Introduction
Osteoblastoma is a rare bone-forming neoplasm
accounting for less than 1% of primary bone tumors. It presents in
young patients and develops most often in the long bones of the
extremities, the talus and posterior elements of the spine
(2). Osteoblastoma is also referred
to as an osteoblastic osteoid tissue-forming tumor, giant osteoid
osteoma, benign osteoblastoma, osteogenic fibroma and a
spindle-cell variant of giant cell tumor. In radiologic
examination, osteoblastoma is typically observed as a radiolucent
lesion surrounded by a thin margin of reactive bone that may have
an expanded aneurysmal appearance. En bloc marginal excision is the
treatment of choice with a risk of recurrence, after marginal
excision of aggresive stage III, of 30–50%.
Case study
A 12-year-old female patient was admitted to the
Pediatric Hospital in Bialystok in June 2003, due to persistent
exophthalmia. The exophthalmos was bilateral, visibly greater on
the right side and had been observed by the patient’s mother for
4–5 months. No other symptoms were reported by the patient, except
for a two-month history of headaches prior to admission. The pain
was localized in the forehead and was not accompanied by nausea or
vomiting.
Written informed consent was obtained from the
patient for publication of this case report and any accompanying
images.
On admission to the hospital and physical
examination, no signs of nasal congestion, nasal dripping or
epistaxis were observed. An ophthalmological examination showed
exophthalmia (R>L), while both the fundus of the eyes and the
visual area were normal.
The findings of the laboratory tests including basal
cell carcinoma, electrolytes, proteinogram, CRP, glucose, urea and
creatinine showed parameters to be within the range of the norm.
