Introduction
Leiomyosarcomas are tumors of the smooth muscle
cells that may originate in any location, but most often arise in
the uterus, gastrointestinal tract and soft tissue. Primary
pulmonary leiomyosarcoma (PPL) is an extremely rare malignant
mesenchymal tumor that appears to originate from the smooth muscle
cells of the bronchial and blood vessel wall. PPL accounts for
<0.5% of all malignant pulmonary tumors (1). A previous study reported a 5-year
survival rate of 60% for PPL (2). The
tumors may be treated using surgical resection, which is the
primary and definitive mode of treatment. Surgical strategies
consist of lobectomy, pneumonectomy and bronchial sleeve resection.
The role of other treatment methods has yet to be defined; however,
radiochemotherapy is recommended in cases of incomplete resection
and malignancy (1). A definitive
diagnosis of PPL is provided following pathological examination of
a tumor sample. Early detection and complete surgical resection of
PPL have been demonstrated to significantly contribute to an
increased survival time of patients with the disease (3). Written informed consent was obtained
from the patient.
Case report
A 48-year-old man was admitted to the First
Affiliated Hospital of Wenzhou Medical University (Wenzhou,
Zhejiang, China) in April 2014 with a cough and the expectoration
of white and yellow sputum, which had been ongoing for 2 months.
The patient had been a smoker for >20 years, but had no family
history of lung cancer. The patient also felt short of breath when
ascending stairs. The patient did not present with a fever,
