Introduction
Pheochromocytoma is a catecholamine-secreting tumor
that originates from chromaffin cells. The majority of these tumors
arise in the adrenal medulla (1), and
when they occur outside the adrenal gland they are known as an
extra-adrenal pheochromocytoma or a paraganglioma (2). Pheochromocytoma is primarily sporadic,
but may additionally be associated with multiple endocrine
neoplasia syndrome (3) and mutations
of mitochondrial complex 2 succinate dehydrogenase enzymes
(4).
Extra-adrenal pheochromocytoma has been previously
reported to account for ~10% of all pheochromocytoma cases during
adulthood and for 30–40% of cases in children (5). These tumors may occur in the superior
para-aortic region, the para-adrenal area and the inferior
para-aortic region, as well as the organ of Zuckerkandl and the
urinary bladder (6–8). Pancreatic localization of extra-adrenal
pheochromocytoma is rare, and to the best of our knowledge, only
one case has been previously reported in the Chinese literature
(9). In the present study, the cases
of three patients (Table I) with
pheochromocytoma involving the pancreas are presented. To the best
of our knowledge, the present report is the first to present three
definite and comprehensive cases of this rare clinical phenomenon.
In addition, the relevant literature is reviewed, with the aim of
improving the understanding of pheochromocytoma, with an emphasis
on sharing patient experiences of diagnosis and surgical
treatment.
 | Table I.Clinical features of three patients
with pancreatic pheochromocytoma. |
Table I.
Clinical features of three patients
with pancreatic pheochromocytoma.
| Variable | Case 1 | Case 2 | Case 3 |
|---|
| Gender | Female | Male | Female |
| Age, years | 55 | 44 | 30 |
| Hypertension | No | No | No |
| Family history | No | No | No |
| Chief complaint | Abdominal pain | Abdominal pain | Neoplasm detected on
routine physical examination |
| Tumor location | Uncinate process | Uncinate process | Pancreatic tail |
| Tumor size, cm | 2.5 | 3.2 | 6 |
| Surgery | Local resection | Local resection | Laparotomy |
| Complications | No | Pancreatic
fistula | Mortality |
| Patient outcome | Alive | Alive | Succumbed |
Case report
Case 1
A 55-year-old female was admitted to the People's
Hospital of Jiangyou (Mianyang, China) in February 2013 with the
symptom of recurrent abdominal pain over a period of >3 years.
On admittance, the patient did not exhibit any weight loss, changes
in appetite or bowel habits, jaundice or non-specific symptoms
associated with hypertension, including palpitations, sweating,
headache, dizziness or fainting. The patient's physical examination
and pre-operative routine examinations did not identify any
significant abnormalities. Serum tumor markers for cancer antigen
19-9, cancer antigen 125 and carcinoembryonic antigen (CEA) were
additionally observed to be within the normal ranges. The patient
underwent ultrasound (US) and computed tomography (CT; LightSpeed
VCT CT system; GE Healthcare Life Sciences, Chalfont, UK), which
revealed a mass in the uncinate process of the pancreas, with a
diameter of ~2.5 cm. Due to suspicions of a pancreatic malignant
tumor, an exploratory laparotomy was performed, which confirmed the
location of the mass in the uncinate process of the pancreas, with
a relatively clear association with the surrounding tissues. A
local resection of the pancreatic tumor was subsequently
successfully performed. Resected tissue was formalin (Beijing Noble
Ryder Technology Co., Ltd., Beijing, China)-fixed, paraffin
(Beijing Noble Ryder Technology Co., Ltd.)-embedded and cut into
4-µm sections. For histological examination, the tissue sections
were stained with hematoxylin and eosin (Haibiao Technology, Co.,
Ltd., Xiamen, China) and observed under a microscope (CX31; Olympus
Coropration, Tokyo, Japan). Pathologists were able to
histologically diagnose the tumor as pancreatic paraganglioma
(extra-adrenal pheochromocytoma originating from the pancreas). The
tumor cells were polygonal, had abundant cytoplasm and contained
basophilic or amphotropic particles The nuclei were round or
irregular, and the tumor cells were arranged in cords or nests.
According to the introperative and histological findings, the tumor
was 2.5 and 3 cm in diameter, respectively. There was no evidence
of distant metastasis, and no direct tissue or arterial or venous
invasion. For immunohistochemical analysis, tissue sections were
incubated with monoclonal mouse anti-human chromogranin A (CgA;
cat. no. ZA0507; 1:50; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China), monoclonal mouse
anti-human synaptophysin (Syn; cat. no. M077629; 1:100; Dako,
Glostrup, Denmark), S-100 (cat. no. ZA0507; 1:100; Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd.), monoclonal rabbit
anti-human vimentin (cat. no. M072529; 1:50; Dako) and monoclonal
mouse anti-human CEA (cat. no. ZA0507; 1:100; Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd.) antibodies at 4°C for 12 h
and visualized under a microscope (CX31; Olympus Corporation).
