Introduction
Cancer is a complex disease demanding improvisation
in the therapeutic avenues for improved efficacy and survival rate
(1). The main reason for the
increasing incidence of cancer is the aging of the worldwide
population. The most documented as well as confirmed causative
agents for cancer included smoking, overweight, viral infections
and lifestyle habits including lack of physical activity (2). According to global cancer statistics, it
is estimated that approximately 14.1 million new cancer cases and
8.2 million mortalities occurred in 2012 worldwide (3).
Lung cancer is the main cause of cancer associated
with mortality among men, and is superior to the incidence of
breast cancer in women (4). For lung
cancer, the primary and main risk factor is the use of tobacco. For
more advanced stages of disease or inoperable tumors, radiotherapy
(RT) with or without chemotherapy (i.e., mainly using cisplatin,
carboplatin, paclitaxel, pemetrexed) remains the main treatment
option, although for EGFR-mutated cases and cases with EML4-ALK
translocation-targeted agents are approved for treatment (5). In non-resectable, locally advanced lung
cancer as well as in cases with metastatic disease (approximately
50% of all diagnosed lung cancer cases), chemotherapy remains the
only available treatment option (6).
Thus, ways to improve the chemotherapeutic response of SCLC are
required, especially for the therapy of refractory cases where the
second-line treatment options are limited.
Previous findings have identified molecular
alterations within human cells that lead to malignant
transformation, termed as cancer hallmarks (7). Additionally, information and concepts
regarding the origin of cancer are also documented in the
literature, but little progress has been made in exploiting
etiology and the mechanisms of disease (8). Most of the anticancer therapies which
constitute the main treatment options, including chemotherapy, were
in fact developed decades ago, when the development of therapeutics
was not yet supported and driven by detailed knowledge of the
genetic, molecular and biochemical, and cellular mechanisms of
cancer pathogenesis (9). More recent
cancer therapy approaches, such as small-molecular-weight drugs or
monoclonal antibodies targeting aberrant growth factor receptors
and RNA interference or gene therapeutics involving the utilization
of viral vectors, have been developed with the aim to target cancer
pathogenesis, but only few percent of these new solutions have been
transferred from the lab to the clinic (10,11).
Although studies have focused on developing new
targeted therapies, conventional ways of treating cancer play a key
role in the clinic. Thus, surgery remains the main treatment of
choice, whenever possible, as it has the highest chance for
complete cure. If surgery is not an option, chemotherapy and/or RT
are considered. Ionizing radiation and most conventional
chemotherapeutic agents cause DNA damage and have a more severe
effect on rapidly proliferating cells. Nevertheless, neither of
these treatment modalities is able to distinguish between tumor and
normal cells, resulting in significant normal tissue toxicity.
Therefore, the development of new strategies, which can be more
accurate in treatment delivery or dose delivery in case of RT, and
which selectively can sensitize tumor cells to enhance the efficacy
of chemotherapy or RT, is imperative and is the main subject theme
of the present study.
DNA damage response (DDR)
Every day each cell of the human body is exposed to
tens of thousands of DNA lesions. Such an amount of DNA damage,
their recognition and repair processes influence cell processes by
inhibition of the progression of the cell cycle, replication or
transcription. When DNA damage is not correctly repaired or left
unrepaired, it leads to the establishment of mutations in the DNA
sequence of the cell or may even cause more serious genomic
aberrations, such as deletions, translocations and aneuploidy,
resulting in genomic instability, which is dangerous for the cell
and the whole organism and may also increase risk of cancer
(12). Some DNA damage appears as a
consequence of physiological processes, e.g., DNA replication,
hydrolytic or non-enzymatic reactions or reactive oxygen species
formation by oxidative respiration, products of lipid peroxidation
or by macrophages or neutrophils during infections and inflammation
(13). DNA damage may also be a
result of environmental agents, such as physical factors, including
ultraviolet (UV) light, ionizing radiation generated during
radioactive compound decay or in therapeutic settings of tumors,
but also after exposure to chemical factors, i.e., cancer-causing
DNA-damaging chemicals, such as those found in cigarette smoke or
aflatoxins in contaminated food (14). Both endogenous processes and exogenous
factors, which attack DNA, lead to the formation of diverse DNA
damage, such as base modifications or loss, DNA interstrand
crosslinks or DNA single- or double-strand breaks (DNA SSBs and
DSBs). This diverse DNA damage can lead to alteration in the DNA
sequence and, thus, DNA rearrangements and/or loss of genetic
information and may therefore cause genomic instability (15). The cell response to DNA damage
includes inhibition of the cell cycle, which allows for repair of
the damage, or leads to the induction of cell death if the damage
cannot be correctly repaired. Of note, inappropriate DNA repair may
cause cell transformation, which in the whole organism can result
in tumor formation, premature aging or inherited defects (13).
In order to counteract this DNA damage and maintain
genomic integrity, cells develop several defense mechanisms known
as the DNA damage response (DDR), and which consist of multiple
signaling networks (16). These
networks involve the detection of DNA lesions and signal
transducers with the help of sensors. These signal transducers in
turn transmit information of the presence of DNA damage, and
downstream effector molecules, which mediate cell cycle arrest,
localized chromatin remodeling, and the promotion of DNA repair
(17–21). Thus, if the DNA damage is correctly
repaired, DDR signaling is inactivated, the cell cycle restarts and
the cell survives. When DNA lesions are not correctly repaired or
cannot be eradicated, persistent DDR signaling causes cell
inactivation by either death (apoptosis) or by senescence, a form
of permanent cell cycle blockade, both of which have antitumor
potential (22,23).