anorexia, hemoptysis, chest pain or weight loss. The vital signs of
the patient were stable. Lung auscultation revealed decreased
breath sounds on the right lung and fine crackling was heard at the
base of the right lung. Other physical examinations were
unremarkable. Blood tests revealed that the patient's white blood
count was 9.9×109 cells/l (normal range,
4.0–10.0×109 cells/l), the C-reactive protein level was
48.8 mg/l (normal range, 0–10 mg/l) and the erythrocyte
sedimentation rate was 45 mm/h (normal range in men, 0–15 mm/h).
Other laboratory examinations were all negative, including those
for blood chemistry, blood tumor markers, and liver and renal
function. Sputum from the patient was negative for acid-fast
bacilli and sputum culture.
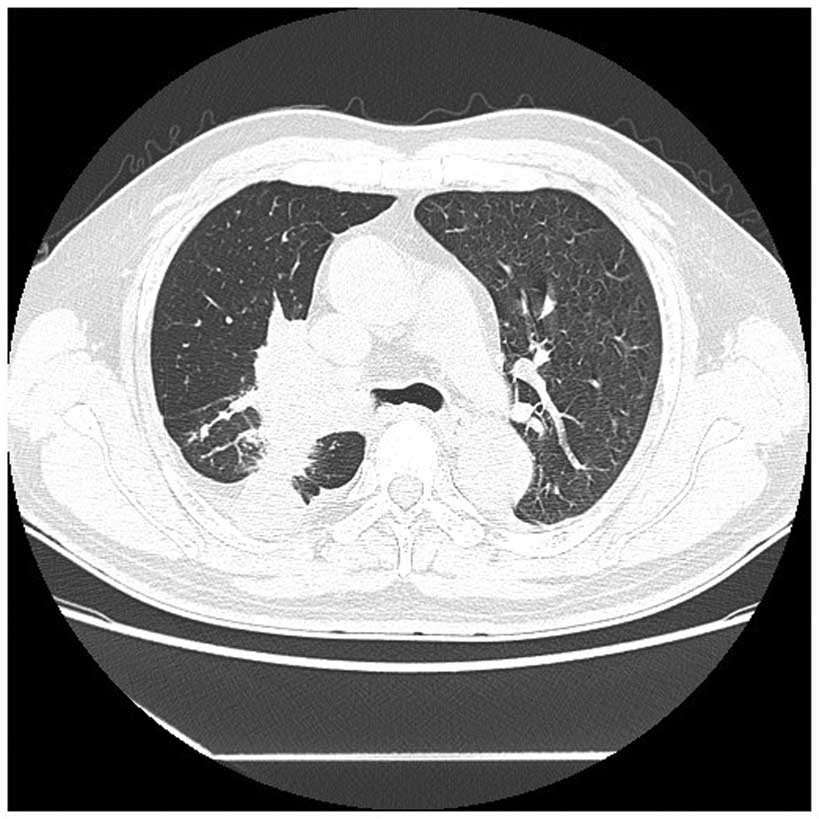
Enhanced computed tomography (CT) scans of the
patient's chest revealed the presence of a 6.6×5.2-cm irregular
heterogeneous mass in the hilar region of the right lung, with
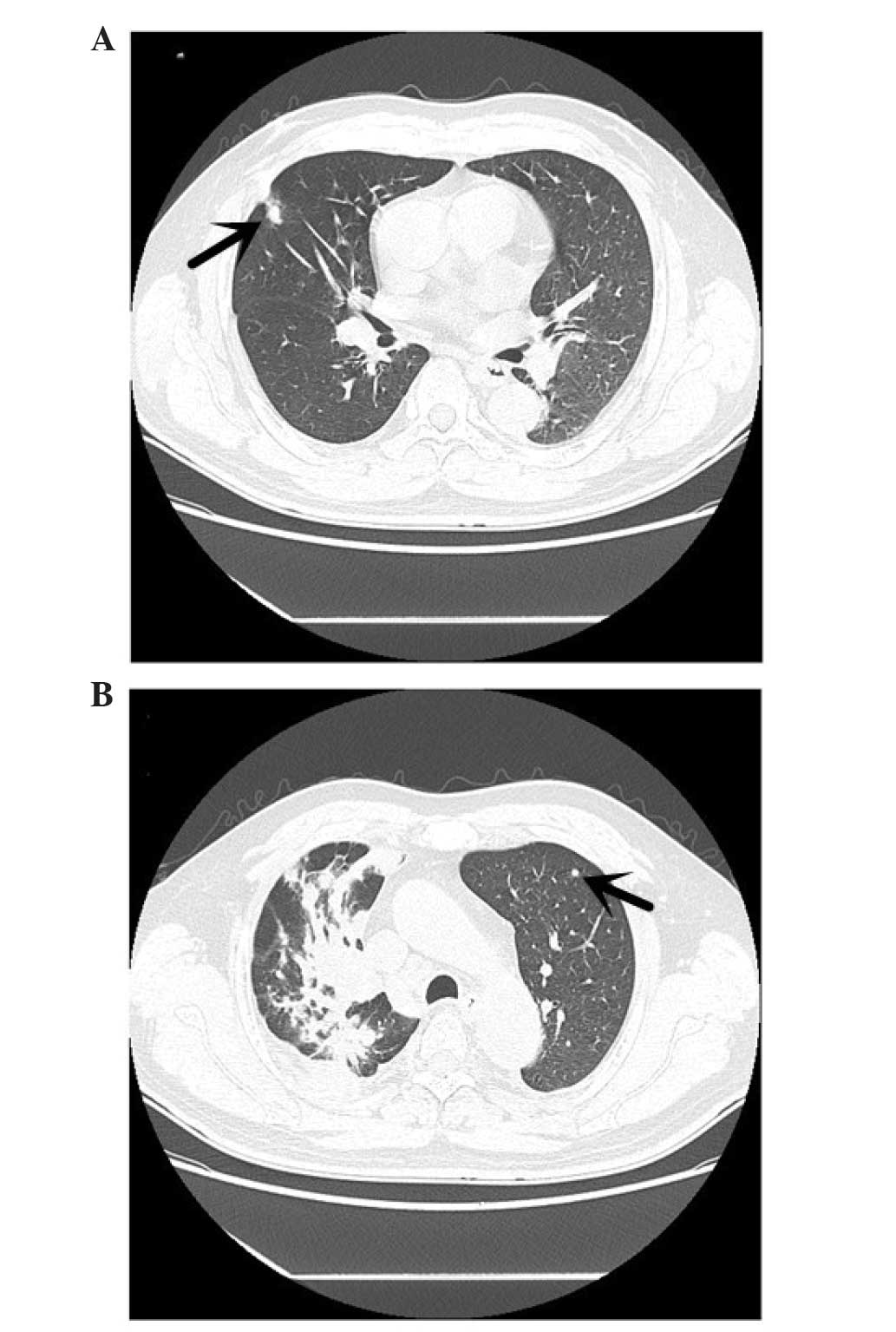
mediastinal lymphadenopathy and intrapulmonary metastasis (Fig. 1). The CT scan demonstrated that there
was a high-density shadow, 8 mm in diameter (Fig. 2A), on the upper lobe of the right lung
and a high-density shadow, 4 mm in diameter, on the upper lobe of
the left lung (Fig. 2B). A whole-body
bone scan using positron emission tomography-CT demonstrated that
the lung cancer had metastasized to the bone, with abnormal
radioactive concentrations observed in the shoulder blades. There
were no metastatic lesions observed on abdominal CT and magnetic
resonance imaging (MRI) of the brain.