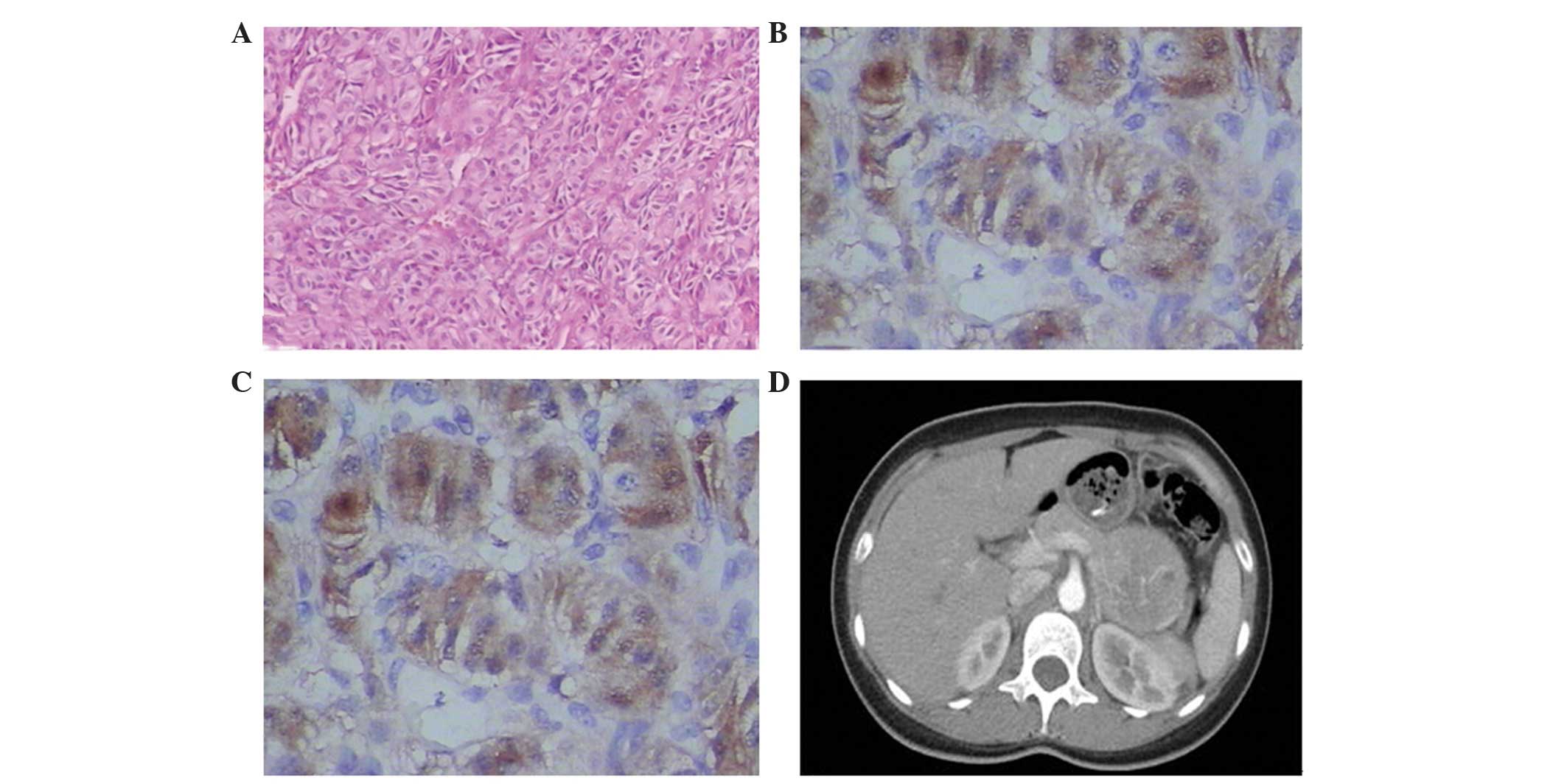
Immunohistochemical examination (Fig. 1A
and B) revealed that the tumor was positive for S-100, CgA,
Syn, vimentin and CEA expression. The patient recovered well
following the procedure and was discharged 11 days
post-surgery.
