major
hallmarks of Alzheimer’s disease. Antioxid Redox Signal.
17:860–877. 2012.PubMed/NCBI
|
|
122
|
Yang HQ, Sun ZK, Ba MW, Xu J and Xing Y:
Involvement of protein trafficking in deprenyl-induced
alpha-secretase activity regulation in PC12 cells. Eur J Pharmacol.
610:37–41. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Tsunekawa H, Noda Y, Mouri A, Yoneda F and
Nabeshima T: Synergistic effects of selegiline and donepezil on
cognitive impairment induced by amyloid beta (25–35). Behav Brain
Res. 190:224–232. 2008.PubMed/NCBI
|
|
124
|
Calderón-Garcidueñas L, Kavanaugh M, Block
M, D’Angiulli A, Delgado-Chávez R, Torres-Jardón R, González-Maciel
A, Reynoso-Robles R, Osnaya N, Villarreal-Calderon R, Guo R, Hua Z,
Zhu H, Perry G and Diaz P: Neuroinflammation, hyperphosphorylated
tau, diffuse amyloid plaques, and down-regulation of the cellular
prion protein in air pollution exposed children and young adults. J
Alzheimers Dis. 28:93–107. 2012.
|
|
125
|
Stozicka Z, Zilka N, Novak P, Kovacech B,
Bugos O and Novak M: Genetic background modifies neurodegeneration
and neuroinflammation driven by misfolded human tau protein in rat
model of tauopathy: implication for immunomodulatory approach to
Alzheimer’s disease. J Neuroinflammation. 7:642010.
|
|
126
|
McGeer EG and McGeer PL: Neuroinflammation
in Alzheimer’s disease and mild cognitive impairment: a field in
its infancy. J Alzheimers Dis. 19:355–361. 2010.
|
|
127
|
Hensley K: Neuroinflammation in
Alzheimer’s disease: mechanisms, pathologic consequences, and
potential for therapeutic manipulation. J Alzheimers Dis. 21:1–14.
2010.
|
|
128
|
Streit WJ: Microglial activation and
neuroinflammation in Alzheimer’s disease: a critical examination of
recent history. Front Aging Neurosci. 2:222010.
|
|
129
|
Saura J, Luque JM, Cesura AM, Da Prada M,
Chan-Palay V, Huber G, Löffler J and Richards JG: Increased
monoamine oxidase B activity in plaque-associated astrocytes of
Alzheimer brains revealed by quantitative enzyme radioautography.
Neuroscience. 62:15–30. 1994. View Article : Google Scholar
|
|
130
|
Menazza S, Blaauw B, Tiepolo T, Toniolo L,
Braghetta P, Spolaore B, Reggiani C, Di Lisa F, Bonaldo P and
Canton M: Oxidative stress by monoamine oxidases is causally
involved in myofiber damage in muscular dystrophy. Hum Mol Genet.
19:4207–4215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zheng H, Amit T, Bar-Am O, Fridkin M,
Youdim MB and Mandel SA: From anti-Parkinson’s drug rasagiline to
novel multitarget iron chelators with acetylcholinesterase and
monoamine oxidase inhibitory and neuroprotective properties for
Alzheimer’s disease. J Alzheimers Dis. 30:1–16. 2012.
|
|
132
|
Dimpfel W and Hoffmann JA: Effects of
rasagiline, its metabolite aminoindan and selegiline on glutamate
receptor mediated signalling in the rat hippocampus slice in vitro.
BMC Pharmacol. 11:22011. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Youdim MB, Maruyama W and Naoi M:
Neuropharmacological, neuroprotective and amyloid precursor
processing properties of selective MAO-B inhibitor antiparkinsonian
drug, rasagiline. Drugs Today (Barc). 41:369–391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Weinreb O, Amit T, Bar-Am O and Youdim MB:
Ladostigil: a novel multimodal neuroprotective drug with
cholinesterase and brain-selective monoamine oxidase inhibitory
activities for Alzheimer’s disease treatment. Curr Drug Targets.
