Introduction
Hepatoblastoma (HB) is an uncommon liver malignancy
in infants and children. It accounts for just over 1% of pediatric
cancers (1), but exhibits increasing
incidence in North America and Europe (2). This disease is most commonly diagnosed
during a child's first 3 years of life (3); only 5% of new HB cases are diagnosed in
children older than 4 years (4). The
disease occurs significantly more frequently in boys than in girls,
for reasons that remain to be determined (4).
HB originates from immature liver precursor cells
(2). Histologically, HB can be
divided into epithelial or mixed epithelial/mesenchymal tissues
(5). The majority of HB cases are
epithelial in origin, and consist of a mixture of embryonal and
fetal cell types (5). Approximately
5% of HB cases are of the small cell undifferentiated subtype
(5). Currently, surgical resection,
adjuvant chemotherapy and liver transplantation are the only
options to treat HB (2). Thus, novel
strategies are required to improve the current understanding of HB
biology and to provide targets for therapy or early detection of
the disease.
Methylation of DNA is the only known genetically
programmed DNA modification process in mammals; it regulates
several biological processes, including gene transcription, X
chromosome inactivation, genomic imprinting and chromatin
modification (6–8). Methylation changes in CpG islands (CGIs)
and CpG shores (low CpG density areas within 2 kb of CGIs), as well
as CpG shelves (low CpG density areas within 2–4 kb of CGIs)
(9), affect gene expression.
Hypomethylation of promoter regions of crucial genes can activate
relevant gene expression and may contribute to tumorigenesis
(10–12).
Several genome-wide analyses had been conducted in
different types of tumors. For instance, Revill et al
revealed sphingomyelin phosphodiesterase 3 as a potent tumor
suppressor gene, which is hypermethylated in primary hepatocellular
carcinoma (13). The present study
aimed to determine whether changes in methylation status could
affect HB by analyzing HB tissue samples and paired distant
noncancerous tissues to profile differentially methylated genes in
the disease.
Materials and methods
HB specimens and clinical
information
Matched pairs of HB and adjacent non-tumor tissues
were obtained from the surgical specimen archives of the Children's
Hospital of Fudan University (Shanghai, China) from 10 patients who
underwent partial hepatectomy, and none of them had received
radiation therapy or chemotherapy prior to tumour biopsy or
resection; their tissue pathology results confirmed HB with >80%
viable tumor cells. The patients did not undergo any prior
chemotherapy or other forms of therapy, and underwent standardized
treatment post-operation. Clinical data were obtained
retrospectively from clinical files (Table I). In total, 6 patients were of the
mixed embryonal/fetal subtype, and the remaining 4 patients were of
the epithelial subtype. The parents of the patients from whom these
samples were obtained provided their written informed consent to
participate in the study, and the Ethics Committee of the
Children's Hospital of Fudan University approved the study as well
as the consent procedure.
 | Table I.Distribution of study subjects with HB
and their serum AFP levels. |
Table I.
Distribution of study subjects with HB
and their serum AFP levels.
| Case | Age
(months)a | Gender | Diagnosis HB
type | AFP
(ng/ml)b | Viral hepatitis | Chip |
|---|
| 1 | 7 | Male | Mixed embryonal/fetal
subtype | 68,490 | None | 1 |
| 2 | 23 | Male | Mixed embryonal/fetal
subtype | >121,000 | None | 2 |
| 3 | 11 | Female | Mixed embryonal/fetal
subtype | >121,000 | None | 3 |
| 4 | 10 | Female | Mixed embryonal/fetal
subtype | >121,000 | None | – |
| 5 | 20 | Female | Mixed embryonal/fetal
subtype | >121,000 | None | – |
| 6 | 7 | Male | Mixed embryonal/fetal
subtype | >121,000 | None | – |
| 7 | 30 | Male | Epithelial type | >121,000 | None | – |
| 8 | 14 | Male | Epithelial
type | >121,000 | None | – |
| 9 | 19 | Male | Epithelial
type | >121,000 | None | – |
| 10 | 7 | Female | Epithelial
type | >121,000 | None | – |
Genomic DNA was extracted from 10 HB primary tumors
(fresh frozen tissue) and adjacent non-tumor tissues, of which, 3
were subjected to Infinium HumanMethylation450 BeadChip analysis
(Table I). Total RNA was extracted
from 10 matched HB tumor and non-tumor pairs, reverse transcribed
to complementary DNA (cDNA) and then subjected to quantitative
polymerase chain reaction (qPCR) analysis of
alpha-fetoprotein (AFP) messenger RNA (mRNA)
expression.
