Introduction
A causal interlink between inflammation and cancer
was first suggested in the 19th century by Rudolf Virchow (1). Accumulating evidence over several
decades has delineated underlying molecular mechanisms between
chronic inflammation and cancer progression (2). Multiple studies have proposed that
inflammation-induced genetic instability promotes tumor progression
(2). Inflammation also causes
defective immune surveillance and responses to anti-cancer
chemotherapy (3). Chronic
inflammation in the tumor microenvironment induces the production
of free radicals such as reactive oxygen and nitrogen species,
leading to oxidative damage and nitration of DNA bases, which
increases the risk of cancer initiation (4). One of the key features of cancer
progression and metastasis is markedly enhanced inflammatory stress
factors in the tumor microenvironment (5).
One of the events linking inflammation and cancer is
an increase in inflammatory cytokines, including tumor necrosis
factor (TNF)α, interleukin (IL)-6 and IL-17, and stress growth
factors such as vascular endothelial growth factor (VEGF), in the
tumor microenvironment (6). The
pro-tumorigenic role of IL-17, an inflammatory cytokine, has also
been implicated in several types of cancer (7). In mice with carcinogen-induced skin
tumors, deficient expression of IL-17 receptor resulted in a lower
tumor incidence and a diminished tumor size (8). Typically, inflammatory cytokines induce
their effect by activating inflammatory transcription factors such
as the Janus kinase (JAK)/signal transducer and activator of
transcription (STAT) signaling pathway (9). Along with cytokines, chronic
inflammation-induced stress molecules such as VEGF are known to be
involved in cell proliferation, cell adhesion, inflammatory cell
recruitment, angiogenesis and cell migration, leading to cancer
progression (10). Specifically, the
functions of VEGF in cancer are not limited to angiogenesis alone
(11). VEGF-mediated signaling occurs
in tumor cells and is known to contribute to key aspects of
tumorigenesis, including cancer initiation and progression through
its interaction with specific receptors, leading to the regulation
of trafficking and secretion of other chemokines and extracellular
matrix proteins (11).
A wide variety of environmental and infectious
agents have been attributed to increased risk of
inflammation-induced cancer progression (12). Dietary high salt intake has been
correlated with an increase in the incidence of cancers (13). Traditionally, high salt levels have
been correlated with cardiovascular disease and inflammatory injury
in arteries (14). In animal models,
high sodium chloride levels have been demonstrated to cause
excessive inflammatory activation, triggering ischemia injury and
end-organ stress mediated by reactive oxygen species and
pro-inflammatory cytokine secretion, leading to irreversible
cardiac cell damage (15,16). Importantly, in the context of breast
cancer, the sodium content of mammary adenocarcinomas has been
reported to be significantly higher than in the normal mammary
epithelium (17). However, in that
study, it was not clear if the increased sodium content observed
was intracellular or extracellular. Human studies on gastric cancer
suggested that excess sodium could cause inflammation and stomach
ulcers, which could lead to gastric cancer (18). It is also important to understand that
high sodium content in mammary adenocarcinomas has been shown to be
significantly higher than that of normal lactating mammary
epithelium (19). Furthermore, a
correlation between expression of sodium symporters and increased
invasion capacity of breast cancer has been previously suggested
(20–22). In the present study, the synergistic
effect of high sodium chloride levels with pro-inflammatory
cytokines towards induction of the expression of VEGF-A, a crucial
inflammatory and angiogenic stress factor that is important in
tumor progression and metastasis, is reported.
Materials and methods
Cell culture
The breast cancer cell line MCF-7 (HTB-22™) and
normal human aortic endothelial cells (NHAECs; PCS-100-011™) were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA) and cultured in cell basal essential medium (30–2003;
ATCC) along with the medium supplements recommended by the
manufacturer. For stimulation studies, breast cancer cells were
treated with varying concentrations (0–1,000 ng/ml) of IL-17
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) or sodium
chloride supplement (0.2 M final concentration; Sigma-Aldrich, St.
Louis, MO, USA) for 48 h. It is important to note that sodium
chloride concentration in regular cell culture basal medium is 0.1
M. Therefore, to perform high salt studies (0.2 M), the basal
medium was supplemented with additional 0.1 M NaCl to achieve a
final concentration of 0.2 M. Specific small interfering (si)RNA
(Santa Cruz Biotechnology, Inc., Dallas, TX, USA) mediated gene
knock-down of STAT3 (sc-29493) and NFAT5 (sc-43968). The knock-down
efficiency was measured by reverse transcription-quantitative
polymerase chain reaction (RT-qPCR), and non-specific siRNA
(sc-37007) was used as a negative control. All experiments were
performed in triplicate.
Western blot analysis
Western blot analysis was performed as described
previously (23,24). All primary and secondary antibodies
were obtained from Santa Cruz Biotechnology, Inc., unless mentioned
otherwise. All primary antibodies were diluted to 1:200 and all
secondary antibodies were diluted to 1:1,000. The following
specific primary antibodies against VEGF-A (sc-152), STAT3
(sc-482), STAT5 (sc-835), NFAT1 (sc-7294), NFAT5 (sc-13035),
vascular cell adhesion protein (VCAM; sc-8304), β1 integrin
(sc-9970) and actin (sc-10731) were utilized. Phosphorylated
proteins were probed with the phospho (p)-specific primary
antibodies Ser-727-p-STAT3 (sc-21876) and Ser-155-p-NFAT5
(SAB4504718) (Sigma-Aldrich). The nitrocellulose membranes were
developed using a chemiluminescence kit (EMD Millipore, Billerica,
MA, USA) and analyzed using Universal Hood II (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Densitometric analysis was
performed using ChemiDoc XRS systems software provided by Bio-Rad
Laboratories, Inc.
Messenger (m)RNA expression
analysis
The expression profiles of intracellular signaling
genes in MCF-7 cells were analyzed using fluorescein
amidite-labeled RT-qPCR primers for VEGF-A (Hs03929036_s1), NFAT5
(Hs00232437_m1), STAT3 (Hs01051722_s1), glyceraldehyde 3-phosphate
dehydrogenase (GAPDH; Hs402869) and actin (Hs4333762T), which were
obtained from Applied Biosystems (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol (25). Briefly, total RNA was extracted from
106 cells using TRIzol reagent (Sigma-Aldrich). RNA
samples were quantified by their absorbance at 260 nm. The RNA was
reverse transcribed, and qPCR was subsequently performed in a final
reaction volume of 50 µl using LightCycler® 480 Probes
Master (Roche Diagnostics, Indianapolis, IN, USA). Each sample was
analyzed in triplicate. Cycling conditions consisted of an initial
denaturation of 95°C for 15 min, followed by 40 cycles of 95°C for
30 sec, followed by annealing/extension at 61°C for 1 min.
Expression data were quantified using the 2−ΔΔCq method
(26) and normalized to GAPDH
expression.
Immunofluorescence microscopy
For staining of VEGF-A, 50,000 MCF7 cells were grown
on coverslips in 24-well plates (19). Cells were fixed with 4%
paraformaldehyde (23). Normal 2%
goat serum in Dulbecco's phosphate-buffered saline (Thermo Fisher
Scientific, Inc.) with 1% bovine serum albumin (Sigma-Aldrich) and
0.1% Tween 20 was used for blocking and washing. The aforementioned
specific anti-VEGF-A primary antibody and a
phycoerythrin-conjugated antibody (cat no. 12-4739-81; eBioscience,
Inc., San Diego, CA, USA) were used for immunofluorescence, with
primary antibodies diluted to 1:10 and secondary antibodies diluted
to 1:40. The images were captured using an Eclipse 80i fluorescence
microscope (Nikon Corporation, Tokyo, Japan) and processed using
Metamorph version 6.3r2 software (Molecular Devices, LLC,
Sunnyvale, CA, USA).
Plasmid constructs and luciferase
reporter assay
For the reporter constructs, the VEGF-A promoter
regions (−2,000 to +50 bp) were amplified from human genomic DNA
(Zyagen, San Diego, CA, USA) by PCR using iProof™ High-Fidelity DNA
Polymerase (Bio-Rad Laboratories, Inc.) (23). The PCR products were subcloned into
the pGL4.11[luc2P] vector (Promega Corporation, Madison, WI, USA)
upstream of a luciferase gene using appropriate restriction sites.
