Introduction
Hypophysitis is an uncommon sellar condition that
presents as inflammatory lesions on structures of the hypophysis,
including the pituitary gland and stalk (1–3).
Histologically, hypophysitis may be classified into four distinct
types: Lymphocytic (LYH), granulomatous (GRH), xanthomatous and
necrotizing (1). LYH is typically
secondary to pregnancy and autoimmune diseases, while GRH may be
associated with granulomatous processes (4). A number of recent studies have reported
IgG4-related hypophysitis and additional mixed subtypes, including
lymphogranulomatous and xanthogranulomatous hypophysitis, may also,
more rarely, occur (5–7).
Clinically, the typical manifestation of
hypophysitis includes headache, hypopituitarism, nausea, vomiting,
diabetes insipidus and potentially visual damage (8). The diagnosis of hypophysitis may be
challenging due to its various forms and the influence of
concomitant diseases (8). The
characteristic features of hypophysitis, which may be observed by
magnetic resonance imaging (MRI), are a thickened pituitary stalk
and an enlarged pyramidal or round-shaped gland (9). Other potential pathological conditions
in the sellar region, including tuberculosis, histiocytosis, fungal
infections and germinoma infiltrative neoplasms, must be taken into
account during differential diagnosis (10).
The optimal therapeutic course for the treatment of
hypophysitis is currently disputed (4). Surgery is an effective method for the
removal of masses and to obtain an accurate pathological diagnosis
of hypophysitis, while high-dose methylprednisolone therapy is an
alternative treatment option that provides hormonal replacement
(7).
In the present report, seven cases of hypophysitis
are described based on biopsies and imaging results, and the
limitations underlying the diagnosis and treatment strategies for
hypophysitis are discussed. Written informed consent was obtained
for all patients.
Case report
Patients and diagnoses
Seven cases of hypophysitis, including two male and
five female patients, were reviewed at the Department of
Neurosurgery, Second Affiliated Hospital (Hangzhou, China) between
January 2009 and December 2011. The mean age (± standard deviation)
of the patients was 45.71±22.16 years. The patients presented with
a range of symptoms, including headache, fever, gradual decrease of
visual acuity, nausea and vomiting. Endocrinological examinations
of the patients revealed varying levels of hormone indices,
including human thyroid-stimulating hormone (h-TSH) and prolactin
(PRL) (Table I).
 | Table I.Clinical and endocrinological summary
of 7 hypophysitis patients. |
Table I.
Clinical and endocrinological summary
of 7 hypophysitis patients.
|
|
|
|
| Hormone level |
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Case | Age, years | Gender | Chief complaint | Increased | Decreased | Etiology | Treatment | Pathological
result |
|---|
| 1 | 66 | F | Headache, visual
damage (1 month) | PRL | h-TSH | Idiopathic | Transsphenoidal
surgery; postoperative regular-dose methylprednisolone therapy (160
mg, daily) | Granulomatous
hypophysitis |
| 2 | 72 | M | Headache, recurrent
fever (2 months) | FSH | PRL | Idiopathic | High-dose
methylprednisolone therapy (500 mg, daily) | NA |
| 3 | 46 | M | Headache, chill,
nausea, vomiting (3 days) |
| h-TSH, PRL, LH, PGN,
TES, COR | Idiopathic | High-dose
methylprednisolone therapy (800, 600 or 400 mg, daily) | NA |
| 4 | 29 | F | Headache (4
months) |
| h-TSH, PRL, TT4, FT4,
COR (8 am), ACTH (8 am) | Pregnancy | Transsphenoidal
pituitary biopsy; methylprednisolone therapy (5 mg, three times a
day) | Lymphocytic
hypophysitis |
| 5 | 42 | F | Visual disorder,
nausea, vomiting (6 months) |
| h-TSH, FT3, TT4, FT4,
COR (8 am), ACTH (8 am) | Idiopathic | Transsphenoidal
surgery | Lymphocytic
hypophysitis |
| 6 | 8 | F | Obesity (2
years) | h-TSH |
| Idiopathic | Levothyroxine sodium
therapy | NA |
| 7 | 57 | F | Headache, progressive
visual damage (4 months) | PRL | h-TSH | Idiopathic | Transsphenoidal
surgery; postoperative regular-dose methylprednisolone therapy (160
mg, daily) | Granulomatous
hypophysitis |
Sagittal and coronal enhanced MRI scans revealed an
enlarged pituitary gland with a pyramidal or round shape (a sellar
lesion with iso- or hypodense signal on a T1-weighted image) and a
thickened pituitary stalk. Following surgical or hormonal therapy,
all cases demonstrated improvements to a varying extent (Fig. 1).