Infinium HumanMethylation450
BeadChip
Samples from 3 patients were analyzed with Infinium
HumanMethylation450 BeadChip (Illumina Inc., San Diego, CA, USA),
which is a cost-effective approach to quickly analyze each
individual's methylome (14). The DNA
was treated with bisulfite and hybridized to arrays according to
the manufacturer's protocol. Raw data were adjusted for dye bias
and quantile-normalized with data derived from Infinium I or
Infinium II assay chemistry probes considered separately (Illumina
Inc., San Diego, CA, USA). This high-throughput platform enabled
quantitative evaluation of methylation levels with
single-nucleotide resolution. The β-values were used to reflect
methylation status, ranging from 0 to 1, where 0 is unmethylated
and 1 is fully methylated.
Gene function analysis
The predicted target genes were input into the
Database for Annotation, Visualization and Integrated Discovery
(http://david.abcc.ncifcrf.gov/), which
uses Gene Ontology (GO) to identify the molecular function of the
profiled genes (15). The Kyoto
Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.ad.jp/kegg/) was used to analyze the
potential functions of these target genes in the pathways.
P<0.05 was used as the cut-off value.
Reverse transcription (RT)-qPCR
Total cellular RNA was isolated from HB and normal
tissues using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and then reverse transcribed using a
PrimeScript RT reagent kit (Perfect Real Time) with gDNA Eraser
(Takara Biotechnology Co., Ltd., Dalian, China) following the
manufacturer's protocol. The expression of AFP was analyzed
by qPCR with a SYBRGreen PCR kit (Takara Biotechnology Co., Ltd.).
The AFP primers used for q-PCR were: Forward,