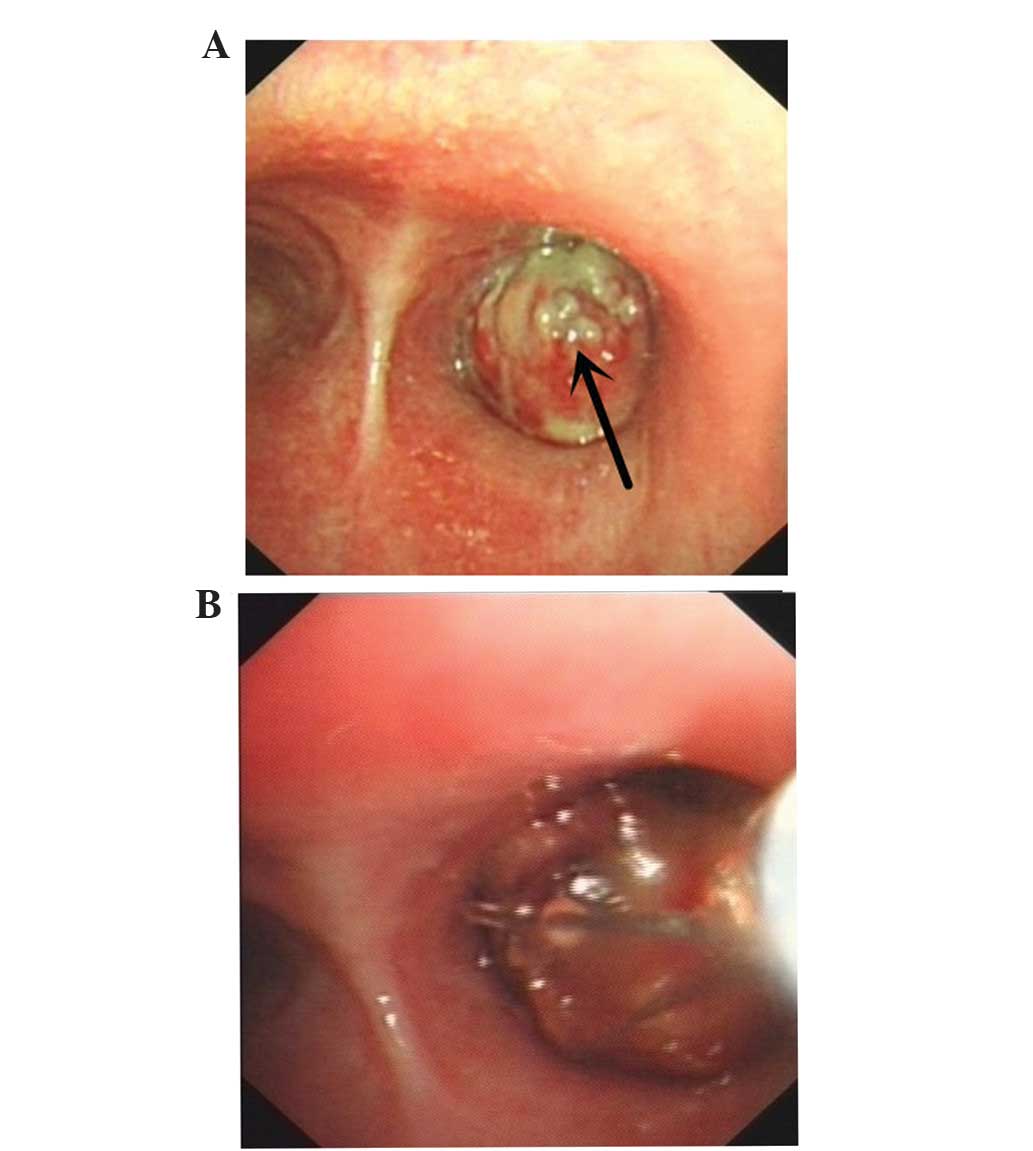
Bronchoscopy revealed visible neoplasms in the lumen
of the respiratory tract, which caused the lumen to become
decreased in diameter (Fig. 3A). A
resection of the right upper lobe tumor of the right total bronchus
using a flexible bronchoscope was successfully performed in the
patient (Fig. 3B). Subsequently, the
patient was administered with systemic chemotherapy targeting the
intrapulmonary and bone metastases 15 days subsequent to resection,
as follows: 110 mg epirubicin intravenously for 1 day; and 2.8 g
ifosfamide intravenously for 3 days. The chemotherapy cycle was 21
days. However, the patient succumbed to the disease 2 weeks after
the second cycle of chemotherapy.
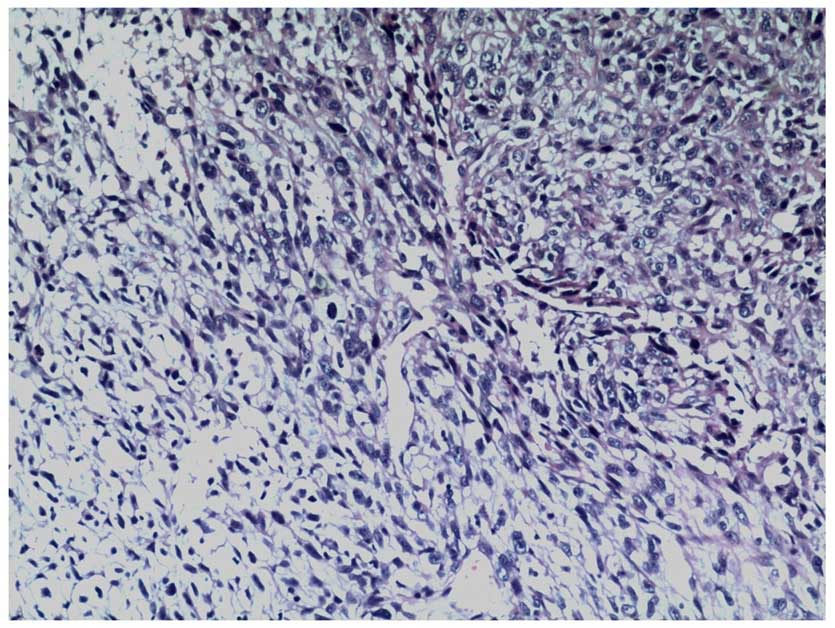
A biopsy of the resected tumor demonstrated that the
tumor was composed of spindle cells, with marked nuclear
pleomorphism and numerous mitotic figures (hematoxylin and eosin
staining; Fig. 4). There was no
epithelial differentiation observed in the tumor.
Immunohistochemical staining demonstrated that the tumor expressed
vimentin, smooth muscle actin, cluster of differentiation 34 (CD34)
and actin, and did not express high and low molecular weight
cytokeratins, epithelial membrane antigen, desmin and protein
S-100. The cell proliferation index of the tumor using antigen
Ki-67 was 30%. There was no epithelial differentiation observed in
the tumor and the overall morphological features favored a
high-grade sarcoma with evidence of smooth muscle differentiation,
indicating a leiomyosarcoma. Thus, the final diagnosis was PPL.