Case 2
A 44-year-old man, who had experienced continuous
upper abdominal pain for 6 months was admitted to the People's
Hospital of Jiangyou in March 2012. The patient demonstrated no
notable abnormalities on physical examination and serum biochemical
tests. In addition, the patient did not have a history of
hypertension or a remarkable family history. CT scanning revealed a
soft-tissue lesion located in the pancreatic uncinate process,
measuring 3.2×3 cm, together with a normal pancreatic duct and
common bile duct. The presence of the lesion was confirmed by
magnetic resonance imaging (MRI; GE 1.5T Signa HDe; GE Healthcare
Life Sciences). Due to concerns over the presence of an undetected
malignancy, a diagnostic laparotomy was performed, which confirmed
that the mass was located in the uncinate process of the pancreas
without distorting the surrounding tissues. During surgery,
fast-frozen pathological testing of the resected mass indicated a
tumor-like lesion, which was suspected to be a high-grade
pancreatic neuroendocrine tumor or cystic adenoma. A histological
diagnosis of pancreatic pheochromocytoma was confirmed by
pathologists following successful local resection of the pancreatic
tumor. The tumor cells of the resected tissue were positive for
S-100, Syn, CgA and inhibin expression (Fig. 1C). The patient experienced the
post-operative complication of a pancreatic fistula, which was
treated with drug therapy (0.1 mg sandostatin subcutaneously
injected 3 times/day for 7 days) and percutaneous drainage, which
prolonged the duration of the patient's hospital stay. The patient
was subsequently discharged almost 2 months after surgery.
Case 3
A 30-year-old woman was admitted the West China
Hospital of Sichuan University (Chengdu, China) in August 2011 due
to the positive identification of a neoplasm in the pancreatic tail
during routine abdominal US. The patient did not exhibit any
typical symptoms associated with this disease, and had no history
of hypertension. A physical examination and tumor-associated marker
evaluation revealed no abnormalities. Following admission to
hospital, abdominal enhanced CT detected a mass, ~6 cm in size,
situated in the tail of the pancreas (Fig. 1D). Considering the rapid growth of the
tumor and its potential malignancy, a laparotomy was suggested and
subsequently performed. However, during surgery, the patient's
heart rate and blood pressure suddenly increased to 140 beats/min
and 180/110 mmHg, respectively, as surgeons attempted to dissect
the neoplasm. The patient's vital signs were unstable and
uncontrollable, which led the surgeons to hypothesize that the
tumor may be a pheochromocytoma of the pancreas. Following an
emergency consultation, the planned surgery was halted and the
patient was transferred immediately to the intensive care unit. The
patient's post-surgical blood plasma test revealed noradrenaline
and adrenaline levels of 318,306 and 346,940 ng/l, respectively
(normal range, <174,357 and <60,104 ng/l, respectively),
which confirmed the diagnosis of a pheochromocytoma of the
pancreas. Due to an unstable post-operative heart rate and blood
pressure, the patient succumbed to heart failure.
Discussion
Owing to excessive secretion of catecholamines,
pheochromocytoma may cause life-threatening symptoms, including
hypertension, cardiac arrhythmias, headaches and palpitations
(1). However, clinical manifestations
of this tumor are variable and non-specific, and in certain cases,
the clinical symptoms are not clear. It has been reported that ~70%
of extra-adrenal pheochromocytoma cases are non-functional and only
~30% of patients may manifest with different presentations;
however, the number of patients who do not present with typical
symptoms is increasing (10,11).
Due to variable and non-specific manifestations, a
diagnosis of extra-adrenal pheochromocytoma is difficult and
dependent on clinician experience (1,2,5). In general, a pre-operative diagnosis of
extra-adrenal pheochromocytoma is established by taking into
account the presence of clinical signs, the determination of
catecholamines and their metabolites in blood or urine, and the
presence of a mass in the extra-adrenal region. Imaging
examinations (US, CT and MRI) are primarily used for locating the
lesion, and additionally provide reference values for final
histological diagnosis. As previously mentioned, pheochromocytoma
derived from the pancreas is seldom observed. Guo et al
(9) reported the first pancreatic
pheochromocytoma in the Chinese literature: The patient was
40-year-old women that had a mass on the left-upper side of the
abdomen for >1 month. The patient did not have abdominal pain,
distension or discomfort, or a history of trauma, fever, headache,
dizziness or high blood pressure. A gigantic pheochromocytoma was
identified in the tail of pancreas, and the patient underwent
surgery, from which the patient recovered well. The three patients
in the present study did not have a history of hypertension nor the
typical symptoms associated with this disease. The tumors of the
first two patients were located in the uncinate process of the
pancreas and were successfully resected. No significant changes in
blood pressure or heart rate were detected during surgery, and the
diagnosis of a pancreatic pheochromocytoma was primarily based on
the pathological and immunohistochemical examination of the
resected tissues following surgery. The third patient did not
experience any discomfort, but presented with a positive indication
of a neoplasm situated in the tail of the pancreas, for which a
distal resection was planned as a treatment strategy. However, the
surgery was halted intraoperatively due to a sudden increase in the
patient's blood pressure and heart rate. Due to a marked
post-operative increase in noradrenaline and adrenaline levels in
the patient's blood plasma, a diagnosis of pheochromocytoma located
in the pancreas was established.