5′-GTGGTCAGTTTGCAGCATTC-3′ and reverse, 5′-AGAGGAGATGTGCTGGATTG-3′
(length, 110 bp). Glyceraldehyde 3-phosphate dehydrogenase mRNA was
used as the internal control, with forward 5′-AAAGCCCACTCCAGCATC-3′
and reverse 5′-TAGCGAGCAGCCCAAAGA-3′ primers. The PCR cycling
conditions were as follows: 95°C for 15 sec, followed by 95°C for 5
sec and 60°C for 31 sec, for 40 cycles. For quantitative results,
AFP expression was represented as fold-change by the
2−ΔΔCq method and statistically analyzed (16).
Statistical analyses
Statistical analyses and graphical depiction of data
were conducted with GraphPad Prism 5.0 (GraphPad Software, Inc., La
Jolla, CA, USA). Results were presented as the mean ± standard
error of the mean and were evaluated with Student's t-test
(two-tailed) unless otherwise specified (paired t-test or
Pearson's correlation). Certain statistical calculations were
performed using SPSS version 19.0 (IBM SPSS, Armonk, NY, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
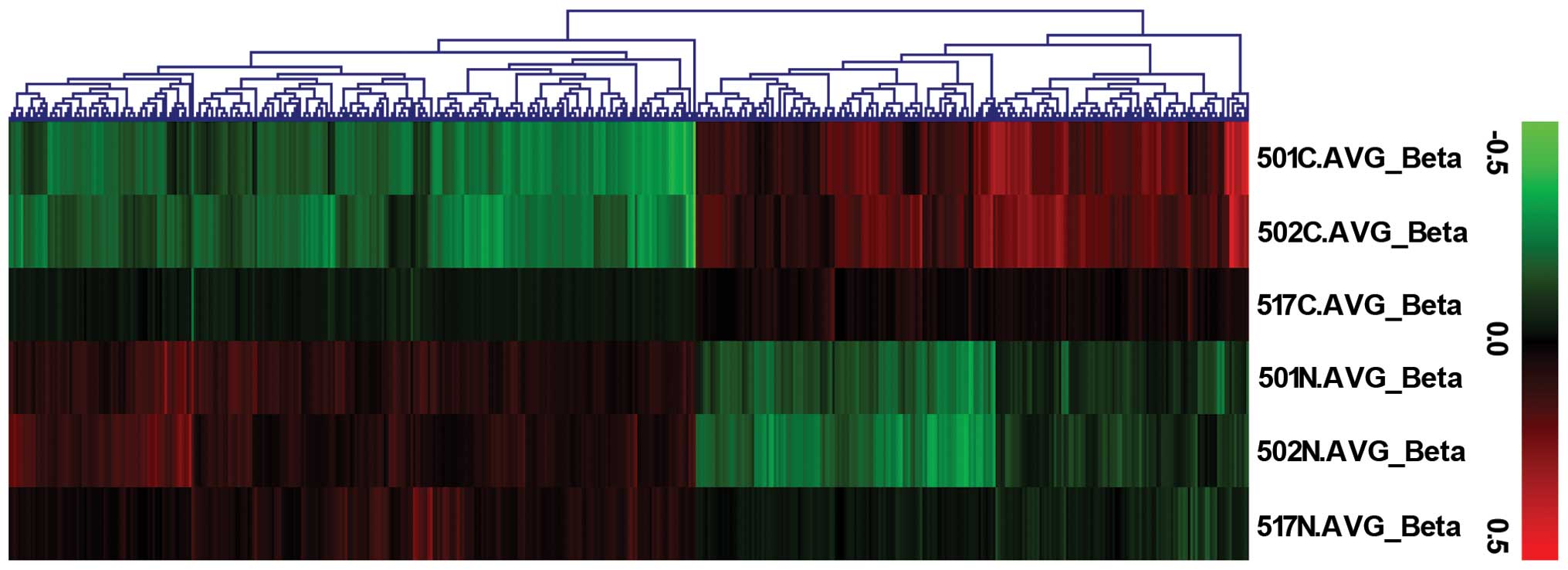
Hierarchical clustering analysis by
Infinium HumanMethylation450 BeadChip and functional annotations of
differentially methylated CpG sites
The Infinium HumanMethylation450 BeadChip was used
to obtain data from an independent cohort of 3 HB-non-tumor tissue
pairs (probe call rate, >99% for all samples). Principal
component analysis revealed separation of HB-non-tumor pairs and
demonstrated the methylation levels of HB tissues to be
significantly lower than those of non-tumor tissues (Fig. 1).
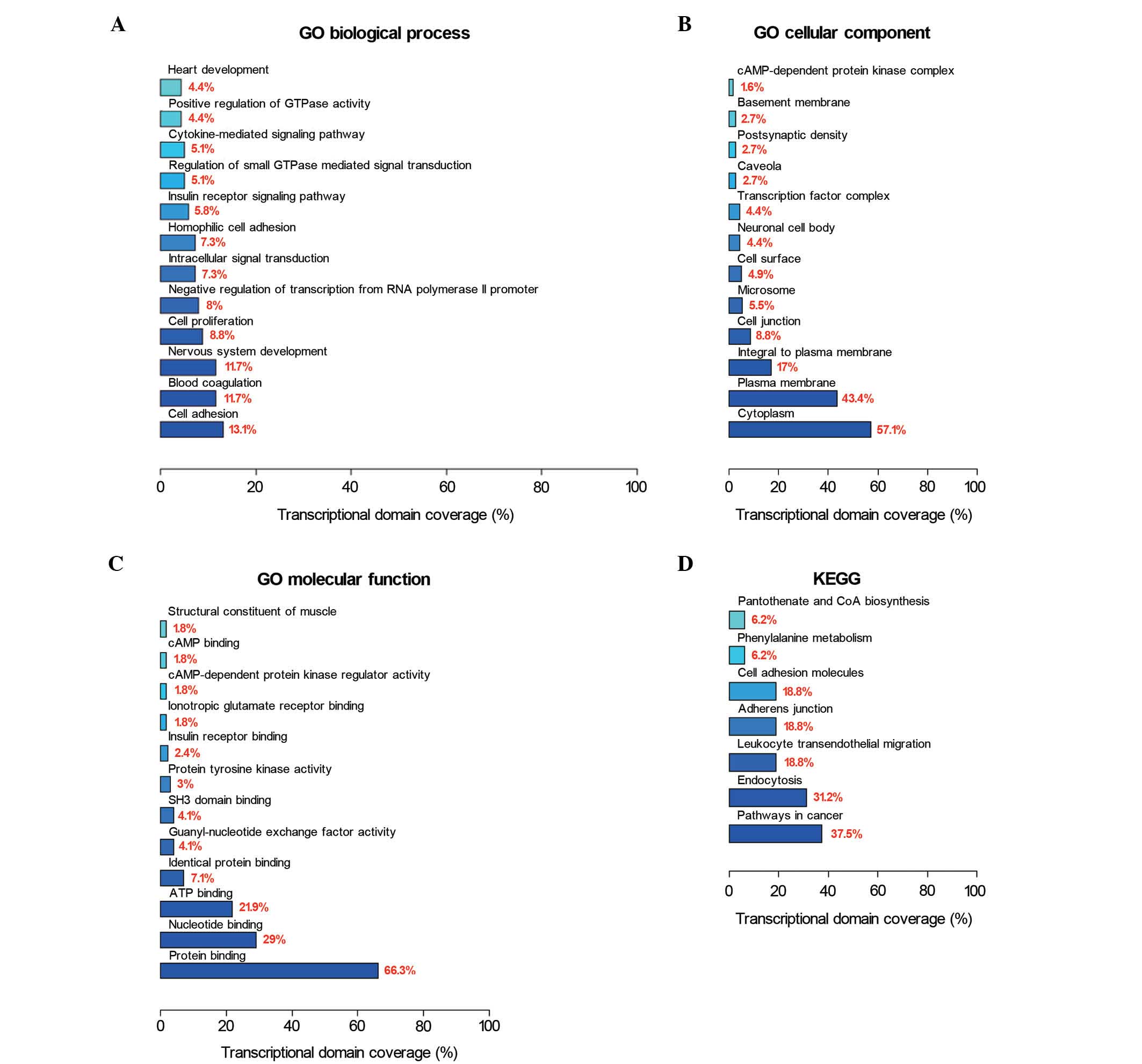
Next, GO and KEGG pathway database categories were
used to analyze the 524 differential methylated genes identified.