Discussion
PPL is a mesenchymal tumor that has been observed to
originate from the smooth muscle cells of the bronchial or blood
vessel wall. PPL accounts for <0.5% of all malignant pulmonary
tumors (1), and is classified by
type: Intraluminal, intrapulmonary and pulmonary vascular (4). The intrapulmonary type of PPL is the
most common, and the pulmonary vascular type originates from the
vascular wall and occurs in the pulmonary artery, where it may
cause stenosis and obstruction (5).
The PPL possessed by the patient in the present study was the
intraluminal type.
The majority of patients with PPL present with
symptoms similar to those observed in other primary pulmonary
tumors, including the presence of a cough, chest pain, dyspnea,
hemoptysis and asthenia (1). In order
to differentiate PPL from bronchogenic carcinoma, an excisional
biopsy is required. Vimentin is present in the majority of
mesenchymal cells and is a good mesenchymal tumor marker, which may
additionally be used in the identification of sarcoma and
carcinoma. Cytokeratin is an epithelial marker. Therefore, vimentin
and cytokeratin may be used together to distinguish between the
majority of epithelial and mesenchymal tumors (6,7). In the
present case, immunohistochemical staining demonstrated that the
tumor expressed vimentin, smooth muscle actin, cluster of
differentiation 34 and actin, and did not express high and low
molecular weight cytokeratins. A diagnosis of PPL should only be
considered if signs of an occult tumor are not noted in any other
bodily location. In women, it is particularly important to observe
if there is a tumor present in the uterus (8). Following a tissue biopsy that confirms
PPL, pre-operative staging of the tumor may be considered.
Generally, this consists of CT of the lungs and MRI of the primary
lesion to determine if the tumor has metastasized, which is
uncommon in leiomyosarcomas and is typically observed in the
advanced stages of the disease (8).
Leiomyosarcomas are characterized grossly by a firm
grey or white surface. Microscopically, malignant spindle cells
with cigar-shaped nuclei, arranged in interweaving fascicles, are
noted (9). Common features that PPL
cells possess are mitotic figures, multinucleation, nuclear atypia,
prominent vascularity, scanty cytoplasm and zonal necrosis.
Calcification, cavitation, pleural effusions and pneumothorax are
not often observed (9). PPL expresses
actin, smooth muscle actin, desmin and vimentin, which may be
detected using immunohistochemistry and antibodies to these
proteins. Generally, leiomyosarcomas do not express
carcinoembryonic antigen, cytokeratin, leukocyte common antigen,
neuroendocrine filament and S100 protein (10). In the present case, analysis of the
resected tumor biopsy demonstrated that the tumor was composed of
spindle cells, with marked nuclear pleomorphism and numerous
mitotic figures. There was no epithelial differentiation observed
in the tumor. Immunohistochemical staining identified expression of
vimentin, smooth muscle actin, CD34 and actin in the tumor,
however, high and low molecular weight cytokeratins, epithelial
membrane antigen, desmin and S100 protein were not expressed.
Therefore, the pathological and immunohistochemical results
observed in the present study are consistent with these
aforementioned characteristics.
Treatment regimens for patients with PPL aim to
achieve local and systemic control of the tumor, while preserving
function and quality of life. If pre-operative staging demonstrates
that there is no evidence of metastases, surgery is recommended,
such as a lobectomy and pneumonectomy, which require resection of
the chest wall, diaphragm or vascular structures (9). If an early complete resection is
performed, the 5-year survival rate of patients is ~50%, and there
have been reports of patient survival 20 years post-resection
(10). Adjuvant radio- or
chemotherapy treatment is recommended in cases of incomplete
resection, unresectable tumors and patients with increased
histological malignancy; however, it has been demonstrated that
radio- and chemotherapy do not extend the survival time of the
patient (11). Prognostic indicators
of PPL consist of tumor size, extent of bronchial invasion and
degree of malignancy (9). In the
present study, malignant spindle cells were positive for S100
antigen, smooth muscle actin and vimentin, and were negative for
anti-cytokeratin 5.2, caldesmon, cytokeratin 5/6, cytokeratin 7,
cytokeratin 20, CD34, c-Kit, desmin, epithelial membrane antigen,
myogenin, pancytokeratin and thyroid transcription factor-1.