Putative transcription factor binding sites were identified with
the Transcription Element Search System (TESS) algorithm
(http://www.cbil.upenn.edu/downloads/).
The transcription factor binding to the VEGF-A
promoter was analyzed by luciferase assay as previously reported
(20,23). Briefly, MCF-7 cells (1×105)
were transfected in 24-well plates with 1 µg pGL4.11[luc2P]
luciferase reporter vector driven by the VEGF-A promoter, or with 2
µg control vector. For transfection of MCF-7 cells,
0.2×105 cells/well were seeded into a 24-well plate and
grown for 24–48 h. Cells were harvested 48 h post-transfection, and
efficiency was measured by qPCR, immunostaining and western
blotting.
Chromatin-immunoprecipitation (ChIP)
and co-immunoprecitation (Co-IP)
ChIP and Co-IP were performed as previously
described (20). For ChIP analysis to
identify the binding regions of NFAT5 and STAT3, ChIP-IT Express
kit (Active Motif, Inc., Carlsbad, CA, USA) was utilized according
to the manufacturer's protocol and as previously described
(23). Control immunoglobulin (Ig)Gs,
anti-NFAT5 or anti-STAT3 (1 µg) were used to immunoprecipitate the
DNA-protein complexes. Specific VEGF-A promoter regions were
amplified using PCR and resolved on 2% agarose gels. PCR cycling
conditions consisted of an initial denaturation step at 95°C for 3
min, followed by 35 cycles of 95°C for 30 sec, annealing at 60°C
for 30 sec and extension at 72°C for 1 min. For NFAT5 and STAT3
Co-IP, proteins obtained from MCF-7 cells were subjected to sodium
dodecyl sulfate polyacrylamide gel electrophoresis (4–12%
polyacrylamide gels) and immunoblotting. NFAT5 and STAT3 were
detected with rabbit anti-NFAT5 and rabbit anti-STAT3 antibodies,
respectively. The primary antibodies were diluted to 1:200 and
secondary antibodies were diluted to 1:1,000
Enzyme-linked immunosorbent assay
(ELISA)
The secretory extracellular VEGF-A in the cell
supernatant was quantitated by ELISA according to the
manufacturer's protocol (R&D Systems, Inc., Minneapolis, MN,
USA) (21). Due to the limitation of
the detection system, the supernatant was diluted 1:1,000 and
quantified with a standard curve using the manufacturer-provided
standards. Detection of the optical density value at 450 nm was
performed using EMax® Plus Microplate Reader (Molecular
Devices, LLC), and data analysis was conducted using SoftMax Pro
GxP software (version 5.4) provided by Molecular Devices, LLC.
Cell migration assay
The migratory ability of NHAECs following
stimulation with the supernatant isolated from various
MCF-7-experimental cell culture conditions was performed using
Cultrex® Cell Migration Assays (cat no. 3465-096-K;
R&D Systems, Inc.) according to the manufacturer's protocol.
Briefly, serum-starved NHAECs were placed in the top chamber, and
fresh 24-h supernatant collected from MCF-7 cells that had been
subjected to various experimental cell culture conditions was
placed in the bottom chamber, separated by a basement
membrane-coated separation membrane. The migration ability was
quantified following dissociation of the cells from the membrane
with the appropriate dissociation buffers (provided in the Cultrex
Cell Migration Assay) and colorimetrically analyzed at 595 nm.
Flow cytometry
The expression of cluster of differentiation (CD)31
on NHAECs was analyzed by flow cytometry using appropriated primary
and fluorescein isothiocyanate-labelled secondary antibodies
(25). Samples were analyzed using a
FACSCalibur/LSR II flow cytometer (BD Biosciences, Franklin Lakes,
NJ, USA), and cell sorting was performed with a FACSVantage cell
sorter (BD Biosciences). Data were analyzed using FACSDiva™
software (version 8.0.1; BD Biosciences). Gates were set according
to isotype controls.
Statistical analysis
Data were expressed as the mean ± standard error of
the mean from four independent experiments. Statistical differences
between the means were analyzed using a paired or unpaired
Student's t test. P<0.05 was considered to indicate a
statistically significant difference. All analyses were conducted
using Origin version 7.0 (OriginLab, Northampton, MA, USA) or
GraphPad version 5 software (GraphPad Software, Inc., LaJolla, CA,
USA).
Results
Enhanced expression of VEGF-A
following co-stimulation of MCF-7 cells with high sodium chloride
and sub-minimal IL-17 levels
Although VEGF was initially considered to be a
vascular permeability factor and an endothelial cell-specific
mitogen, multiple lines of evidence convincingly suggest its
critical role as an autocrine-signaling factor in solid tumors,
including breast cancer (22).
Previous studies have reported that IL-17 induces the expression of
VEGF-A in solid tumors (27). In line
with this evidence, preliminary experiments conduced by the present
authors on MCF-7 breast cancer cells demonstrated a significantly
higher expression of VEGF-A following stimulation with IL-17 at
concentrations >100 ng/ml, but minimal response (<20%) at
concentrations <10 ng/ml (data not shown). To determine the
specific role of high sodium chloride levels (referred to as high
salt) in a pro-inflammatory microenvironment, initial cell
viability studies were performed, which demonstrated <30% cell
viability at NaCl concentrations >0.25 M, while >95% cell
viability was maintained at 0.15 and 0.2 M NaCl in the culture
medium (data not shown). Therefore, in all the experiments
described in the present study, 0.2 M NaCl was used for high salt
condition, and 1 ng/ml IL-17 was used for sub-minimal IL-17
concentration.
As VEGF-A is a key inflammatory stress molecule and
biomarker, experiments were performed in the present study to
determine the role of high salt synergising with the
pro-inflammatory cytokine effect of IL-17 on the expression of
VEGF-A protein. For that purpose, the breast cancer cell line MCF-7
was treated with either high salt (0.2 M NaCl), IL-17 (1 ng/ml) or
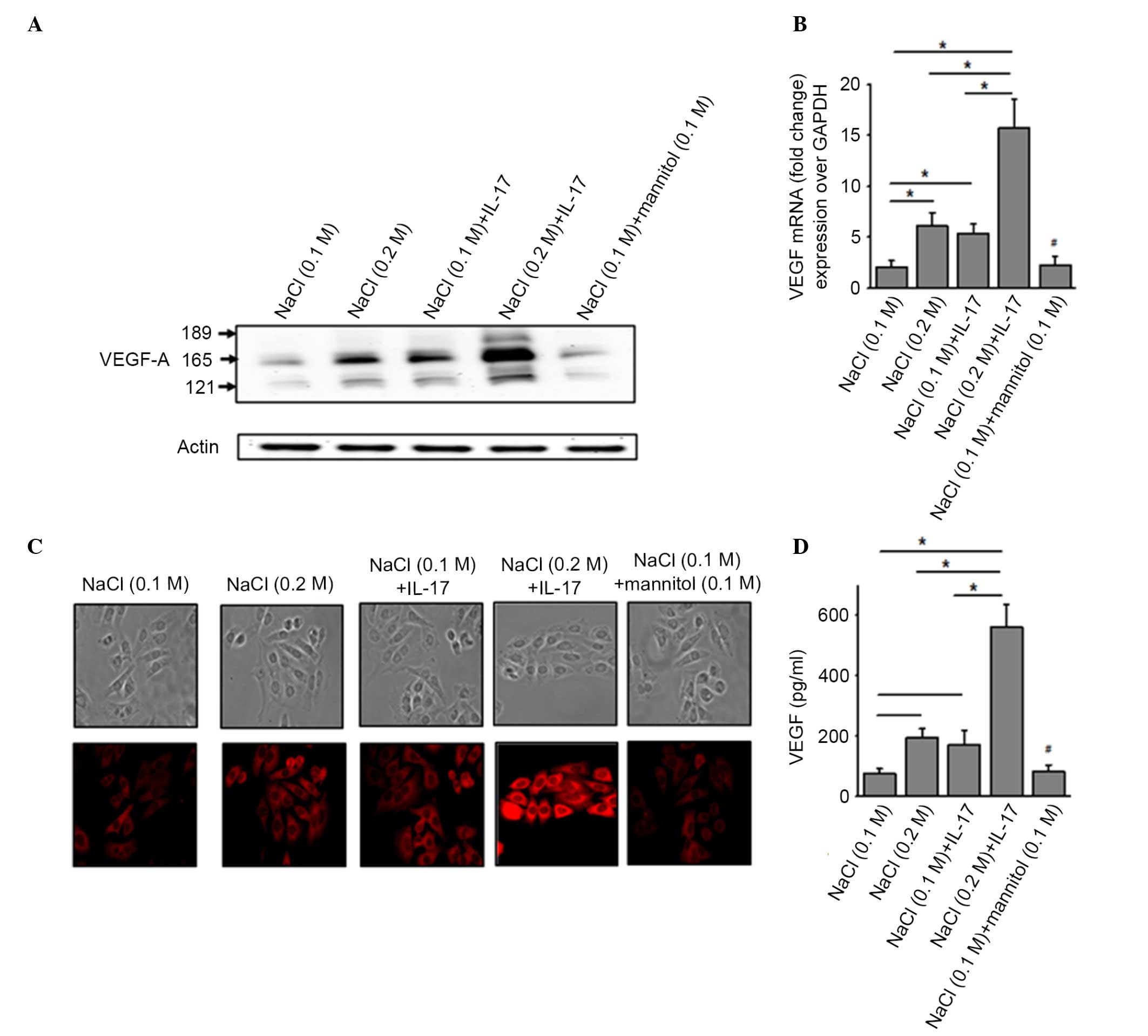
a combination of both. As shown in Fig.