Cell adhesion, blood coagulation and nervous system development
were observed to be the three most affected biological processes;
while cytoplasm, plasma membrane and integral plasma membrane
structures were observed to be the three most affected cellular
components; and protein binding, nucleotide binding and adenosine
triphosphate binding were observed to be the three most affected
molecular functions (Fig. 2A-C).
Pathway-based analyses revealed significant enrichment for genes in
cancer pathways (Fig. 2D).
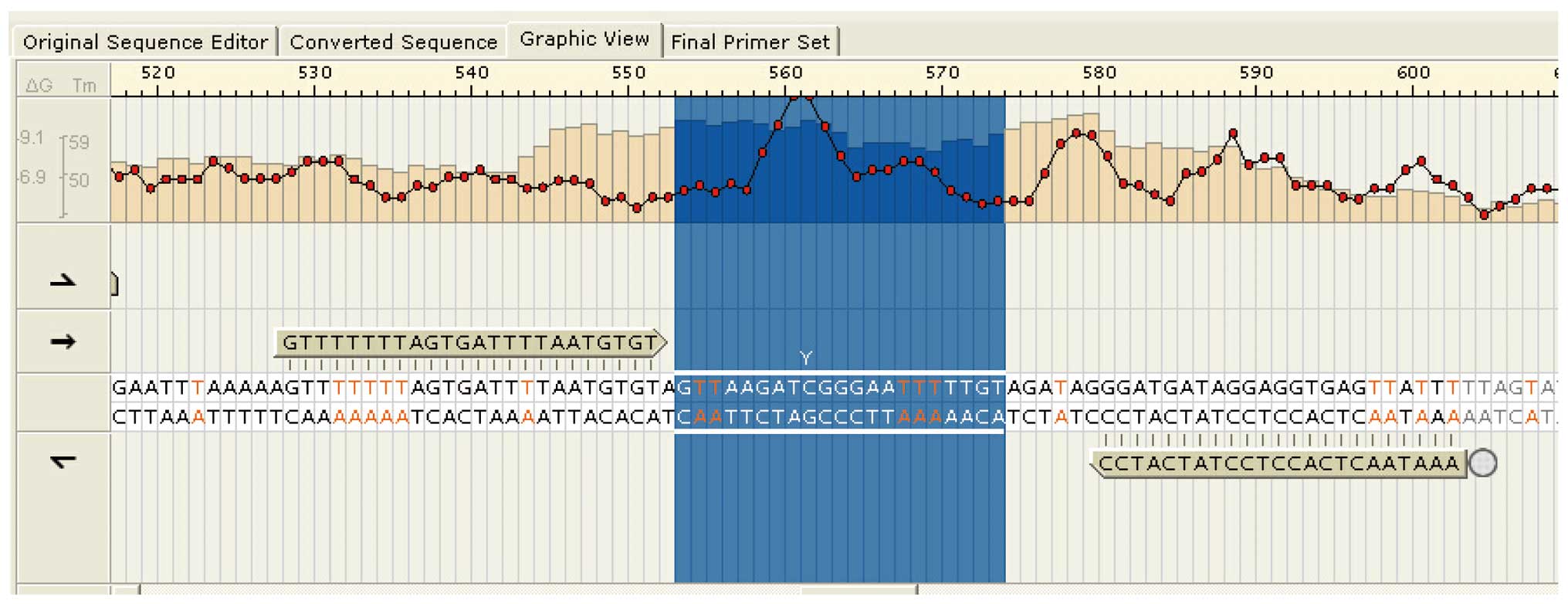
Aberrant methylation of AFP in HB
The methylation microarray indicated enriched
methylation of positions near the transcription start site of
AFP (Fig. 3). A representative
illustration of AFP methylation levels in a matched
HB-non-tumor tissue pair is shown in Fig.