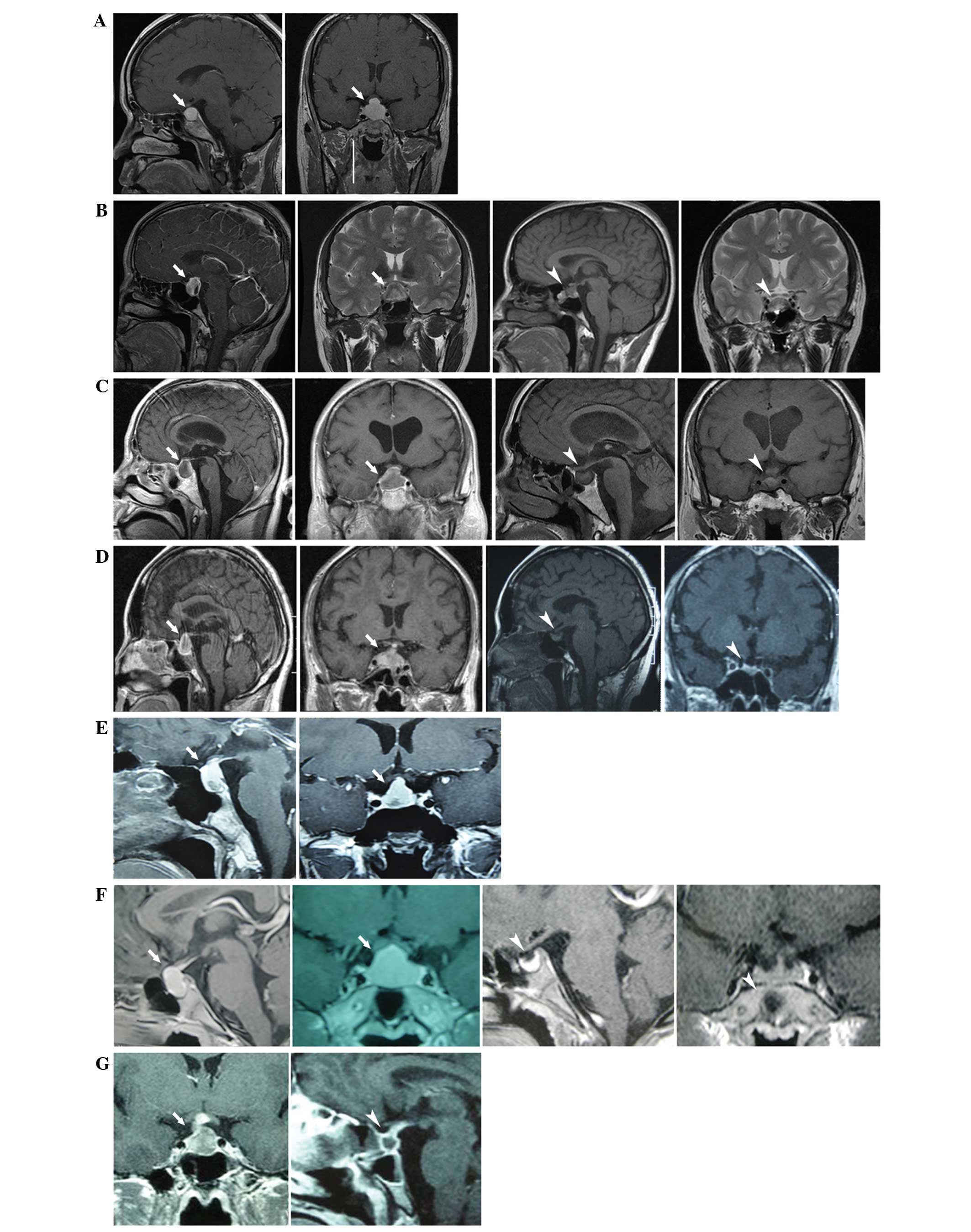 | Figure 1.MRI revealed a thickened stalk, and
diffuse enlargement and enhancement of the pituitary gland. White
tailed arrows indicate the position of the mass, whereas white
arrowheads indicate the post-therapeutic condition. (A) Case 1,
(left) pre-operative sagittal and (right) coronal MRI images. (B)
Case 2, (left) pre-operative and (right) postoperative images
showing pituitary gland recovery following high-dose glucocorticoid
therapy. (C) Case 3, (left) pre-operative images and (right)
postoperative images, showing a reduction in mass size. (D) Case 4,
(left) pre-biopsy images and (right) postoperative images following
hormone treatment, showing a reduction in mass size. (E) Case 5,
pre-operative sagittal and coronal MRI images. (F) Case 6 (left)
pre-theraputic images and (right) following hormone treatment,
showing a reduction in pituitary gland size. (G) Case 7, (left)
preoperative image and (right) image showing a contractible mass
following therapy. MRI, magnetic resonance imaging. |
Of the 4 patients that underwent transsphenoidal
surgery or biopsy, 3 were diagnosed with lymphocytic hypophysitis,
and 1 female patient was histopathologically diagnosed with
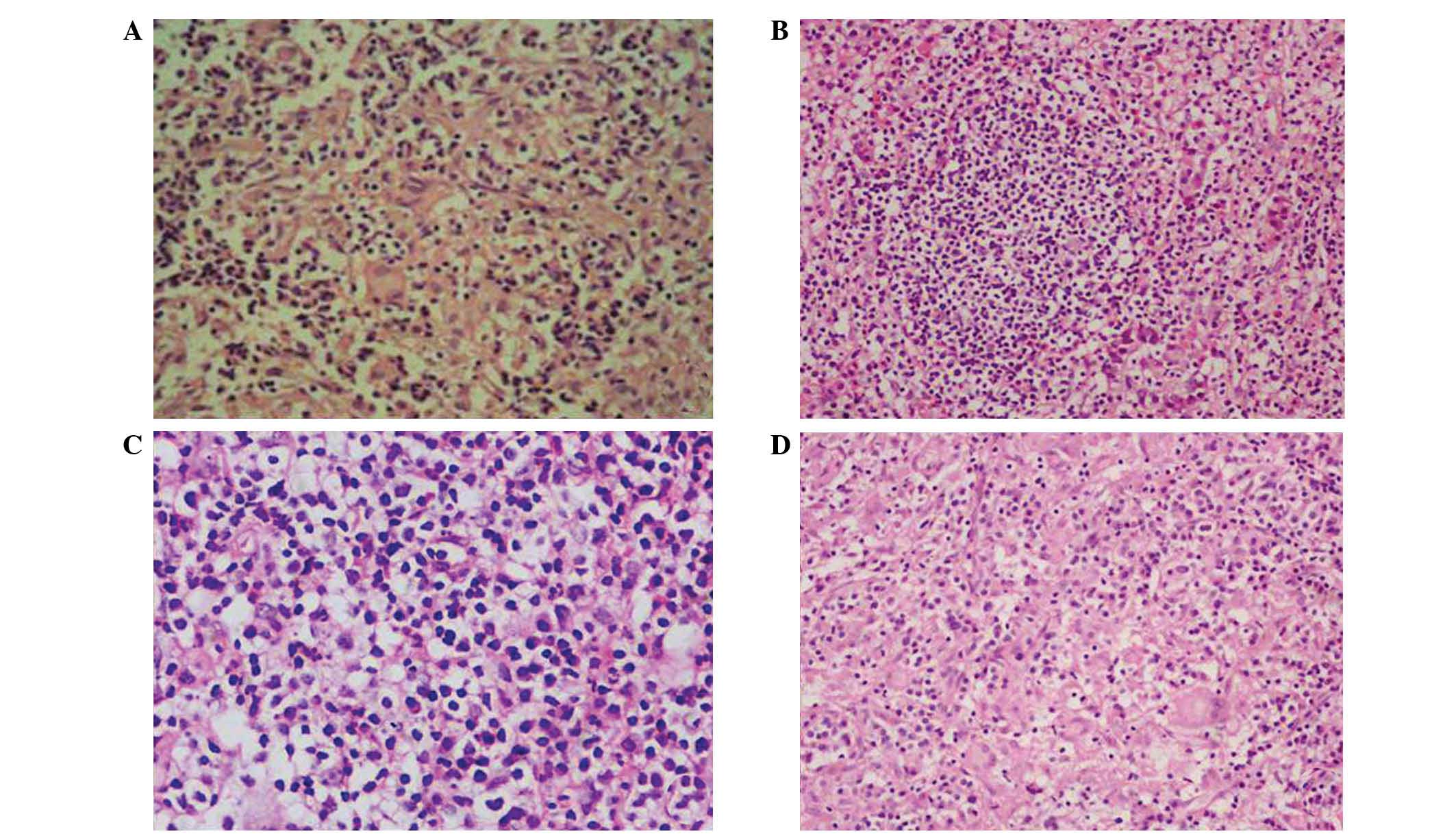
granulomatous hypophysitis (Fig. 2).
Additionally, the morphological characteristics of hypophysitis