Notably, thyroid hormones and TSH levels were also within the range
of the norm. Laboratory tests were followed by radiologic
examinations, plain radiography, computed tomography (CT) and
magnetic resonance imaging (MRI).
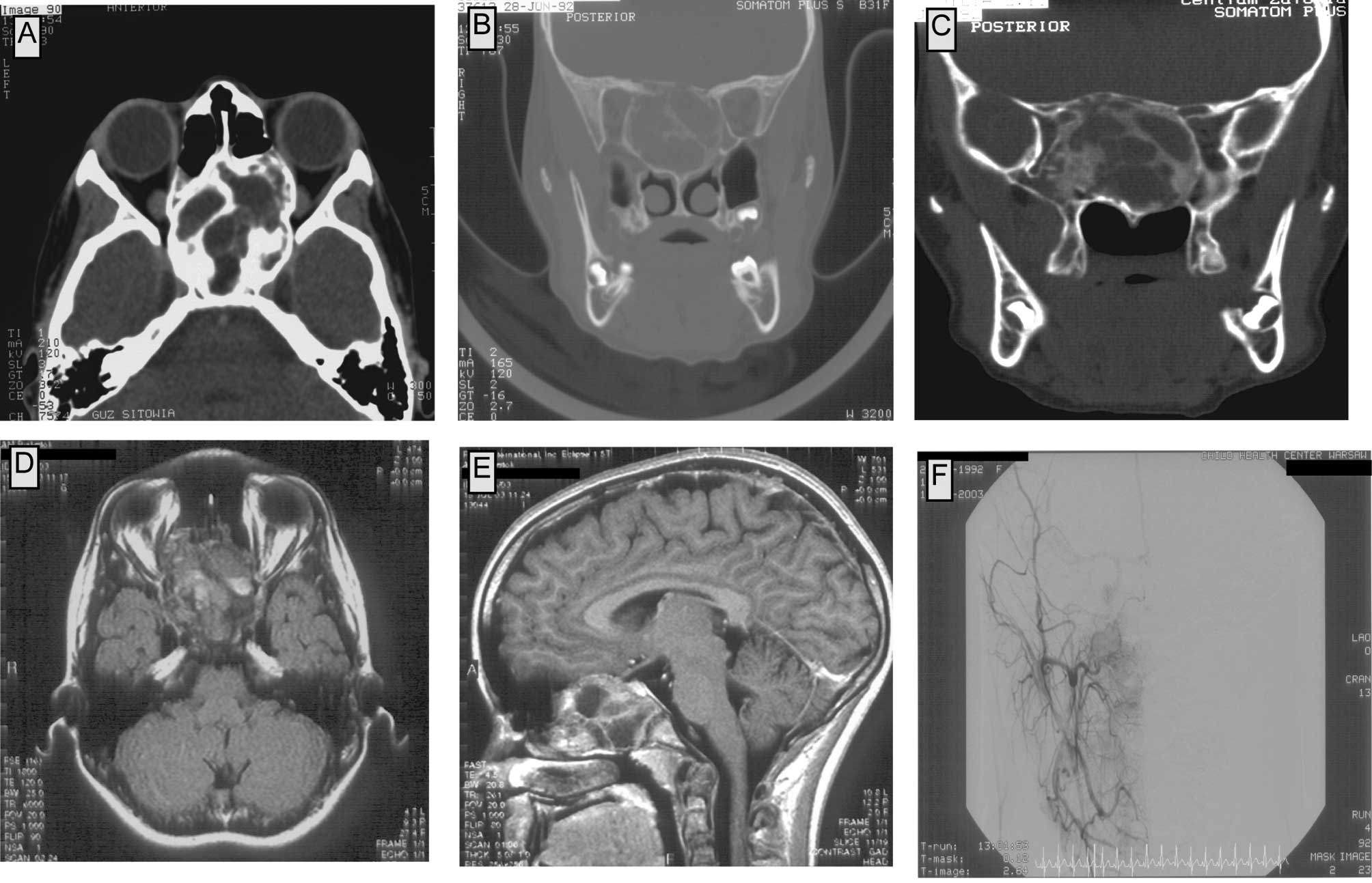
In plain radiography, a pathological mass in the
area of the sella turcica and the sphenoid sinus was observed. CT
scans showed a tumor filling the upper-posterior part of the nasal
cavity, the posterior ethmoids and the sphenoid sinus. Medial walls
of the orbits were modelled by the tumor; on the right side the
tumor compressed the medial and inferior rectus muscle and reached
the maxillary sinus. The corpus of the sphenoid bone was expanded;
the tumor reached the frontal lobes, but did not infiltrate into
the neural tissue of the brain. The upper-posterior part of the
tumor reached the cavernous sinus bilaterally. The tumor consisted
of numerous bony trabeculae and areas of calcification. No contrast
enhancement in the tumor was observed in the CT scans (Fig. 1A–D).
The MRI showed a tumor with an approximate diameter
of 55×35×55 mm, involving the corpus of the sphenoid bone, the
ethmoids bilaterally and the upper part of the nasal cavity. In the
frontal and temporal region, the lesion invaded the cranial base,
but no signs of brain infiltration were found (Fig. 1E and F). To define the diagnosis, a
biopsy was taken from the lumen of the sphenoid sinus on July 31,
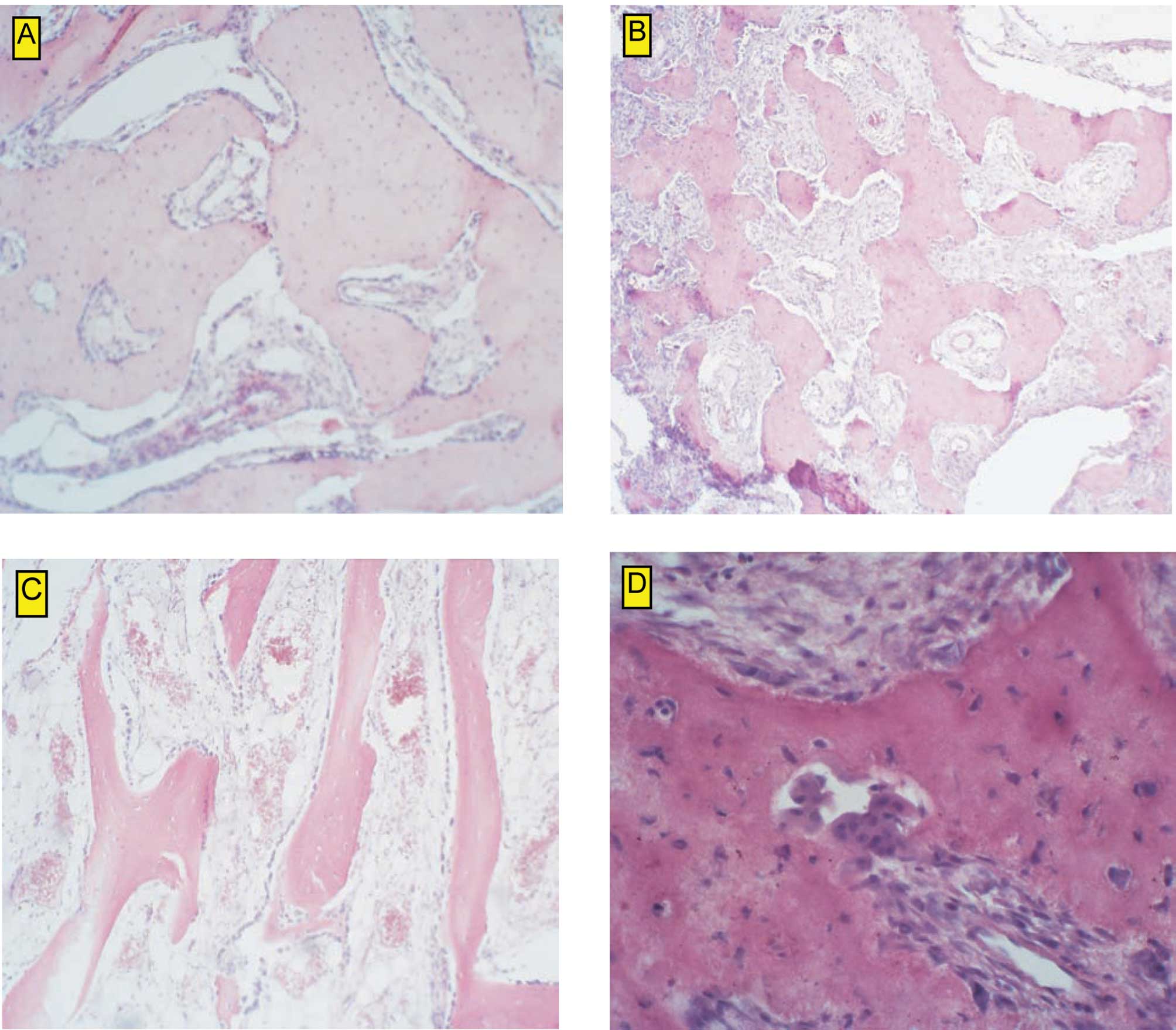
2003. Upon histological examination, large cell heterogeneity was
observed. Among osseus trabeculae with well-mineralized osteoid and
osteocytes in the osseous lacunae, very poorly mineralized osteoids
were noted. The tumor was described as a heterogeneous mass with a