4. As HB tissues had lower AFP methylation levels than
non-tumor liver tissues, aberrant methylation may be a
tumor-specific event in HB.
Correlation of AFP mRNA expression and
percentage of DNA methylation
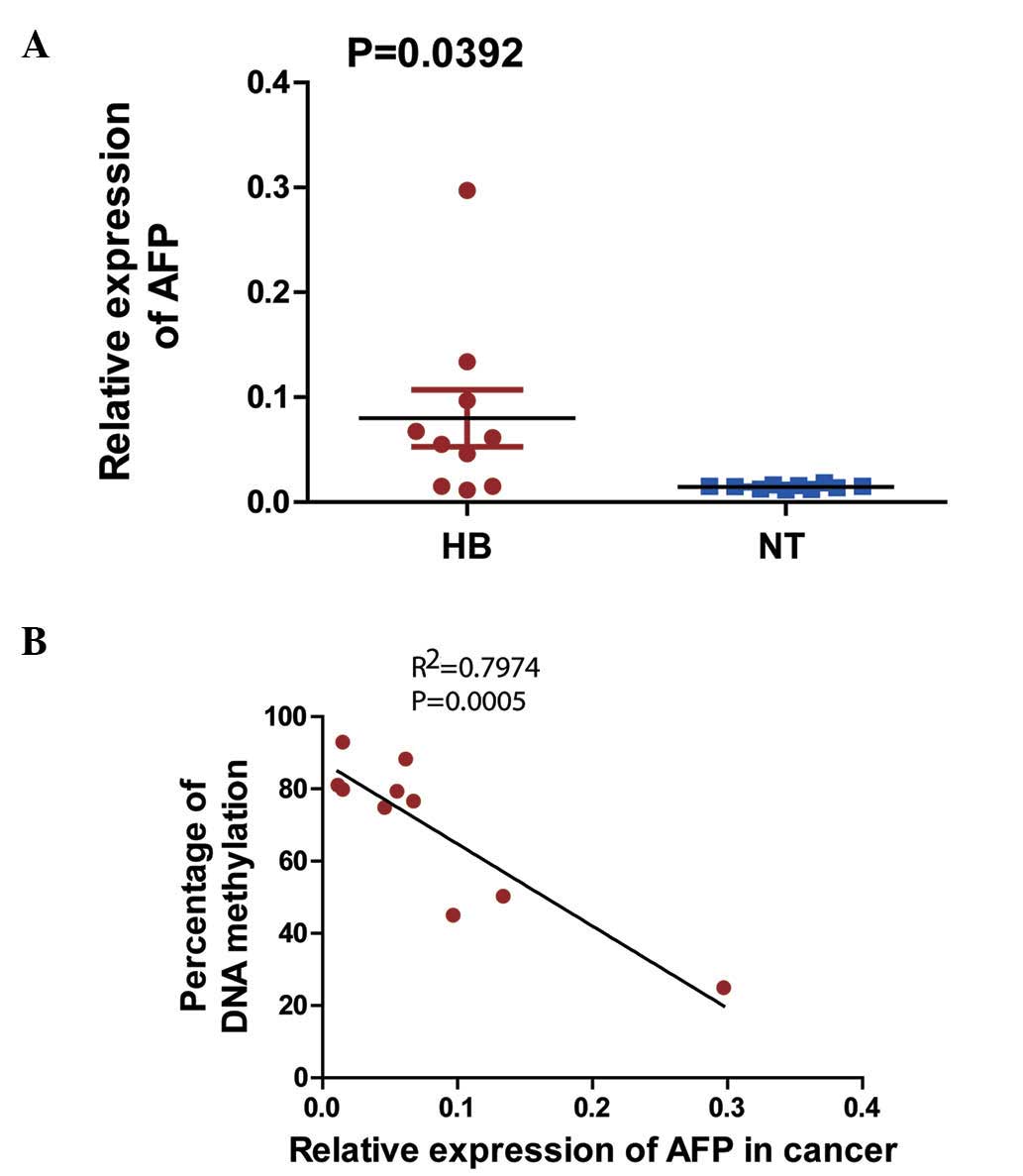
The expression levels of the AFP gene in 10
HB-non-tumor liver tissue pairs were observed to be significantly
higher in the HB tissues than in their non-tumor counterparts
(Fig. 5A). The correlations between
AFP mRNA expression and DNA methylation percentage were
tested using the linear Pearson's R correlation
coefficients, which were interpreted using the scale provided by
Salkin (R=1-0.8, very strong; R=0.8–0.6, strong;
R=0.6–0.4, moderate; R=0.4–0.2, weak; and
R=0.2–0.0, very weak or not correlated) (17) (Fig. 5B),
and were observed to be negatively correlated (R=0.89;
P<0.05).