were markedly different from those of pituitary adenomas or other
sellar masses. Typical features of hypophysitis include a thickened
pituitary stalk, and diffuse enlargement and enhancement of the
pituitary gland on MRI, while other masses exhibit asymmetric
enlargement or invasion of the surrounding tissue. When viewed
under a microscope, the hypophysitis tissue exhibited a more
tensile structure compared with adenomas, which meant that
resection was more difficult (Fig.
3).
Outcome and follow-up
Of the 3 patients that underwent transsphenoidal
surgery for the removal of sellar masses, a biopsy to identify
hypophysitis with fast-frozen pathology was performed in only 1
case, as the patient's tissues did not demonstrate characteristics
of a pituitary adenoma. The remaining patients were administered
glucocorticoid therapy and experienced positive outcomes. All
patients were followed up for a two year period; pituitary function
recovered well in all cases and no recurrences were reported.
Discussion
Diagnosis
Hypopituitarism occurs in the majority of patients
with hypophysitis; therefore, an analysis of levels of pituitary
hormones in such patients may be useful for evaluation of the
characteristics of hypophysitis, even if there are no obvious
changes in pituitary hormone levels in certain hypophysitis cases
(11). In contrast to the majority of
pituitary adenoma patients, hypophysitis patients typically
experience a series of marginal changes in the levels of pituitary
hormones. It is notable that the majority of the 7 cases
investigated in the present study exhibited variations in PRL and
h-TSH levels, but no measurable changes in the levels of any other
pituitary hormones. Therefore, PRL and h-TSH may be potential
indices for presurgical diagnosis. In addition, a number of
researchers have identified that appropriate measurements of
electrolyte levels and osmolality in the urine and serum are able
to assist in identifying the occurrence of diabetes insipidus, a
secondary effect of hypophysitis (12,13). The
present cases did not demonstrate disequilibrium of electrolytes or
osmolality in the urine or serum. However, it is possible that the
measurement methods utilized were not sensitive enough to diagnose
diabetes insipidus. Considering that infectious diseases are the
primary cause of hypophysitis, laboratory tests and cultures
associated with infectious diseases, particularly tuberculosis and
fungal infections, are necessary (14). Although autoimmunity has been proposed
as a field that may aid in the diagnosis of hypophysitis, previous
studies have identified that anti-pituitary antibodies possess
insufficient specificity and sensitivity, negating the potential
for measuring autoimmune responses as a laboratory standard in the
diagnosis of hypophysitis (15–18).