1A, western blot analysis of the cellular extract from MCF-7
cells demonstrated an increased expression of VEGF-A following
co-treatment with high salt and sub-minimal IL-17. This was further
verified by intracellular immunocytochemical staining of MCF-7
cells for VEGF-A (Fig. 1B).
Quantitative mRNA analysis of VEGF-A (Fig. 1C) demonstrated that, under basal
conditions, there was minimal expression of VEGF-A mRNA transcript
(2.1±0.6-fold) compared with the GAPDH control transcript. However,
following treatment with either high salt or sub-minimal IL-17
alone, there was a significant elevation of VEGF-A mRNA
transcription (6.1±1.3 and 5.4±0.8-fold, respectively, P<0.05).
Co-treatment with both high salt and sub-minimal IL-17 demonstrated
a 15.7±2.9-fold increased VEGF-A transcript expression (P<0.05)
over the basal conditions. These data were further confirmed by
ELISA-based quantitative analysis of the supernatant from the
aforementioned conditions to measure the secretion of VEGF-A
protein by MCF-7 cells (Fig. 1D).
Importantly, treatment with equimolar mannitol concentrations (0.1
M NaCl + 0.1 M mannitol) demonstrated no enhanced expression of
VEGF-A (2.5±0.9-fold, P>0.70) over basal conditions. These data
indicate that high salt and IL-17 exert a synergistic effect.
Furthermore, as the synergistic effect was higher than the
individual conditions alone, these results strongly suggest a
possibility of two different signaling mechanisms separately
induced by high salt and IL-17.
Transcription factors NFAT5 and STAT3
induce VEGF-A expression following co-stimulation with high salt
and IL-17
Based on the aforementioned findings (Fig. 1), experiments were performed to
determine the transcription factors involved in the signaling
events mediated by high salt and IL-17 that led to increased
expression of VEGF-A. The present authors previously demonstrated
that STAT3 mediates IL-17-induced pro-inflammatory signaling
(23). NFAT5, also known as
tonicity-responsive enhancer binding protein (TonEBP), which is a
mediator of osmotic stress and immune regulation, has also been
observed to be upregulated in breast cancer (28). Therefore, various transcription
factors were initially analyzed by RT-qPCR, and NFAT5 and STAT3
demonstrated significantly enhanced expression following treatment
with high salt and IL-17. The other transcription factors tested
that did not induce a significant change under the current
experimental conditions were NFAT1-4 and STAT1, −2, −4 and −6 (data
not shown). Western blot and phosphoblot studies (Fig. 2A) demonstrated enhanced expression and
phosphorylation of NFAT5 in MCF-7 cells following treatment with
high salt (0.2 M NaCl) for 60 min, while stimulation with
sub-minimal IL-17 (1 ng/ml) enhanced the expression and
phosphorylation of STAT3. However, co-treatment with high salt and
sub-minimal IL-17 led to increased expression and phosphorylation
of both NFAT5 and STAT3. Quantitative analysis by RT-qPCR
demonstrated that there was a 3.4-fold upregulation of total and
phosphorylated NFAT5 (Fig. 2B and C)
following treatment with high salt, which did not increase further
upon co-treatment with both high salt and sub-minimal IL-17.
Similarly, there was a 3-fold upregulation in total and
phosphorylated STAT3 levels (Fig. 2D and
E) following treatment with sub-minimal IL-17, which did not
increase further upon co-treatment with high salt and sub-minimal
IL-17. These data support the earlier assertion that high salt and
IL-17 potentially induce VEGF-A expression through two different
signaling pathways.
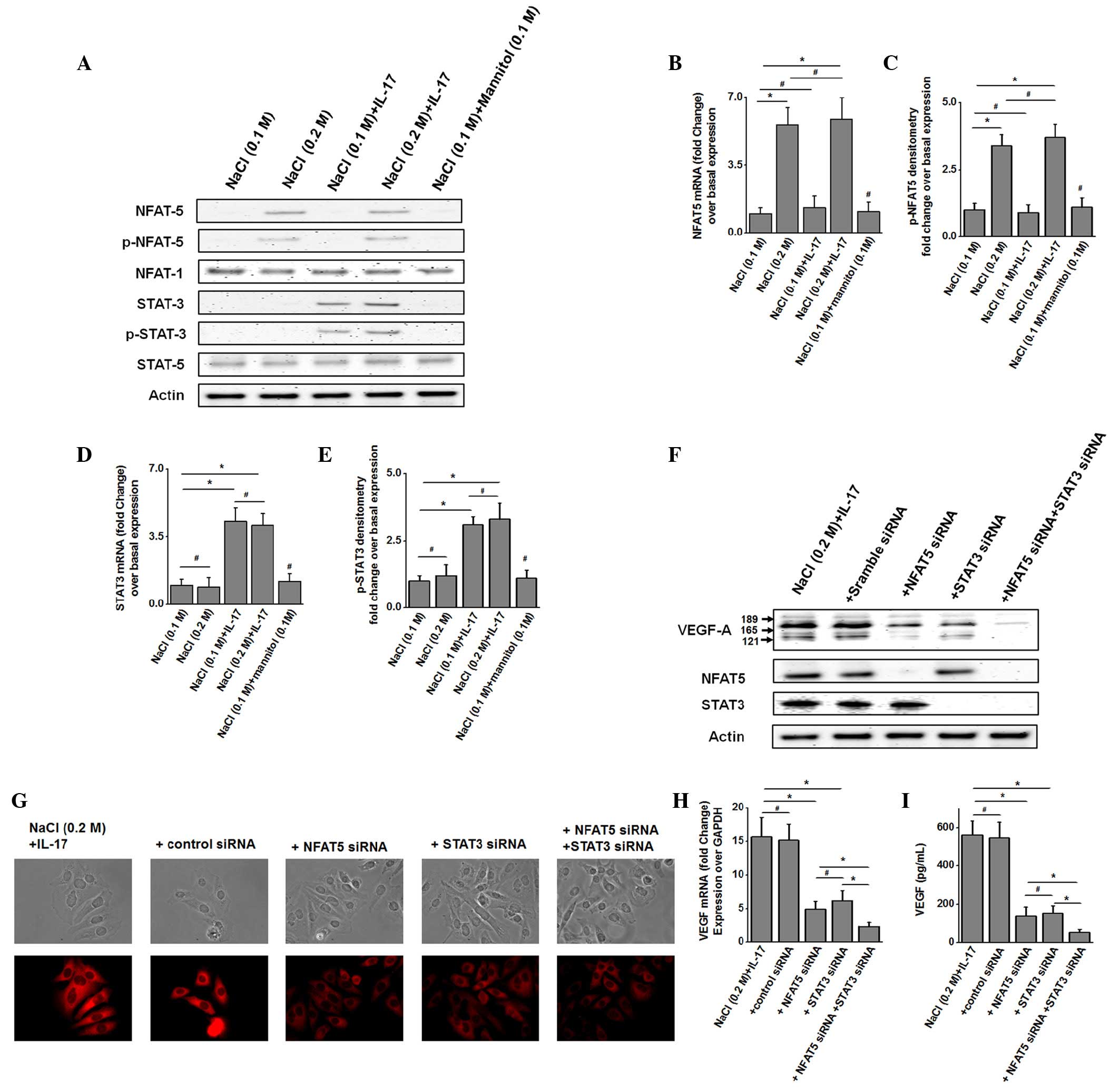 | Figure 2.NFAT5 and STAT3 transcription factors
were induced in MCF-7 cells following stimulation with high salt
and sub-minimal IL-17, respectively. (A) Western blot analysis was
performed to determine the protein expression levels of NFAT5 (170
kDa), p-NFAT5 (170 kDa), NFAT1 (140 kDa), STAT3 (86 kDa), p-STAT3
(86 kDa), STAT5 (92 kDa) and actin (43 kDa), both total and active
phosphorylated forms, after 30 min of stimulation with high salt,
sub-minimal IL-17 or both. Quantitative mRNA expression of (B)
NFAT5, (C) STAT3, (D) p-NFAT5 and (E) p-STAT3 by RT-qPCR was
analyzed after 60 min of stimulation with high salt, sub-minimal
IL-17 or both. Quantitation was performed with the
2−ΔΔCq method normalized to GADPH expression. (F)
Western blot analysis of VEGF-A expression in MCF-7 cells following
stimulation with high salt and IL-17, along with NFAT5 and STAT3
knock-down by specific siRNA. Scrambled siRNA was used as negative
control. Specific NFAT5 and STAT3-siRNA treatment resulted in
decreased expression of NFAT5 and STAT3 transcription factors,
respectively. (G) Immunocytostaining of MCF-7 cells for VEGF-A