Discussion
Although HB is an uncommon cancer in the general
population, it is the most common liver tumor in children (4). Its etiology is still unknown, but it has
been associated with familial adenomatous polyposis (18), low and high birth weights (19) and constitutional trisomy 18 (20). Mutations in numerous components of the
wingless-related integration site/β-catenin signaling pathway are
also often present in HB tissues (21).
Although the mechanisms responsible for HB
development have been extensively studied for the last two decades,
the pathogenesis of this disease is still vague, and the majority
of its altered gene expression and regulation remain to be
delineated. The present authors recently sequenced the HB exome and
identified a novel oncogene (caprin family member 2) and three
tumor suppressors (speckle-type POZ protein, olfactory receptor
family 5 subfamily I member 1 and cell division cycle 20B) that
influence HB cell growth (22). A
link between long non-coding RNA and HB was also detected (23).
Hypermethylation or hypomethylation of gene promoter
regions can result in their transcriptional silencing or activation
(24). Although genome-wide copy
number alterations in HB have been studied, little is known about
genome-wide methylation changes (4).
The present study is the first to explore whole-genome DNA
methylation status in HB. In the present study, Infinium
HumanMethylation450 BeadChip, a new platform for high-throughput
DNA methylation analysis, was used (25). The cluster analysis revealed markedly
less methylation in HB tissues than in normal liver tissues in the
three sets of paired tissues analyzed. HB tissues are relatively
low in genome-wide DNA methylation with preponderant CpG sites, and
highly methylated genes may also have more numerous CpG sites
(26). The present results revealed
the DNA in HB tissues to be less methylated than in normal liver
tissues. This may explain how the disease begins: A number of
oncogenes are activated by demethylation, resulting in rapid tumor
growth.
Serum AFP screening for HB has been a
clinical practice for a long time (27). Additionally, high mRNA levels of
AFP are reportedly associated with HB (27). To further understand the association
between HB and AFP methylation, a methylation microarray was
used in the present study to specifically detect AFP
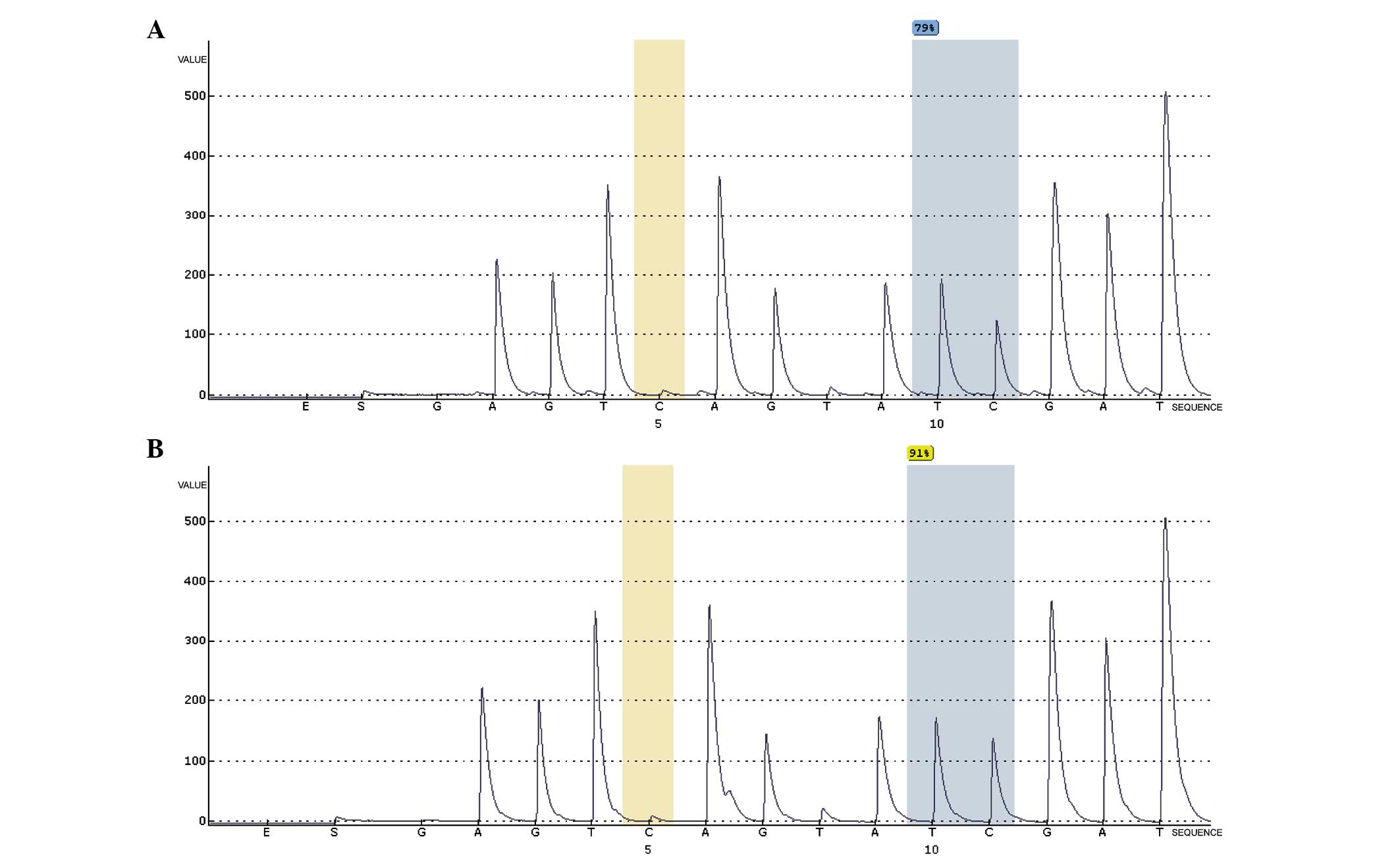
methylation status. A methylation-enriched position near the
AFP transcription start site [g10778295 (TSS1500)] was
identified, which was hypomethylated in HB. Pyrosequencing
demonstrated the methylation level of the AFP locus
cg10778295 to be lower in cancer tissues (79%) than in normal liver