variable content of multinuclear cells. No myelogenic tissue was
observed among the mature and immature trabeculae. On the basis of
the histological examination, the tumor was classified as
aggressive osteoblastoma (Fig.
2B–D).
The above-mentioned patient was treated surgically.
Prior to surgery, an angiography was performed, as osteoblastoma
may be a well-vascularized tumor (3–5).
Additionally, the tumor location itself indicated the possibility
of its connection to large vessels, which had to be excluded
preoperatively. In the angiography, very poor vascularization of
the tumor was observed. The tumor vessels originated from the right
facial artery with a net of tiny vessels. Additionally, the tumor
received a blood supply from the left internal carotid artery
(Fig. 2A).
The patient underwent surgery on December 1, 2003.
Bilateral fronto-orbital craniotomy was performed. The procedure
revealed that the tumor had not infiltrated the brain, but expanded
from the sphenoid bone to the ethmoids, entered the medial walls of
the two orbits and reached the clivus. It was firmly attached to
the bony structures and dura. The tumor was removed together with
the dura mater close to the cribriform plate. A patch of fascia
lata was used to close the defect in the dura mater. The nasal
cavity was separated with an additional piece of fascia lata and
with the pedicle flap of the periostium. Fatty tissue was placed in
the space of the frontal sinus, and the frontal bone was
restored.
Lumbar CSF drainage was applied for 8 days
postoperatively. No CSF leak (rhinorrhoea) was observed. The wound
healed as expected. The ophthalmological examination revealed no
vision impairment. The patient was discharged from the hospital on
December 11, 2003.