13:483–494. 2012.
|
|
135
|
Weinreb O, Amit T, Bar-Am O and Youdim MB:
A novel anti-Alzheimer’s disease drug, ladostigil neuroprotective,
multimodal brain-selective monoamine oxidase and cholinesterase
inhibitor. Int Rev Neurobiol. 100:191–215. 2011.
|
|
136
|
Alafuzoff I, Helisalmi S, Heinonen EH,
Reinikainen K, Hallikainen M, Soininen H and Koivisto K: Selegiline
treatment and the extent of degenerative changes in brain tissue of
patients with Alzheimer’s disease. Eur J Clin Pharmacol.
55:815–819. 2000.PubMed/NCBI
|
|
137
|
Birks J and Flicker L: Selegiline for
Alzheimer’s disease. Cochrane Database Syst Rev. CD0004422003.
|
|
138
|
Wilcock GK, Birks J, Whitehead A and Evans
SJ: The effect of selegiline in the treatment of people with
Alzheimer’s disease: a meta-analysis of published trials. Int J
Geriatr Psychiatry. 17:175–183. 2002.
|
|
139
|
Birks J and Flicker L: Selegiline for
Alzheimer’s disease. Cochrane Database Syst Rev. CD0004422000.
|
|
140
|
Filip V and Kolibás E: Selegiline in the
treatment of Alzheimer’s disease: a long-term randomized
placebo-controlled trial. Czech and Slovak Senile Dementia of
Alzheimer Type Study Group. J Psychiatry Neurosci. 24:234–243.
1999.
|
|
141
|
Lawlor BA, Aisen PS, Green C, Fine E and
Schmeïdler J: Selegiline in the treatment of behavioural
disturbance in Alzheimer’s disease. Int J Geriatr Psychiatry.
12:319–322. 1997.
|
|
142
|
Sano M, Ernesto C, Klauber MR, Schafer K,
Woodbury P, Thomas R, Grundman M, Growdon J and Thal LJ: Rationale
and design of a multicenter study of selegiline and
alpha-tocopherol in the treatment of Alzheimer disease using novel
clinical outcomes. Alzheimer’s Disease Cooperative Study. Alzheimer
Dis Assoc Disord. 10:132–140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Schneider LS, Olin JT and Pawluczyk S: A
double-blind crossover pilot study of l-deprenyl (selegiline)
combined with cholinesterase inhibitor in Alzheimer’s disease. Am J
Psychiatry. 150:321–323. 1993.PubMed/NCBI
|
|
144
|
Youdim MB: The path from anti Parkinson
drug selegiline and rasagiline to multifunctional neuroprotective
anti Alzheimer drugs ladostigil and m30. Curr Alzheimer Res.
3:541–550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zheng H, Fridkin M and Youdim MB:
Site-activated chelators derived from anti-Parkinson drug
rasagiline as a potential safer and more effective approach to the
treatment of Alzheimer’s disease. Neurochem Res. 35:2117–2123.
2010.PubMed/NCBI
|
|
146
|
Zheng H, Weiner LM, Bar-Am O, Epsztejn S,
Cabantchik ZI, Warshawsky A, Youdim MB and Fridkin M: Design,
synthesis, and evaluation of novel bifunctional iron-chelators as
potential agents for neuroprotection in Alzheimer’s, Parkinson’s,
and other neurodegenerative diseases. Bioorg Med Chem. 13:773–783.
2005.PubMed/NCBI
|
|
147
|
Gökhan-Kelekçi N, Yabanoǧlu S, Küpeli E,
Salgin U, Ozgen O, Uçar G, Yešilada E, Kendi E, Yešilada A and
Bilgin AA: A new therapeutic approach in Alzheimer disease: some
novel pyrazole derivatives as dual MAO-B inhibitors and
antiinflammatory analgesics. Bioorg Med Chem. 15:5775–5786.
2007.PubMed/NCBI
|
|
148
|
Yamada M and Yasuhara H: Clinical
pharmacology of MAO inhibitors: safety and future. Neurotoxicology.
25:215–221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Yu PH: Pharmacological and clinical
implications of MAO-B inhibitors. Gen Pharmacol. 25:1527–1539.
1994. View Article : Google Scholar : PubMed/NCBI
|