tissues (91%). Finally, RT-qPCR demonstrated AFP mRNA
expression in HB to be much higher than in normal liver tissue in
an independent cohort of 10 adjacent HB-non-tumor tissues pairs.
The expression of AFP mRNA was significantly negatively
correlated with its methylation status.
In conclusion, the current study of methylation in
HB tissues is a proof-of-principle that methylation status probably
affects HB development and progression. HB is an uncommon malignant
liver neoplasm of children, and its etiology, pathophysiology and
molecular mechanisms are largely unknown. Further studies are
required to fully understand and control this disease. Although the
current study demonstrated only that high AFP expression is
associated with epigenetic modification, this topic warrants
further investigation.
Acknowledgements
The present study received financial support from
the National Natural Science Foundation of China (Beijing, China;
grant nos. 81370472 and 81300517), the Shanghai City Health Bureau
for Youth Scientific Fund Project (Shanghai, China; grant no.
20134y100), the Science Foundation of Shanghai (Shanghai, China;
grant nos. 11JC1401300, 13ZR1451800 and 15ZR1404200) and the State
Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer
Institute (Shanghai, China; grant no. SKLORG #90-12-01).
References
|
1
|
Herzog CE, Andrassy RJ and Eftekhari F:
Childhood cancers: Hepatoblastoma. Oncologist. 5:445–453. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
von Schweinitz D: Hepatoblastoma: Recent
developments in research and treatment. Semin Pediatr Surg.
21:21–30. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
De Ioris M, Brugieres L, Zimmermann A,
Keeling J, Brock P, Maibach R, Pritchard J, Shafford L, Zsiros J,
Czaudzerna P and Perilongo G: Hepatoblastoma with a low serum
alpha-fetoprotein level at diagnosis: The SIOPEL group experience.
Eur J Cancer. 44:545–550. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Litten JB and Tomlinson GE: Liver tumors
in children. Oncologist. 13:812–820. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Haas JE, Feusner JH and Finegold MJ: Small
cell undifferentiated histology in hepatoblastoma may be
unfavorable. Cancer. 92:3130–3134. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geiman TM and Robertson KD: Chromatin
remodeling, histone modifications and DNA methylation-how does it
all fit together? J Cell Biochem. 87:117–125. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reik W and Walter J: Genomic imprinting:
Parental influence on the genome. Nat Rev Genet. 2:21–32. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chow J and Heard E: X inactivation and the
complexities of silencing a sex chromosome. Curr Opin Cell Biol.