Pituitary imaging data is able to reveal the
characteristic features of hypophysitis. For example, an MRI scan
is able to reveal enlargement of the pituitary gland with a
pyramidal or round shape, and a thickened pituitary stalk that
extends toward the hypothalamus in the suprasellar area (19). An extension of the pituitary gland
into the cavernous sinus is observed by imaging in cases of
granulomatous hypophysitis (20).
Mucosal thickening in the bilateral sphenoid sinus has additionally
been reported in certain granulomatous hypophysitis cases, while
markedly homogeneous contrast enhancement of the pituitary gland is
considered to be a characteristic of lymphocytic hypophysitis
(21). Experimental corticosteroid
therapy has been discussed as a potential diagnostic tool (2).
Transsphenoidal biopsy has provided the most precise
classification of hypophysitis types and allowed for conclusive
histopathological diagnoses; however, the biopsy procedure may be
traumatic for patients (7,22).
Treatment
In addition to hypopituitarism, diabetes insipidus
and other medical issues, a large number of hypophysitis patients
present with pituitary masses, visual disorders and severe
headaches (14). The cases reviewed
in the present study demonstrated these additional characteristic
symptoms. Certain previous studies have suggested that resecting
the sellar mass as much as possible is a suitable treatment
(7,23,24).
Furthermore, a fast-frozen pathological diagnosis performed during
surgery, and a postoperative histopathological diagnosis, can be
undertaken using surgical samples (22,23).
Additionally, patients who do not exhibit a strong response to
corticosteroid therapy or experience a mass recurrence require
standard surgical treatment (22).
However, even if surgery is able to sufficiently remove the sellar
mass, it is necessary to evaluate potential complications that may
arise due to surgery (10). Low doses
of radiotherapy have been used on a number of patients exhibiting
resistant hypophysitis (25); two
patients exhibiting recurrent disease following standard
transsphenoidal surgery were treated with low-dose stereotactic
radiotherapy. No adverse effects were reported following radiation
treatment and mass size was reduced in both cases, while the
symptoms were resolved and pituitary function was recovered. A
gamma knife has additionally been utilized in certain cases
(26). In a patient with
histopathologically diagnosed lymphocytic hypophysitis, who
underwent transsphenoidal surgery, the disease had proven difficult
to resect and following the withdrawal of corticosteroid therapy
symptoms returned and mass enlargement was observed. Subsequently,
the patient underwent gamma knife treatment, corticoid therapy was
stopped and symptoms did not recur (26).
In patients that are unsuitable for surgery due to
pregnancy or autoimmune diseases, a high dose of corticosteroid
medication may be used to treat hypophysitis (4). Prediagnosis should be based on clinical
symptoms, laboratory testing and imaging data to determine the most
appropriate medical management for the patient (2). The 3 cases in the present study that
were administered high-dose methylprednisolone or levothyroxine
sodium therapy demonstrated a positive prognosis. Additionally, the
patients that underwent transsphenoidal surgery demonstrated
positive outcomes with postoperative regular-dose
methylprednisolone therapy. Thus, corticosteroid therapy may be
regarded as an effective treatment method for hypophysitis caused
by an autoimmune disorder. In addition, pituitary hormone
replacement may be used to treat hypophysitis (10). Diabetes insipidus is typically treated
effectively with desmopressin (27).