following treatment with both high salt and sub-minimal IL-17, and
specific siRNA knock-down of NFAT5 and STAT3. (H) VEGF-A mRNA
expression was analyzed by RT-qPCR in MCF-7 cells following
stimulation with high salt and IL-17, along with NFAT5 and STAT3
knock-down by specific siRNA. Quantitation was performed with the
2−ΔΔCq method normalized to GADPH expression. (I)
Enzyme-linked immunosorbent assay-based analysis of VEGF-A in the
supernatant collected from MCF-7 cells following stimulation with
high salt and IL-17, along with NFAT5 and STAT3 knock-down by
specific siRNA. Data are represented as mean values ± standard
error of the mean from four independent experiments. Statistical
significance was analyzed by the Student's t test, *P<0.05;
#P>0.05. VEGF, vascular endothelial growth factor;
mRNA, messenger RNA; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; IL, interleukin; NFAT, nuclear factor of activated
T-cells; STAT, signal transducer and activator of transcription;
p-, phosphorylated; siRNA, small interfering RNA; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
To confirm that high salt and IL-17 induce separate
signaling pathways that result in the induction of VEGF-A
expression, siRNA knockdown experiments specific for NFAT5 and
STAT3 were performed. Western blot analysis (Fig. 2F) demonstrated that siRNA knock-down
of NFAT5 and STAT3 individually, following co-treatment with high
salt and sub-minimal IL-17, led to reduced intracellular expression
of VEGF-A. As expected, combined knock-down of both NFAT5 and STAT3
synergistically diminished VEGF-A expression. This was further
verified by immunocytochemical analysis (Fig. 2G). Quantitative ELISA-based analysis
of VEGF-A secretion (Fig. 2I)
demonstrated that NFAT knock-down reduced VEGF expression to 137±49
pg/ml, while STAT3 knock-down reduced VEGF expression to 152±41
pg/ml in high salt and sub-minimal IL-17-co-treated MCF-7 cells,
where VEGF-A expression was 562±73 pg/ml prior to treatment. Taken
together, these data confirm that high salt and IL-17 induce two
different signaling mechanisms leading to enhanced expression of
the inflammatory stress molecule VEGF-A.
NFAT5 and STAT3 bind to the VEGF-A
promoter region at −1,471 and −840 bp upstream of the open reading
frame
To determine the role of high salt and IL-17 in
VEGF-A gene transcription, the luciferase reporter construct
containing the −2,000 to +50 bp region of the human VEGF-A gene
promoter was transfected into MCF-7 breast cancer cells. These
luciferase-transfected cells were treated with either high salt,
sub-minimal IL-17 or both, and luciferase activity was recorded. An
increase in VEGF-A reporter activity was observed following
individual stimulation with high salt (7.2-fold, Fig. 3A) and sub-minimal IL-17 (6.4-fold,
Fig. 3B) over control null-luciferase
vector-transfected cells. Further co-treatment with both high salt
and sub-minimal IL-17 produced a 25.2-fold increase (Fig. 3C) in luciferase activity compared with
untreated cells. These data were in line with the results described
in Fig. 2, thus supporting the
hypothesis that high salt and IL-17 work synergistically to enhance
VEGF-A expression.
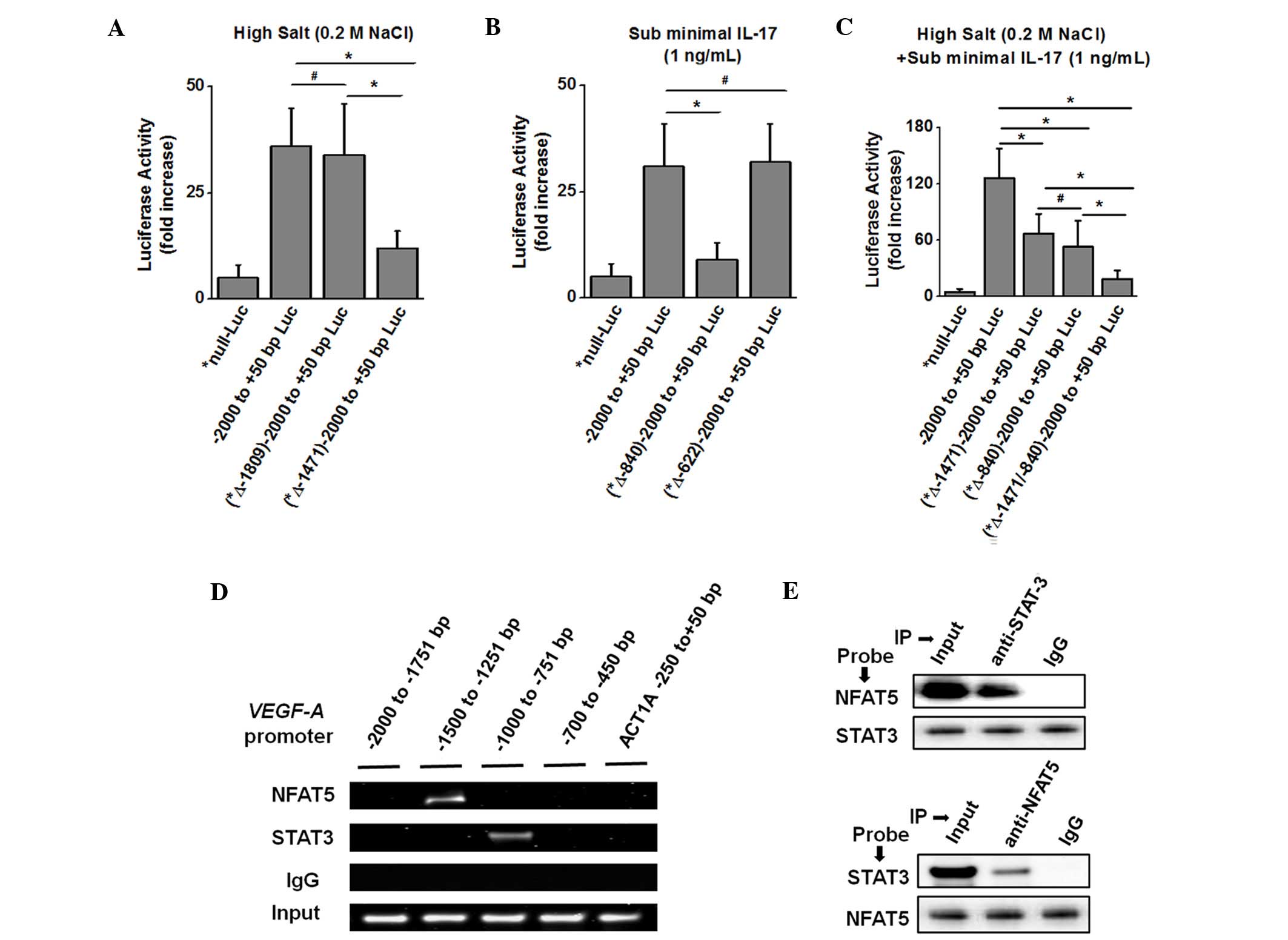 | Figure 3.Binding sites of the transcription
factors NFAT5 and STAT3 on the VEGF-A promoter to induce downstream
gene transcription. (A) Transcription Element Search System-based
computational analysis of −2,000 bp upstream of the VEGF-A open
reading frame. Putative binding domains for NFAT5 and STAT3 at
−1,809, −1,471, −840 and −622 bp were predicted to contain
consensus sequences on the VEGF-A promoter. MCF-7 cells were
transfected with a luciferase reporter driven by constructs of the
VEGF-A promoter (−2,000 to +50 bp; mutation *Δ-1,812-11 bp/-2,000
to +50 bp; and mutation *Δ-1,474-73 bp/-2,000 to +50 bp) construct,
and stimulated with high salt. (B) MCF-7 cells were transfected
with luciferase reporters driven by constructs of the VEGF-A
promoter (−2,000 to +50 bp; mutation *Δ-843-42 bp/-2,000 to +50 bp;
and mutation *Δ-625-24 bp/-2,000 to +50 bp) construct, and
stimulated with IL-17. (C) MCF-7 cells were transfected with
luciferase reporters driven by constructs of the VEGF-A promoter
(−2,000 to +50 bp; mutation *Δ-1,474-73 bp/-2,000 to +50 bp;
mutation *Δ-843-42 bp/-2,000 to +50 bp; or both mutations)
construct, and stimulated with high salt and IL-17. Luciferase
activity was measured 48 h after transfection and normalized to a
Renilla luciferase internal control. The numbers indicate
fold-change over the control vector. (D) VEGF-A promoter with
binding sites for NFAT5 and STAT3. The four black horizontal bars
represent the regions amplified by polymerase chain reaction with