21:359–366. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bibikova M, Barnes B, Tsan C, Ho V,
Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et
al: High density DNA methylation array with single CpG site
resolution. Genomics. 98:288–295. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Doi A, Park IH, Wen B, Murakami P, Aryee
MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al:
Differential methylation of tissue- and cancer-specific CpG island
shores distinguishes human induced pluripotent stem cells,
embryonic stem cells and fibroblasts. Nat Genet. 41:1350–1353.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z,
Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al:
The human colon cancer methylome shows similar hypo- and
hypermethylation at conserved tissue-specific CpG island shores.
Nat Genet. 41:178–186. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ogoshi K, Hashimoto S, Nakatani Y, Qu W,
Oshima K, Tokunaga K, Sugano S, Hattori M, Morishita S and
Matsushima K: Genome-wide profiling of DNA methylation in human
cancer cells. Genomics. 98:280–287. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Revill K, Wang T, Lachenmayer A, Kojima K,
Harrington A, Li J, Hoshida Y, Llovet JM and Powers S: Genome-wide
methylation analysis and epigenetic unmasking identify tumor
suppressor genes in hepatocellular carcinoma. Gastroenterology.
145:1424–1435, e1-e25. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sandoval J, Heyn H, Moran S, Serra-Musach
J, Pujana MA, Bibikova M and Esteller M: Validation of a DNA
methylation microarray for 450,000 CpG sites in the human genome.
Epigenetics. 6:692–702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Johnson MR, Wang KS, Smith JB, Heslin MJ
and Diasio RB: Quantitation of dihydropyrimidine dehydrogenase
expression by real-time reverse transcription polymerase chain
reaction. Anal Biochem. 278:175–184. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perek B, Malinska A, Stefaniak S,
Ostalska-Nowicka D, Misterski M, Zabel M, Suri A and Nowicki M:
Predictive factors of late venous aortocoronary graft failure:
Ultrastructural studies. PLoS One. 8:e7062882013. View Article : Google Scholar
|
|
18
|
Kingston JE, Herbert A, Draper GJ and Mann
JR: Association between hepatoblastoma and polyposis coli. Arch Dis
Child. 58:959–962. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ross JA: Hepatoblastoma and birth weight:
Too little, too big, or just right? J Pediatr. 130:516–517.
1997.PubMed/NCBI
|
|
20
|
Mamlok V, Nichols M, Lockhart L and Mamlok
R: Trisomy-18 and hepatoblastoma. Am J Med Genet. 33:125–126. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takayasu H, Horie H, Hiyama E, Matsunaga
T, Hayashi Y, Watanabe Y, Suita S, Kaneko M, Sasaki F, Hashizume K,
et al: Frequent deletions and mutations of the beta-catenin gene
are associated with overexpression of cyclin D1 and fibronectin and
poorly differentiated histology in childhood hepatoblastoma. Clin
Cancer Res. 7:901–908. 2001.PubMed/NCBI
|
|
22
|
Jia D, Dong R, Jing Y, Xu D, Wang Q, Chen
L, Li Q, Huang Y, Zhang Y, Zhang Z, et al: Exome sequencing of
hepatoblastoma reveals novel mutations and cancer genes in the Wnt
pathway and ubiquitin ligase complex. Hepatology. 60:1686–1696.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dong R, Jia D, Xue P, Cui X, Li K, Zheng
S, He X and Dong K: Genome-wide analysis of long noncoding RNA
(lncRNA) expression in hepatoblastoma tissues. PLoS One.
9:e855992014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baylin SB and Ohm JE: Epigenetic gene
silencing in cancer-a mechanism for early oncogenic pathway
addiction? Nat Rev Cancer. 6:107–116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morris TJ, Butcher LM, Feber A,
Teschendorff AE, Chakravarthy AR, Wojdacz TK and Beck S: ChAMP:
450k chip analysis methylation pipeline. Bioinformatics.
30:428–430. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tomlinson GE and Kappler R: Genetics and
epigenetics of hepatoblastoma. Pediatr Blood Cancer. 59:785–792.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Clericuzio CL, Chen E, McNeil DE, O'Connor
T, Zackai EH, Medne L, Tomlinson G and DeBaun M: Serum
alpha-fetoprotein screening for hepatoblastoma in children with
Beckwith-Wiedemann syndrome or isolated hemihyperplasia. J Pediatr.
143:270–272. 2003. View Article : Google Scholar : PubMed/NCBI
|