Pheochromocytoma originates in chromaffin cells
derived from primitive neural crest cells, which may react and
stain with chromic salts (8).
Furthermore, pheochromocytoma or paraganglioma in the pancreas, to
a certain extent, is a type of pancreatic neuroendocrine tumor
(pNET), which means that the tumor cells of resected tumors may
demonstrate positive reactions for CgA and Syn following
immunohistochemical staining (12).
With regard to the first two cases presented here, this
pathological feature is of significant value for histological
diagnosis of pancreatic pheochromocytoma.
Surgery is the only curative treatment for pNET and
pancreatic pheochromocytoma (13). If
pheochromocytoma of the pancreas is suspected, surgery should be
suggested. Prior to this, pre-operative assessments should be
managed for a certain period of time in case of sudden large
increases in blood pressure and heart rate. Exploratory laparotomy
is a method of diagnosis, but if there are not enough pre-operative
preparations, surgery may lead to sudden hypertension resulting in
patient mortality, as occurred in the third case reported in the
present study. For surgeons who encounter an unexpected
hypertensive crisis during pancreatic tumor surgery, an undiagnosed
pheochromocytoma of the pancreas should be immediately considered.
The surgery should be halted, and appropriate preparations should
then be made for a safer second attempt at the surgery.
References
|
1
|
Werbel SS and Ober KP: Pheochromocytoma.
Update on diagnosis, localization, and management. Med Clin North
Am. 79:131–153. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sandur S, Dasgupta A, Shapiro JL, Arroliga
AC and Mehta AC: Thoracic involvement with pheochromocytoma: A
review. Chest. 115:511–521. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neumann HP, Berger DP, Sigmund G, Blum U,
Schmidt D, Parmer RJ, Volk B and Kirste G: Pheochromocytomas,
multiple endocrine neoplasia type 2, and von Hipple-Lindau disease.
N Engl J Med. 329:1531–1538. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gimm O, Armanios M, Dziema H, Neumann HP
and Eng C: Somatic and occult germ-line mutations in SDHD, a
mitochondrial complex II gene, in nonfamilial pheochromocytoma.
Cancer Res. 60:6822–6825. 2000.PubMed/NCBI
|
|
5
|
Atiyeh BA, Barakat AJ and Abumrad NN:
Extra-adrenal pheochromocytoma. J Nephrol. 10:25–29.
1997.PubMed/NCBI
|
|
6
|
Schwartz EL, Mao P, Hernried O, Born EE
and Waldmann EB: Catecholamine-secreting paraganglioma. The problem
of classification. Arch Intern Med. 135:978–985. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stackpole RH, Melicow MM and Uson AC:
Pheochromocytoma in children. Report of 9 cases and review of the
first 100 published cases with follow-up studies. J Pediatr.
63:314–330. 1963.PubMed/NCBI
|
|
8
|
Madani R, Al-Hashmi M, Bliss R and Lennard
TW: Ectopic pheochromocytoma: Does the rule of tens apply? World J
Surg. 31:849–854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo XY, Wang XJ and Yang JY: A gigantic
pheochromocytoma in the tail of the pancreas. He Bei Yi Yao.
13:16282009.(In Chinese).
|
|
10
|
Birrenbach T, Stanga Z, Cottagnoud P and
Stucki A: Unexpected metastatic pheochromocytoma - an unusual
presentation. Eur J Intern Med. 19:60–62. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Parmer RJ and Zinder O: Catecholaminergic
pathways, chromaffin cells, and human disease. Ann NY Acad Sci.
971:497–505. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Panzuto F, Severi C, Cannizzaro R, Falconi
M, Angeletti S, Pasquali A, Corleto VD, Annibale B, Buonadonna A,
Pederzoli P and Delle Fave G: Utility of combined use of plasma
levels of chromogranin A and pancreatic polypeptide in the
diagnosis of gastrointestinal and pancreatic endocrine tumors. J
Endocrinol Invest. 27:6–11. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Halfdanarson TR, Rabe KG, Rubin J and
Petersen GM: Pancreatic neuroendocrine tumors (PNETs): Incidence,
prognosis and recent trend toward improved survival. Ann Oncol.
19:1727–1733. 2008. View Article : Google Scholar : PubMed/NCBI
|