Therefore, the results of the present study are consistent with the
existing literature (10).
In conclusion, PPL is a rare tumor that grows
rapidly. It may be challenging to differentiate PPL from other
pulmonary tumors due to the lack of specific manifestations. A
pre-operative diagnosis of PPL is considered following the results
of a sputum smear, lung biopsy or bronchoscopic examination;
however, diagnosing PPL from these methods may also be challenging.
In order to differentiate a primary pulmonary leiomyosarcoma from
bronchogenic carcinoma, an excisional biopsy is required.
Therefore, the most reliable methods for detection and diagnosis of
PPL are chest radiography and post-operative pathological
examination. The present case emphasizes the important role of
pathological and immunohistochemical results in differentiating
between PPL and bronchogenic carcinoma. The goal of treatment is to
obtain local and systemic control of the sarcoma, while preserving
functioning and quality of life. If preoperative evaluation reveals
no evidence of metastases, then treatment is surgical. However, if
preoperative evaluation reveals evidence of metastasis, the
radiation therapy, chemotherapy or a combination of the two is
required. An increased awareness of PPL leading to an early
diagnosis and the performance of a complete surgical resection with
adjuvant radio- and chemotherapy in selected patients may improve
the prognosis of patients with PPL.
Acknowledgements
The authors would like to thank the Records System
and Medical Imaging Division of the First Affiliated Hospital of
Wenzhou Medical University for collecting the data and figures used
by the present study.
References
|
1
|
Rozada R, Vila A and Sosa L: Primary
leiomyosarcoma of the lung. Arch Bronconeumol. 46:338–339. 2010.(In
Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Janssen JP, Mulder JJ, Wagenaar SS, Elbers
HR and van den Bosch JM: Primary sarcoma of the lung: A clinical
study with long-term follow-up. Ann Thorac Surg. 58:1151–1155.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen W, Chen J, Wei S, Wang X, Li X and
Zhou Q: Primary pulmonary leiomyosarcoma. J Chin Med Assoc.
77:49–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu HQ, Ren H, Miao Q, Wang Z, Zhang Z and
Xu L: Pulmonary leiomyosarcoma. Chin Med Sci J. 12:129–131.
1997.PubMed/NCBI
|
|
5
|
Pain JA and Sayer RE: Primary
leiomyosarcoma of the pulmonary artery. Eur J Respir Dis.
65:139–143. 1984.PubMed/NCBI
|
|
6
|
Hartel PH, Fanburg-Smith JC, Frazier AA,
Galvin JR, Lichy JH, Shilo K and Franks TJ: Primary pulmonary and
mediastinal synovial sarcoma: A clinicopathologic study of 60 cases
and comparison with five prior series. Mod Pathol. 20:760–769.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nath D, Arava S, Joshi P, Madan K and
Mathur S: Primary pulmonary leiomyosarcoma of lung: An unusual
entity with brief review. Indian J Pathol Microbiol. 58:338–340.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ramanathan T: Primary leiomyosarcoma of
the lung. Thorax. 29:482–489. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cordes BG, Collins BT, McDonald JW, Khosla
A and Salimi Z: Fine needle aspiration biopsy of primary
leiomyosarcoma arising from a pulmonary vein. Acta Cytol.
43:523–526. 1999.PubMed/NCBI
|
|
10
|
Arnold LM III, Burman SD and O-Yurvati AH:
Diagnosis and management of primary pulmonary leiomyosarcoma. J Am
Osteopath Assoc. 110:244–246. 2010.PubMed/NCBI
|
|
11
|
Shaw RR, Paulson DL, Kee JL and Lovett VF:
Primary pulmonary leiomyosarcomas. J Thorac Cardiovasc Surg.
41:430–436. 1961.
|