Due to the possibility of secondary side effects associated with
high-dose glucocorticoid therapy, alternative methods of immune
modulation, including azathioprine treatment, have been considered
(28).
In conclusion, the most appropriate therapy course
for the treatment of hypophysitis remains controversial. Surgical
treatment is beneficial for resolving the effects of an enlarged
pituitary gland, which may press on the optical or oculomotor
nerve, to prevent high intracranial pressure and visual damage
(14). However, for patients who are
unsuitable for surgery, including those that are pregnant or in
poor health, glucocorticoid therapy is the first choice, as total
removal of the pituitary masses may functionally disable the
pituitary gland (8). In addition,
immune modulatory agents may be considered as an alternative
therapy to glucocorticoids for recurrent or resistant cases
(2). Furthermore, high-dose
glucocorticoid therapy may be an effective primary treatment for
hypophysitis due to its sensitivity and low rate of traumatic side
effects (10,29).
References
|
1
|
Caturegli P, Newschaffer C, Olivi A, et
al: Autoimmune hypophysitis. Endocr Rev. 26:599–614. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carmichael JD: Update on the diagnosis and
management of hypophysitis. Curr Opin Endocrinol Diabetes Obes.
19:314–321. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gutenberg A, Buslei R, Fahlbusch R,
Buchfelder M and Bruck W: Immunopathology of primary hypophysitis,
Implications for pathogenesis. Am J Surg Pathol. 29:329–338. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheung CC, Ezzat S, Smyth HS and Asa SL:
The spectrum and significance of primary hypophysitis. J Clin
Endocrinol Metab. 86:1048–1053. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Landek-Salgado MA, Leporati P, Lupi I,
Geis A and Caturegli P: Growth hormone and proopiomelanocortin are
targeted by autoantibodies in a patient with biopsyproven
IgG4-related hypophysitis. Pituitary. 15:412–419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bando H, Iguchi G, Fukuoka H, et al: A
diagnostic pitfall in IgG4-related hypophysitis: Infiltration of
IgG4-positive cells in the pituitary of granulomatosis with
polyangiitis. Pituitary. 18:722–730. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Imber BS, Lee HS, Kunwar S, Blevins LS and
Aghi MK: Hypophysitis A single-center case series. Pituitary.
18:630–641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fukuoka H: Hypophysitis. Endocrinol Metab
Clin North Am. 44:143–149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Unlu E, Puyan FO, Bilgi S and Kemal
Hamamcioglu M: Granulomatous, hypophysitis. Presentation and MRI
appearance. J Clin Neurosci. 13:1062–1066. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Laws ER, Vance ML and Jane JA Jr:
Hypophysitis. Pituitary. 9:331–333. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gazioğlu N: Lymphocytic and granulomatous
hypophysitis: E xperience with nine cases. Neurosurgery.
46:12682000. View Article : Google Scholar
|
|
12
|
Huang CH, Chou KJ, Lee PT, Chen CL, Chung
HM and Fang HC: A case of lymphocytic hypophysitis with masked
diabetes insipidus unveiled by glucocorticoid replacement. Am J
Kidney Dis. 45:197–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lupi I, Manetti L, Raffaelli V, Lombardi
M, Cosottini M, Iannelli A, Basolo F, Proietti A, Bogazzi F,
Caturegli P and Martino E: Diagnosis and treatment of autoimmune
hypophysitis, A short review. J Endocrinol Invest. 34:e245–252.
2011.
|
|
14
|
Leung GK, Lopes MB, Thorner MO, Vance ML
and Laws ER Jr: Primary hypophysitis. A single-center experience in
16 cases. J Neurosurg. 101:262–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
De Bellis A, Pane E, Bellastella G, Sinisi
AA, Colella C, Giordano R, Giavoli C, Lania A, Ambrosio MR, Somma
C, et al: Italian Autoimmune Hypophysitis Network Study: Detection
of antipituitary and antihypothalamus antibodies to investigate the
role of pituitary or hypothalamic autoimmunity in patients with
selective idiopathic hypopituitarism. Clin Endocrinol (Oxf).