specific primers for the −2,000 to −1,751 bp, −1,500 to −1,251 bp,
−1,000 to −751 bp and −700 to −450 bp regions of the VEGF-A
promoter, and the negative control −250 to +50 bp region of the
ACT1a promoter. Chromatin was immunoprecipitated with anti-NFAT5,
anti-STAT3 or isotype control immunoglobulin G antibodies from
MCF-7 cells following stimulation with both high salt and IL-17.
The first three lanes represent chromatin-immunoprecipitation,
while the fourth lane represents input chromatin. (E)
Co-immunoprecitation of the protein-complex extracted by anti-STAT3
and anti-NFAT5 antibodies, and western blot analysis to probe with
the opposite antibody (upper panel, probed with NFAT5 antibody and
protein complex pulled-down with anti-STAT3 antibody; lower panel,
probed with STAT3 antibody and protein complex pulled-down with
anti-NFAT5 antibody). Data are represented as mean values ±
standard error of the mean from four independent experiments.
Statistical significance was analyzed with the Student's t test,
*P<0.05; #P>0.05. Luc, luciferase; IL,
interleukin; ACT1a, actin-1a; NFAT, nuclear factor of activated
T-cells; STAT, signal transducer and activator of transcription;
Ig, immunoglobulin; VEGF, vascular endothelial growth factor. |
To specifically identify the putative DNA binding
sequences for NFAT5 and STAT3 on the VEGF-A promoter, a
computational analysis of the −2,000 to +50 bp region of the VEGF-A
promoter was performed using TESS. This analysis identified two
putative DNA binding sites for NFAT5 (TGGAAA at −1,471 and −1,809
bp) and two putative DNA binding sites for STAT3 (TTCCCAAA/TTTCCAAA
at −840 and −622 bp) (Fig. 3A). To
determine whether NFAT5 and STAT3 regulate VEGF-A expression in
MCF-7 cells, these cells were transfected with the mutant VEGF-A
promoter reporter construct and treated with high salt, IL-17 or
both (Fig. 3A-C). As shown in
Fig. 3A, following treatment with
high salt, the putative NFAT5 binding site mutant construct
(*-1,474–73, GA to *TC) exhibited a significant decrease (≤66% loss
of activity) in reporter activity compared with the native VEGF-A
reporter activity. However, the other putative NFAT5 binding site
mutant construct (*-1,812–11, GA to *TC) did not display any change
in VEGF-A promoter activity, thus suggesting that the NFAT5 binding
domain is located at −1,471 bp on the VEGF-A promoter. Similarly,
following treatment with sub-minimal IL-17 (Fig. 3B), the putative STAT3 (TTCCCAAA or
TTTCCAAA) binding mutant construct (*-843–842, CA to *TG) exhibited
a significant decrease (≤71% loss of activity) in reporter activity
compared with the native VEGF-A reporter activity, while the other
putative STAT3 binding mutant construct (*-625-24, CA to *TG) did
not exhibit any change in VEGF-A promoter activity, thus suggesting
that the putative STAT3 binding domain is located at −1,471 bp. As
expected, these two mutants demonstrated the highest loss of
activity following co-treatment with high salt and sub-minimal
IL-17 (Fig. 3C), thus strongly
suggesting a synergistic mechanism of action between NFAT5 and
STAT3 transcription factors in regulating VEGF-A expression.
To determine whether NFAT5 and STAT3 bind to
adjacent DNA binding sites on the VEGF-A promoter to form
transcriptional-activation DNA-protein complexes, following high
salt, IL-17 or combined treatment, MCF-7 cells were
immunoprecipitated with anti-NFAT5, anti-STAT3 or control IgGs. The
results of ChIP and PCR analysis using primers specific for the
VEGF-A promoter regions −2,000 to −1,751 bp, −1,500 to −1,251 bp,
−1,000 to −751 bp, −700 to −450 bp, and control primers for the
−250 to +50 bp region of the actin promoter (Fig. 3E, black bars), demonstrated that NFAT5
and STAT3 bind to the VEGF-A promoter at the −1,500 to −1,251 bp
and −1,000 to −751 bp regions, respectively (Fig. 3D). The DNA binding pattern of NFAT5
and STAT3 strongly correlates with the locations of the consensus
binding sites on the VEGF-A promoter determined by luciferase
reporter activity (Fig. 3A-C). ChIP
with control IgGs did not enrich VEGF-A promoter regions,
demonstrating the specificity for these transcription factors. No
binding was observed in PCRs conducted with primers specific for
the −250 to +50 bp region of the actin promoter, which lacks these
binding sites (Fig. 3D). Furthermore,
the protein-protein complexes were immunoprecipitated (Fig. 3E) and probed with anti-STAT3 or
anti-NFAT5 antibodies on western blotting. This supported the ChIP
findings that NFAT5 and STAT3 were complexed together on the VEGF-A
promoter. Taken together, these data clearly demonstrate that the
transcription factors NFAT5 and STAT3 synergistically interact and
are part of a larger transcription-regulatory complex enhancing the
VEGF-A gene expression.