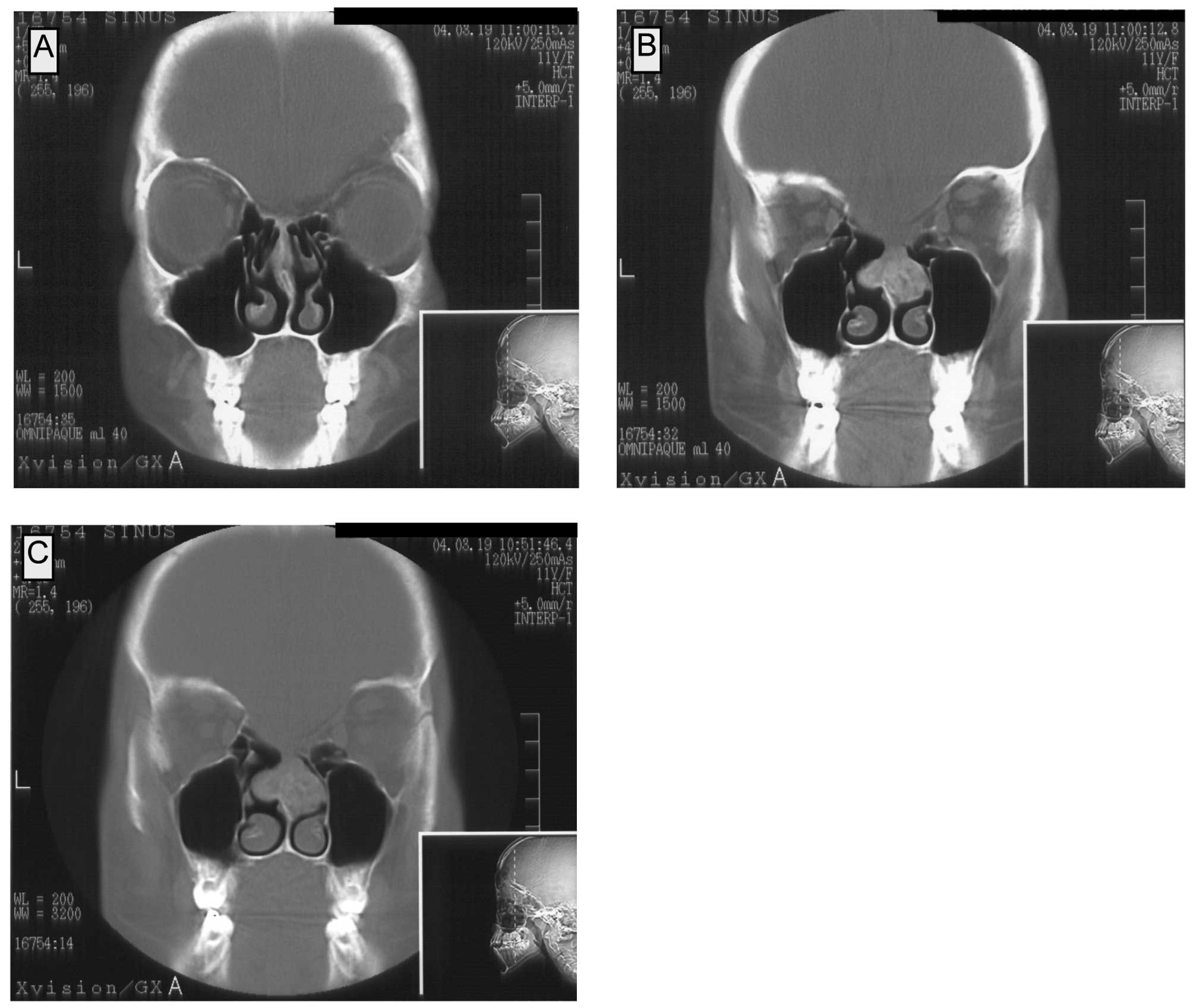
The follow-up CT examination on March 23, 2004
showed a pathological mass in the area of the nasal septum which
was connected to both the median nasal turbinates. The patient had
no symptoms of nasal congestion. In the CT, the lesion showed
enhancement after contrast administration. Numerous calcifications
were observed, at a mean density of 50–200 and 120–230 H before and
after the contrast administration, respectively. In the coronal
projections, the maximal dimensions of the lesion were 25×22 mm
(Fig. 3A–C).
As a result, the patient underwent surgery on June
2, 2004. Intranasal partial resection of the septum was performed.
The histopathological examination showed the complete resection of
the tumor. The tumor was classified as osteoblastoma.