75:361–366. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lupi I, Manetti L, Raffaelli V, Grasso L,
Sardella C, Cosottini M, Iannelli A, Gasperi M, Bogazzi F,
Caturegli P and Martino E: Pituitary autoimmunity is associated
with hypopituitarism in patients with primary empty sella. J
Endocrinol Invest. 34:e240–e244. 2011.PubMed/NCBI
|
|
17
|
Smith CJ, Bensing S, Burns C, Robinson PJ,
Kasperlik-Zaluska AA, Scott RJ, Kämpe O and Crock PA:
Identification of TPIT and other novel autoantigens in lymphocytic
hypophysitis, Immunoscreening of a pituitary cDNA library and
development of immunoprecipitation assays. Eur J Endocrinol.
166:391–398. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leporati P, Landek-Salgado MA, Lupi I,
Chiovato L and Caturegli P: IgG4-related hypophysitis, A new
addition to the hypophysitis spectrum. J Clin Endocrinol Metab.
96:1971–1980. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Levine SN, Benzel EC, Fowler MR, Shroyer
JV 3rd and Mirfakhraee M: Lymphocytic adenohypophysitis: Clinical,
radiological, and magnetic resonance imaging characterization.
Neurosurgery. 22:937–941. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vasile M, Marsot-Dupuch K, Kujas M,
Brunereau L, Bouchard P, Comoy J and Tubiana JM: Idiopathic
granulomatous hypophysitis, Clinical and imaging features.
Neuroradiology. 39:7–11. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goyal M, Kucharczyk W and Keystone E:
Granulomatous hypophysitis due to Wegener's granulomatosis. AJNR Am
J Neuroradiol. 21:1466–1469. 2000.PubMed/NCBI
|
|
22
|
Shi J, Zhang JM, Wu Q, Chen G, Zhang H and
Bo WL: Granulomatous hypophysitis, Two case reports and literature
review. J Zhejiang Univ Sci B. 10:552–558. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Honegger J, Fahlbusch R, Bornemann A,
Hensen J, Buchfelder M, Müller M and Nomikos P: Lymphocytic and
granulomatous hypophysitis, Experience with nine cases.
Neurosurgery. 40:713–722; discussion. 722–723. 1997. View Article : Google Scholar
|
|
24
|
Buxton N and Robertson I: Lymphocytic and
granulocytic hypophysitis: A single centre experience. Br J
Neurosurg. 15:242–245; discussion 245–246. 2001. View Article : Google Scholar
|
|
25
|
Selch MT, DeSalles AA, Kelly DF, Frighetto
L, Vinters HV, Cabatan-Awang C, Wallace RE and Solberg TD:
Stereotactic radiotherapy for the treatment of lymphocytic
hypophysitis. Histopathology. J Neurosurg. 99:591–596. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ray DK, Yen CP, Vance ML, Laws ER, Lopes B
and Sheehan JP: Gamma knife surgery for lymphocytic hypophysitis. J
Neurosurg. 112:118–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leroy C, Karrouz W, Douillard C, Do Cao C,
Cortet C, Wémeau JL and Vantyghem MC: Diabetes insipidus. Ann
Endocrinol (Paris). 74:496–507. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lecube A, Francisco G, Rodríguez D, Ortega
A, Codina A, Hernández C and Simó R: Lymphocytic hypophysitis
successfully treated with azathioprine. First case report. J Neurol
Neurosurg Psychiatry. 74:1581–1583. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Khare S, Jagtap VS, Budyal SR, Kasaliwal
R, Kakade HR, Bukan A, Sankhe S, Lila AR, Bandgar T, Menon PS and
Shah NS: Primary (autoimmune) hypophysitis, A single centre
experience. Pituitary. 18:16–22. 2015. View Article : Google Scholar : PubMed/NCBI
|