Enhanced cell migration following
activation of NHAECs with VEGF-A supernatant
As VEGF-A has been implicated in tumor cell
migration, the potential upregulation of cell migration and surface
proteins that mediate cell migration in NHAECs was determined
(29). MCF-7 cells were
pre-stimulated for 48 h with high salt, sub-minimal IL-17 or both,
and later washed. Supernatant collected from 24 h post-stimulation
cultured cells under basal medium conditions was used to determine
the migration efficiency of NHAECs. Cell migration was studied
utilizing a migration chamber with NHAECs coated in the top chamber
with serum-starved medium, while the bottom chamber was filled with
supernatant collected from 24 h post-stimulated MCF-7 cells. As
shown in Fig. 4A and B, following
treatment of NHAECs for 24 h with supernatant of MCF-7 cells
subjected to combined pre-stimulation with high salt and IL-17,
there was increased migration of NHAECs [optical density (OD),
0.62±0.19] compared with individual stimulation with only high salt
(OD, 0.28±0.09) or sub-minimal IL-17 (OD, 0.21±0.07). It is
important to note that there was a significant inhibition of NHAEC
migration when blocked with anti-VEGF-A monoclonal IgG2b antibody
(1:1,000 dilution; cat no. MAB293; R&D Systems) addition to the
supernatant collected from high salt and IL-17-co-treated MCF-7
cells. This supports the hypothesis that cell migration was
mediated by VEGF-A secreted from pre-stimulated MCF-7 cells.
Furthermore, analysis of the expression of the migratory molecules
VCAM (Fig. 4C), β1 integrin (Fig. 4D) and CD31 (Fig. 4E) demonstrated enhanced expression of
these migratory molecules in NHAECs upon treatment with supernatant
from pre-stimulated MCF-7 cells with both high salt and sub-minimal
IL-17. These findings confirm that high salt synergised with the
pro-inflammatory cytokine-mediated expression of the stress factor
VEGF-A.
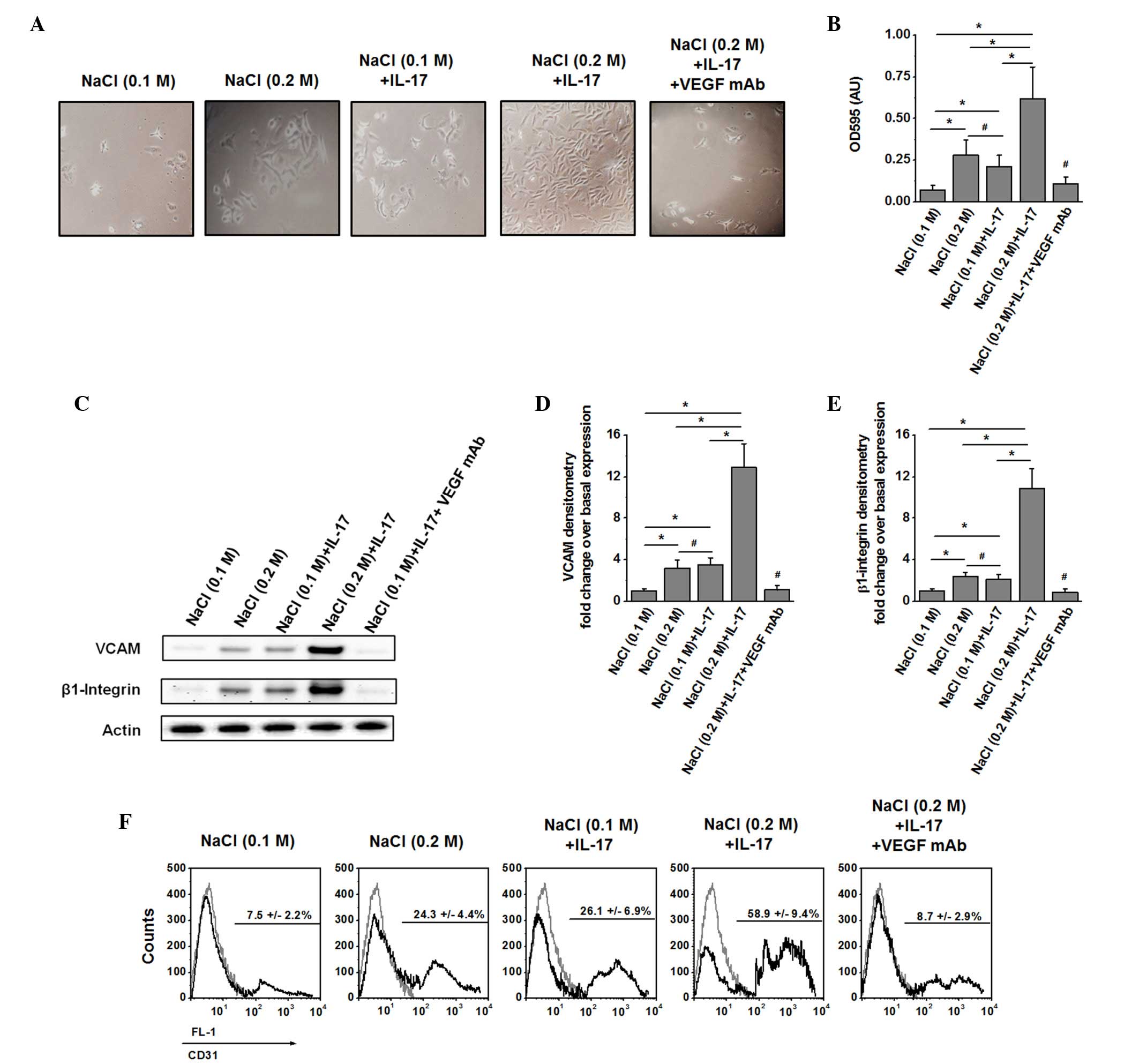 | Figure 4.Induction of cell migration and
migration-specific protein expression following activation of
NHAECs with VEGF-A-rich supernatant. (A) Cell migration analysis
after 24 h of culture in the migration chambers. The top chamber
was coated with NHAECs placed in serum-starved medium. The lower
chamber was filled with fresh 24-h supernatant from MCF-7 cells
following pre-stimulation for 48 h with high salt, IL-17 or both.
To determine the specific role of VEGF-A, in the fifth panel, the
lower chamber was filled with supernatant (as described above) with
both high salt and IL-17, along with VEGF-A neutralization with
immunoglobulin G2b monoclonal antibody at 1:1,000 dilution. (B)
Optical density at 595 nm of the cells collected from the bottom
chamber following 24 h of culture (C) Western blot analysis of
NHAECs probed for VCAM (110 kDa) and β1 integrin (130 kDa)
following treatment with supernatant from MCF-7 cells stimulated
with high salt, IL-17 or both for 24 h. (D) Densitometry analysis
of the western blotting results for VCAM normalized to basal
expression. (E) Densitometry analysis of the western blotting
results for β1 integrin normalized to basal expression. (F) Flow
cytometry analysis of the expression of CD31 on NHAECs subjected to
the stimulation conditions mentioned above. FL-1 refers to the
single fluorescence channel maintained for
phosphatidylethanolamine-labeled probing of CD31. The isotype
control is represented by the grey line, while CD31 is represented
by the black line. Data are presented as mean values ± standard
error of the mean from four independent experiments. Statistical
significance was analyzed by the Student's t test, *P<0.05;
#P>0.05. IL, interleukin; VEGF, vascular endothelial
growth factor; mAb, monoclonal antibody; OD, optical density; AU,
arbitrary unit; VCAM, vascular cell adhesion protein; CD, cluster
of differentiation; NHAECs, normal human aortic endothelial
cells. |
Discussion
Although high salt has been traditionally associated
with cardiovascular diseases, recent evidence suggests that high
salt levels also increase the risk of cancer (13). The tumor microenvironment is known to
have high concentrations of several inflammatory cytokines,
including IL-6, IL-17 and TNFα (30).
High salt is a potent inducer of chronic inflammatory response
through the activation of reactive oxygen and nitrogen species
(31). Furthermore, chronic
inflammation is considered a key initiator for several solid
tumors, including breast cancer (2).
As early as in the 1980s, high salt and sodium transporters in the
tumor tissue were suggested to play a critical role in breast
cancer progression and metastasis (17). More recently, a direct inflammatory
effect of salt on immune cells such as T-cells and macrophages has
been reported (32,33). However, the exact molecular mechanisms
by which high salt induces inflammation in the tumor
microenvironment are not yet defined.