The patient had subsequent follow-up naso-pharyngeal
endoscopy in November 2004, as she complained of impaired nasal
breathing for approximately one month. During endoscopic
examination, hypertrophy of soft tissue was observed bilaterally in
the nasopharynx. The tissue was covered with macroscopically normal
epithelium. Tissue samples were taken for histopathological
examination, and no elements of osteoblastoma tissue were found.
Since this lesion was atypical for the recurrence of osteoblastoma,
an MDP scintigraphy was also performed. The scintigraphy showed a
symmetrical flow of the contrast to the cranial base bony elements
in the vascular phase. A typical, slightly asymetrical perfusion of
the tissues in the operated area in the static phase was observed.
No high intake of the contrast, which is typical for recurrent
osteoblastoma, was found. Surgical revision of the nasal cavity and
nasopharynx was performed. No tissue typical for osteoblastoma was
observed by the surgeon. Bilaterally, in the nasopharynx, the
massive hypertrophy of a tissue resembling an adenoid was found and
resected. The histological examination confirmed that the
hypertrophied tissue was an adenoid. The patient was discharged
from the hospital on December 1, 2004 and still remains under
observation.
The latest MRI examination was performed on February
9, 2009 (Fig. 4A) and, thus far, no
recurrence has been observed. Subsequently, on April 28, 2009, an
MDP scintigraphy was performed and confirmed the disease-free state
of the patient (Fig. 4B). The
disease-free survival is presently 61 months.
Discussion
Osteoblastoma is a benign tumor of the bone.
Nevertheless, it may cause local bone destruction. Its speed of
growth and symptomatology, as a consequence, can be diverse
(6–8). Clinically and morphologically,
osteoblastoma is set between benign osteoid osteoma and malignant
osteosarcoma. Based on its histologic similarity to osteoid
osteoma, in 1954, Dallin and Johnson (9) proposed that this tumor be called
‘giant osteoid osteoma’.
This is because osteoblastoma, unlike osteoid
osteoma, can exceed certain established dimensions. According to
different investigators, the borderline diameter of the lesion is 1
(10,11), 1.5 (12) or 2 cm (6). The name osteoblastoma was introduced
by Lichtenstein (13) and Jaffe
(14) in 1956.
Despite the fact that osteoblastomas are benign
tumors, they have been divided into three groups according to their
various clinical progress and diverse symptomatology as well as
different histopathological findings. In 1986, Enneking (8) established the benign bone lesion
classification for latent, active and aggressive tumors.
Latent tumors are usually asymptomatic and their
diagnosis is usually incidental. In most cases, a latent tumor is
discovered during the diagnostic process for a different
disease.
Active tumors gradually exceed their dimensions and
can be mildly symptomatic. They are usually discovered in the event
of a pathological fracture, patient complaint of chronic pain or
mechanical impairments. Aggressive osteoblastomas, although benign,
are characterized by rapid growth. Therefore, they become
symptomatic readily, and the signs of the disease are similar to
the ones mentioned above.
Different modes of management involving osteoid
osteoma and osteoblastoma have been assessed. They include
radiotherapy, chemotherapy (15,16)
and percutaneous radio-frequency ablation (3). The methods mentioned above may be
useful particularly in patients with recurrent aggressive tumors or
in patients with surgically unresectable tumors. Nevertheless,
surgery remains the optimal mode of treatment of these lesions.
With each surgical procedure, the tumor must be removed with a
clean resection margin. The width of this margin varies depending
on the histological finding in the biopsy specimen (broader margin
in the case of aggressive osteoblastoma).
Osteoblastoma is a very rare tumor. It is
approximately 20 times less common than osteosarcoma (6) and accounts for approximately 1% of all
primary bone tumors (6,17). In the largest series studied, the
mean age at presentation was 20.4 years, the male to female ratio
was 2:1 and the size of the tumors ranged from 1 to 11 cm (17).
Osteoblastoma may affect any bone. Nevertheless,
most frequently it presents in the vertebral column and long
tubular bones. In the spine, the posterior elements are affected
more frequently. In the long bones, osteoblastoma affects mainly
the diaphysis and metaphysis, but rarely the epiphysis. Other
locations include the clavicles, scapulas, ribs, small bones of the
hand and feet, and the skull. The skull itself is a very uncommon
site for osteoblastoma. However, when found in the skull, it may
affect both the calvarial and facial bones. There have been only a
few reports on osteoblastoma affecting the sphenoid or temporal
bone (1,4,5,11,18).