The role of osmotic stress in inflammatory processes
has been extensively suggested in the scientific literature
(34). Culture of human peripheral
blood cells under hyperosmotic conditions (330–410 mOsm/kg
H2O) has been shown to induce expression of
pro-inflammatory cytokines such as IL-1 and IL-8 (35). Notably, patients with Crohn's disease,
an inflammatory bowel disease, present with substantially higher
osmolality of the faecal fluid compared with control subjects (490
vs. 340 mOsm/kg H2O), which strongly correlates with the
intestinal histopathological score (36). In the tumor microenvironment, osmotic
stress is considered to trigger receptor tyrosine kinases such as
epidermal growth factor receptor, resulting in activation of the
erbB-2/neu proto-oncogene (37). All
the above lines of evidence suggest that hypernatremia or high
sodium induce osmotic stress, which in-turn leads to an
inflammatory response. The current study attempted to evaluate the
direct inflammatory effect of high salt on hyperosmolarity-induced
events (by performing control equimolar mannitol studies), in order
to demonstrate that high salt in the cell microenvironment could
directly induce expression of VEGF, a known inflammatory biomarker
(10). However, the current results
do not completely preclude a high salt-mediated,
osmotic-stress-induced VEGF expression, which requires further
studies.
Although VEGF was originally discovered as a
vascular and endothelial-activating factor, several studies have
conclusively suggested its critical role in tumor cell progression
and metastasis (10). Several
isoforms of VEGF have been reported, including VEGF121,
VEGF165 and VEGF189, (38). In addition, cell-specific VEGF
isoforms, including VEGF-A (present in the majority of cell types)
(10), VEGF-B (present in neurons and
retina) (39) and VEGF-C (present in
macrophages) (40) have been
reported. It is important to note that anti-VEGF monoclonal
antibody-based therapeutic strategies are currently employed to
treat cancers (41). It has also been
suggested that direct stimulation of tumor cells by VEGF may
inhibit apoptosis and increase their resistance to conventional
chemotherapy and radiotherapy administered in cancer treatment
(42). Therefore, understanding the
mechanisms of expression of VEGF is crucial for anti-cancer
therapeutic success. It has been well documented that VEGF exerts
autocrine and paracrine pro-cancer effects in breast cancer through
activation of cancer-specific AKT/phosphoinositide 3-kinase
signaling mechanisms (43). In the
present study, the potential role of high salt in the presence of
pro-inflammatory cytokines towards induction of VEGF has been
investigated. Notably, the present results support the previous
reports that inflammatory stimulus induces VEGF expression in MCF-7
breast cancer cells (Fig. 1).
Therefore, the present authors propose a novel role for high salt
in the tumor microenvironment as an important mediator of VEGF
expression. Furthermore, this expressed VEGF was observed to be
critical for endothelial cell migration (Fig. 4). Taken together, the present data
suggest that high salt plays a key inflammatory role in
VEGF-mediated cancer cell proliferation.
The transcription factor NFAT5 is the most recently
identified member of the NFAT family (44). Originally, NFAT5 was termed TonEBP or
osmotic response element-binding protein (34). In humans, previous studies addressing
the function of NFAT5 were primarily focused on the kidney medulla,
since renal cells are physiologically exposed to highly elevated
interstitial osmolalities (34).
NFAT5 is considered to exert an osmoprotective function in the
kidney (33). However, a number of
studies supported the notion that NFAT5 is important in immune
responses and lymphocyte activation (45). A recent study by Remo et al
(28), utilizing in silico
modeling and gene expression analysis on breast cancer patients
(n=197), reported an enhanced expression of NFAT5 in inflammatory
breast cancer. In line with that study, the present findings have
demonstrated that high salt induces VEGF expression through
specific upregulation of the transcription levels of NFAT5
(Figs. 2 and 3), a known key molecule in breast cancer
(27). In addition, the present study
identified a putative NFAT5 binding domain on the VEGF-A promoter
at −1,471 bp upstream of the VEGF-A coding gene (Fig. 3). These data clearly suggest that high
salt mediates a direct inflammatory response in the cancer
microenvironment.
The present study has demonstrated that high salt
synergises with IL-17, a pro-inflammatory cytokine, towards the
expression of the inflammatory molecule VEGF-A (27). The role of IL-17 in cancer progression
is well established. Several reports, including a previous study by
the present authors, have demonstrated that IL-17 exerts its
inflammatory response through activation of the STAT3 transcription
factor signaling pathway (23). STATs
comprise a family of cytoplasmic transcription factors that mediate
intracellular signaling, which is usually generated by membrane
receptor-ligand interactions (45).
Numerous studies have demonstrated constitutive activation of STAT3
in a wide variety of human tumors (46). There is an increasing body of evidence
suggesting that aberrant STAT3 signaling promotes initiation and
progression of human cancers (46).
Suppression of STAT3 activation has been reported to induce tumor
cell death by apoptosis (45). In
line with previous reports, the present authors have identified a
putative STAT3 binding domain (Fig.
3) at −840 bp upstream of the VEGF-A coding gene. Furthermore,
the results of the luciferase reporter assay conducted in the
present study strongly suggest that high salt exerts its
synergistic effect with the pro-inflammatory cytokine IL-17 through
activation of two independent but synchronous signaling pathways
via NFAT5 and STAT3, in order to induce the expression of VEGF-A.
However, the current study has limitations due to the experimental
use and analysis of only one breast cancer cell line. Nonetheless,
the authors consider that the conclusions of the present study are
applicable to other solid tumors, which requires further validation
in other tumor cell lines (particularly breast cancer), along with
animal model studies.
In conclusion, in spite of significant advances,
current anti-cancer therapies have a great scope for improvement
(6). The current data suggest an
important role for NFAT5 and STAT3 signaling in high salt-mediated
cancer cell proliferation and migration. The authors propose a
low-salt diet, and supplementing anti-VEGF therapy with anti-NFAT5
and anti-STAT3 therapies, as a future direction for efficient
cancer therapy. However, further clinical and basic biomedical
studies are required to validate these observations.
Acknowledgements
The present study was supported by the Vanderbilt
University (Nashville, TN, USA) and Tennessee State University
(Nashville, TN, USA) Cancer Partnership (National Institutes of
Health, Bethesda, MA, USA; grant no. 5U54CA163066). The authors
would like to thank the members of the Department of Biological
Sciences of Tennessee State University for their kind support on
the present project.
References
|
1
|
Balkwill F and Mantovani A: Inflammation
and cancer: Back to Virchow? Lancet. 357:539–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Colotta F, Allavena P, Sica A, Garlanda C
and Mantovani A: Cancer-related inflammation, the seventh hallmark
of cancer: Links to genetic instability. Carcinogenesis.
30:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hussain SP, Hofseth LJ and Harris CC:
Radical causes of cancer. Nat Rev Cancer. 3:276–285. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dranoff G: Cytokines in cancer
pathogenesis and cancer therapy. Nat Rev Cancer. 4:11–22. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang SH, Mirabolfathinejad SG, Katta H,
Cumpian AM, Gong L, Caetano MS, Moghaddam SJ and Dong C: T helper
17 cells play a critical pathogenic role in lung cancer. Proc Natl
Acad Sci USA. 111:5664–5669. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He D, Li H, Yusuf N, Elmets CA, Athar M,
Katiyar SK and Xu H: IL-17 mediated inflammation promotes tumor
growth and progression in the skin. PLoS One. 7:e321262012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gaffen SL: Structure and signalling in the
IL-17 receptor family. Nat Rev Immunol. 9:556–567. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goel HL and Mercurio AM: VEGF targets the
tumour cell. Nat Rev Cancer. 13:871–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chung J, Bachelder RE, Lipscomb EA, Shaw
LM and Mercurio AM: Integrin (alpha 6 beta 4) regulation of eIF-4E
activity and VEGF translation: A survival mechanism for carcinoma
cells. J Cell Biol. 158:165–174. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stephenson GD and Rose DP: Breast cancer
and obesity: An update. Nutr Cancer. 45:1–16. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Strnad M: Salt and cancer. Acta Med
Croatica. 64:159–161. 2010.(In Croatian). PubMed/NCBI
|
|
14
|
Zhang F, Tang Z, Hou X, Lennartsson J, Li
Y, Koch AW, Scotney P, Lee C, Arjunan P, Dong L, et al: VEGF-B is
dispensable for blood vessel growth but critical for their
survival, and VEGF-B targeting inhibits pathological angiogenesis.
Proc Natl Acad Sci USA. 106:6152–6157. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mozaffari MS, Patel C, Ballas C and
Schaffer SW: Effects of excess salt and fat intake on myocardial
function and infarct size in rat. Life Sci. 78:1808–1813. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ketonen J and Mervaala E: Effects of
dietary sodium on reactive oxygen species formation and endothelial
dysfunction in low-density lipoprotein receptor-deficient mice on
high-fat diet. Heart Vessels. 23:420–429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sparks RL, Pool TB, Smith NK and Cameron
IL: Effects of amiloride on tumor growth and intracellular element
content of tumor cells in vivo. Cancer Res. 43:73–77.
1983.PubMed/NCBI
|
|
18
|
Tsugane S, Sasazuki S, Kobayashi M and
Sasaki S: Salt and salted food intake and subsequent risk of
gastric cancer among middle-aged Japanese men and women. Br J
Cancer. 90:128–134. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tiriveedhi V, Upadhya GA, Busch RA, Gunter
KL, Dines JN, Knolhoff BL, Jia J, Sarma NJ, Ramachandran S,
Anderson CD, et al: Protective role of bortezomib in steatotic
liver ischemia/reperfusion injury through abrogation of MMP
activation and YKL-40 expression. Transpl Immunol. 30:93–98. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sarma NJ, Tiriveedhi V, Crippin JS,
Chapman WC and Mohanakumar T: Hepatitis C virus-induced changes in
microRNA 107 (miRNA-107) and miRNA-449a modulate CCL2 by targeting
the interleukin-6 receptor complex in hepatitis. J Virol.
88:3733–3743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tiriveedhi V, Takenaka M, Sarma NJ, Gelman
AG and Mohanakumar T: Anti-major histocompatibility complex-induced
obliterative airway disease: Selective role for CD4 and CD8 T cells
in inducing immune responses to self-antigens. J Heart Lung
Transplant. 32:714–722. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barr MP, Gray SG, Gately K, Hams E, Fallon
PG, Davies AM, Richard DJ, Pidgeon GP and O'Byrne KJ: Vascular
endothelial growth factor is an autocrine growth factor, signaling
through neuropilin-1 in non-small cell lung cancer. Mol Cancer.
14:452015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Amara S, Lopez K, Banan B, Brown SK,
Whalen M, Myles E, Ivy MT, Johnson T, Schey KL and Tiriveedhi V:
Synergiztic effect of pro-inflammatory TNFα and IL-17 in periostin
mediated collagen deposition: Potential role in liver fibrosis. Mol
Immunol. 64:26–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tiriveedhi V, Gelman AE and Mohanakumar T:
HIF-1α signaling by airway epithelial cell K-α1-tubulin: Role in
fibrosis and chronic rejection of human lung allografts. Cell
Immunol. 273:59–66. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tiriveedhi V, Takenaka M, Ramachandran S,
Gelman AE, Subramanian V, Patterson GA and Mohanakumar T: T
regulatory cells play a significant role in modulating MHC class I
antibody-induced obliterative airway disease. Am J Transplant.
12:2663–2674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rao X, Huang X, Zhou Z and Lin X: An
improvement of the 2^(−delta delta CT) method for quantitative
real-time polymerase chain reaction data analysis. Biostat
Bioinforma Biomath. 3:71–85. 2013.PubMed/NCBI
|
|
27
|
Chung AS, Wu X, Zhuang G, Ngu H, Kasman I,
Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, et al: An
interleukin-17-mediated paracrine network promotes tumor resistance
to anti-angiogenic therapy. Nat Med. 19:1114–1123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Remo A, Simeone I, Pancione M, Parcesepe
P, Finetti P, Cerulo L, Bensmail H, Birnbaum D, Van Laere SJ,
Colantuoni V, et al: Systems biology analysis reveals NFAT5 as a
novel biomarker and master regulator of inflammatory breast cancer.
J Transl Med. 13:1382015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Zang QS, Liu Z, Wu Q, Maass D,
Dulan G, Shaul PW, Melito L, Frantz DE, Kilgore JA, et al:
Regulation of VEGF-induced endothelial cell migration by
mitochondrial reactive oxygen species. Am J Physiol Cell Physiol.
301:C695–C704. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu H, Ouyang W and Huang C: Inflammation,
a key event in cancer development. Mol Cancer Res. 4:221–233. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Azak A, Huddam B, Gonen N, Yilmaz SR,
Kocak G and Duranay M: Salt intake is associated with inflammation
in chronic heart failure. Int Cardiovasc Res J. 8:89–93.
2014.PubMed/NCBI
|
|
32
|
Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S,
Kishi Y, Regev A and Kuchroo VK: Induction of pathogenic TH17 cells
by inducible salt-sensing kinase SGK1. Nature. 496:513–517. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jantsch J, Schatz V, Friedrich D, Schröder
A, Kopp C, Siegert I, Maronna A, Wendelborn D, Linz P, Binger KJ,
et al: Cutaneous Na+ storage strengthens the antimicrobial barrier
function of the skin and boosts macrophage-driven host defense.
Cell Metab. 21:493–501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Neuhofer W: Role of NFAT5 in inflammatory
disorders associated with osmotic stress. Curr Genomics.
11:584–590. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shapiro L and Dinarello CA: Hyperosmotic
stress as a stimulant for proinflammatory cytokine production. Exp
Cell Res. 231:354–362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schilli R, Breuer RI, Klein F, Dunn K,
Gnaedinger A, Bernstein J, Paige M and Kaufman M: Comparison of the
composition of faecal fluid in Crohn's disease and ulcerative
colitis. Gut. 23:326–332. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
King CR, Borrello I, Porter L, Comoglio P
and Schlessinger J: Ligand-independent tyrosine phosphorylation of
EGF receptor and the erbB-2/neu proto-oncogene product is induced
by hyperosmotic shock. Oncogene. 4:13–18. 1989.PubMed/NCBI
|
|
38
|
Pagès G and Pouysségur J: Transcriptional
regulation of the vascular endothelial growth factor gene-a concert
of activating factors. Cardiovasc Res. 65:564–573. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Poesen K, Lambrechts D, Van Damme P,
Dhondt J, Bender F, Frank N, Bogaert E, Claes B, Heylen L, Verheyen
A, et al: Novel role for vascular endothelial growth factor (VEGF)
receptor-1 and its ligand VEGF-B in motor neuron degeneration. J
Neurosci. 28:10451–10495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wiig H, Schröder A, Neuhofer W, Jantsch J,
Kopp C, Karlsen TV, Boschmann M, Goss J, Bry M, Rakova N, et al:
Immune cells control skin lymphatic electrolyte homeostasis and
blood pressure. J Clin Invest. 123:2803–2815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Harmey JH and Bouchier-Hayes D: Vascular
endothelial growth factor (VEGF), a survival factor for tumour
cells: Implications for anti-angiogenic therapy. Bioessays.
24:280–283. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gorski DH, Beckett MA, Jaskowiak NT,
Calvin DP, Mauceri HJ, Salloum RM, Seetharam S, Koons A, Hari DM,
Kufe DW and Weichselbaum RR: Blockage of the vascular endothelial
growth factor stress response increases the antitumor effects of
ionizing radiation. Cancer Res. 59:3374–3378. 1999.PubMed/NCBI
|
|
43
|
Pidgeon GP, Barr MP, Harmey JH, Foley DA
and Bouchier-Hayes DJ: Vascular endothelial growth factor (VEGF)
upregulates BCL-2 and inhibits apoptosis in human and murine
mammary adenocarcinoma cells. Br J Cancer. 85:273–288. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mancini M and Toker A: NFAT proteins:
Emerging roles in cancer progression. Nat Rev Cancer. 9:810–820.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Berga-Bolaños R, Drews-Elger K, Aramburu J
and López-Rodríguez C: NFAT5 regulates T lymphocyte homeostasis and
CD24-dependent T cell expansion under pathologic hypernatremia. J
Immunol. 185:6624–6635. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View Article : Google Scholar : PubMed/NCBI
|