In conclusion, surgical excision remains the
treatment of choice for benign bone tumors of the skull base
region, particularly in children.
Acknowledgements
A.M.C. was supported by a Fulbright Junior Research
Grant, The Kosciuszko Foundation Scholarship and the Ministry of
Science and Higher Education of The Republic of Poland Grant (no. N
N401 2327 33). Author contributions: W.K. made substantial
contributions to the conception, design and acquisition of data,
and was involved in drafting the manuscript or revising it
critically for important intellectual content. A.O. was responsible
for surgical treatment and neurosurgical follow-up. A.S. made
substantial contributions to the conception, design and acquisition
of data, or analysis and interpretation of data. A.M.C. made
substantial contributions to the conception and design of the
manuscript and was involved in drafting the manuscript or revising
it critically for important intellectual content. K.W. was
responsible for histopathological examination and evaluation. A.K.
gave final approval of the version to be published.
References
|
1
|
Ciappetta P, Salvati M, Raco A, Artico M
and Gagliardi FM: Benign osteoblastoma of the sphenoid bone.
Neurochirurgia. 34:97–100. 1991.
|
|
2
|
Berry M, Mankin H, Gebhardt M, Rosenberg A
and Hornicek F: Osteoblastoma: a 30-year study of 99 cases. J Surg
Oncol. 98:179–183. 2008.PubMed/NCBI
|
|
3
|
Rosenthal DI, Hornicek FJ, Torriani M,
Gebhardt MC and Mankin HJ: Osteoid osteoma: percutaneous treatment
with radiofrequency energy. Radiology. 229:171–175. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ohkawa M, Fujiwara N, Tanabe M, et al:
Benign osteoblastoma of the temporal bone. AJNR Am J Neuroradiol.
18:324–326. 1997.
|
|
5
|
Potter C, Conner GH and Sharkey FE: Benign
osteoblastoma of the temporal bone. Am J Otol. 4:318–322.
1983.PubMed/NCBI
|
|
6
|
Ortman F: Osteoblastoma. eMedicine.
2003.
|
|
7
|
Dorfman H and Czerniak B: Benign
Osteoblastic tumors. Bone Tumors. Mosby; St. Louis: pp. 85–127.
1998
|
|
8
|
Enneking WF: A system of staging
musculoskeletal neoplasms. Clin Orthop Relat Res.
9:241986.PubMed/NCBI
|
|
9
|
Dahlin DC and Johnson EW Jr: Giant osteoid
osteoma. J Bone Joint Surg Am. 36A:559–572. 1954.PubMed/NCBI
|
|
10
|
Krus S and Skrzypek-Fakhoury E:
Patomorfologia Kliniczna. PZWL; Warszawa: 1996
|
|
11
|
Doshi SV, Frantz TD and Korol HW: Benign
osteoblastoma of the temporal bone: case report and literature
review. Am J Otolaryngol. 22:211–214. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kransdorf MJ, Stull MA, Gilkey FW and
Moser RP Jr: Osteoid osteoma. Radiographics. 11:671–696. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lichtenstein L: Benign osteoblastoma; a
category of osteoid- and bone-forming tumors other than classical
osteoid osteoma, which may be mistaken for giant-cell tumor or
osteogenic sarcoma. Cancer. 9:1044–1052. 1956. View Article : Google Scholar
|
|
14
|
Jaffe HL: Benign osteoblastoma. Bull Hosp
Joint Dis. 17:141–151. 1956.
|
|
15
|
Berberoglu S, Oguz A, Aribal E and Ataoglu
O: Osteoblastoma response to radiotherapy and chemotherapy. Med
Pediatr Oncol. 28:305–309. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singer JM and Deutsch GP: The successful
use of radiotherapy for osteoblastoma. Clin Oncol (R Coll Radiol).
5:124–125. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lucas DR, Unni KK, McLeod RA, O’Connor MI
and Sim FH: Osteoblastoma: clinicopathologic study of 306 cases.
Hum Pathol. 25:117–134. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Williams RN and Boop WC Jr: Benign
osteoblastoma of the skull. Case report. J Neurosurg. 41:769–772.
1974. View Article : Google Scholar : PubMed/NCBI
|