DDR signaling in cancer and as a target for
cancer therapy
DDR as a barrier against cancer. During DDR, cells
make decision to repair inflicted DNA damage or to descend to
death. Thus, DDR is considered as the first barrier against the
malignant process (24). Loss of
genetic stability, which is a hallmark of tumorigenesis (25), is driven by DNA damage and errors that
are incorporated during DNA repair (26). In addition, tumors often harbor
genetic/epigenetic defects that, consequently impair factors in DDR
signaling pathways, i.e., p53, ATM, Chk2, γH2AX, causing further
activation of proto-oncogenes and inhibition of tumor suppressor
genes, respectively (27). Studies on
DDR have revealed plenty of links between oncogenesis and inherited
changes in the genome. Additionally, cells defective in DDR/DNA
repair mechanisms generally present increased sensitivity towards
DNA damaging agents and this leads to a high probability of
cancer.
The role of DDR in the protection against cancer
development is also supported by the reported genetic defects in
some DDR components and the increased cancer incidence in
individuals carrying these aberrations. One example is defects in
the NER components in Xeroderma pigmentosum (XP) syndrome. The XP
syndrome is associated with impaired capacity to repair point
mutations such as those inflicted by UV and, accordingly,
individuals with XP deficiency have a 1,000-fold higher probability
of incidence of UV-induced skin cancer and increased
neurodegeneration and premature aging (28). Other inherited human syndromes linked
to DDR, which are rare diseases, are chromosome aberrations in ATM
in Ataxia telangiectasia (AT), in MRE11 in AT-like disorder, or in
NBS1 in Nijmegen breakage syndrome (NBS). Patients with these
syndromes have a higher predisposition to cancer (especially
lymphomas), immunodeficiency, radiation hypersensitivity, and often
also neurological complications and premature ageing (29–31).
Syndromes connected with defects in HR repair include hereditary
breast/ovarian cancers caused by defects in BRCA1 and BRCA2
(32), but also cancer prone
chromosomal instabilities, i.e., the Werner, Bloom and Rothmund
Thomson syndromes, with involved RecQ-like helicases (33). Mouse models with deficiency in HR and
NHEJ give severe mutant phenotypes, indicating the importance of
the two DNA DSB repair mechanisms (34).
DDR signaling as a target for cancer therapy.
Overexpression or loss of specific factors in DNA repair machinery,
result in altered functions of HR and NHEJ repair pathways in
tumors (35). Deregulations in DNA
repair promote genomic instability and malignancy but tumor cells
survive with the help of their acquired potentials to survive DNA
damage inflicted by chemotherapy and RT (36). Therefore, inhibition of DDR and/or DNA
repair pathways has become an attractive strategy to overcome
resistance to DNA damaging therapy and small DNA repair inhibitors
have been developed to be used as a single-agent therapy or, more
often, in tandem with DNA damaging treatments (37–39).
Abrogation of cell cycle checkpoints
Inactivation of the tumor suppressor p53 (40), by chromosomal aberration (deletion),
inactivating mutation or overexpression of p53-negative regulator,
MDM2, results in impaired G1 checkpoint control of tumors.
Accordingly, tumor cells with inactivation of p53 function depend
on S and G2 phase checkpoints to repair DNA damage and survive
(41). Based on these observations,
one of the strategies to overcome altered function of DDR in tumors
is an abrogation of the remaining, intact checkpoints leading to
enhanced tumor cell death (42).
Thus, results from preclinical studies have shown that abrogation
of the S and G2 checkpoints with small molecule inhibitors,
specific enzymes, and RNA interference towards Chk1 (43), may impair the DNA repair response to
DNA damaging chemotherapy in a tumor selective way (44), which also have formed the basis for
clinical trials with Chk1/2 inhibitors (45).
PARP inhibitors (PARPi). The pharmacological
inhibition of the DNA repair pathways may significantly increase
the cell death-inducing capacity of DNA damaging agents.
Combinations of cytotoxic agents with DNA repair inhibitors are
under preclinical investigations or have already been introduced to
the clinic as ongoing clinical trials. However, some aspects
including normal tissue reaction to inhibition of DNA repair, and
selectivity of the potential drugs against carcinogenesis including
DNA repair pathways are involved in tumor cell response to therapy,
but also tumor DNA repair pathway redundancy when exposed to
certain chemotherapeutic drugs (46).
One of the more advanced DNA repair inhibiting
strategies is to attack PARP-1, a component of base-excision repair
of apurinic sites (47,48). Additionally, PARPi in combination with
temozolomide, platinum chemotherapy (cisplatin, carboplatin) are
now in clinical trials, but concerns are raised because of toxicity
of the combined treatment regimen towards normal cells (49). PARPi have also been tested as a
monotherapy. The rationale for this use of PARPi, is based on
synthetic lethality, a concept proposed by Helleday et al
(39), to be a genetic phenomenon in
which the combination of two otherwise non-lethal mutations lead to
a non-viable cell. Man-made destructive phenotypes are indicative
of an interaction between the products of the two mutant genes
within the cell. This concept was introduced when PAPRi were used
in patients with inherited breast and ovarian cancers that lacked
wild-type and carried mutated BRCA1 and/or BRCA2
genes, resulting in impaired HR repair and increased sensitivity to
PARP inhibition (50). The obtained
preclinical results have given support for clinical trials with
PARPi as monotherapy in breast and ovarian cancer patients carrying
BRCA1 or BRCA2 mutations. However, not all patients with BRCA
mutations respond to this new, targeted therapy and resistance to
such treatment is also reported (47). Of note, deficiency in other HR repair
proteins than BRCA, presents enhanced sensitivity to PARPi,
suggesting a broad spectrum of their utility, alone or even in
combination with other inhibitors (51,52).
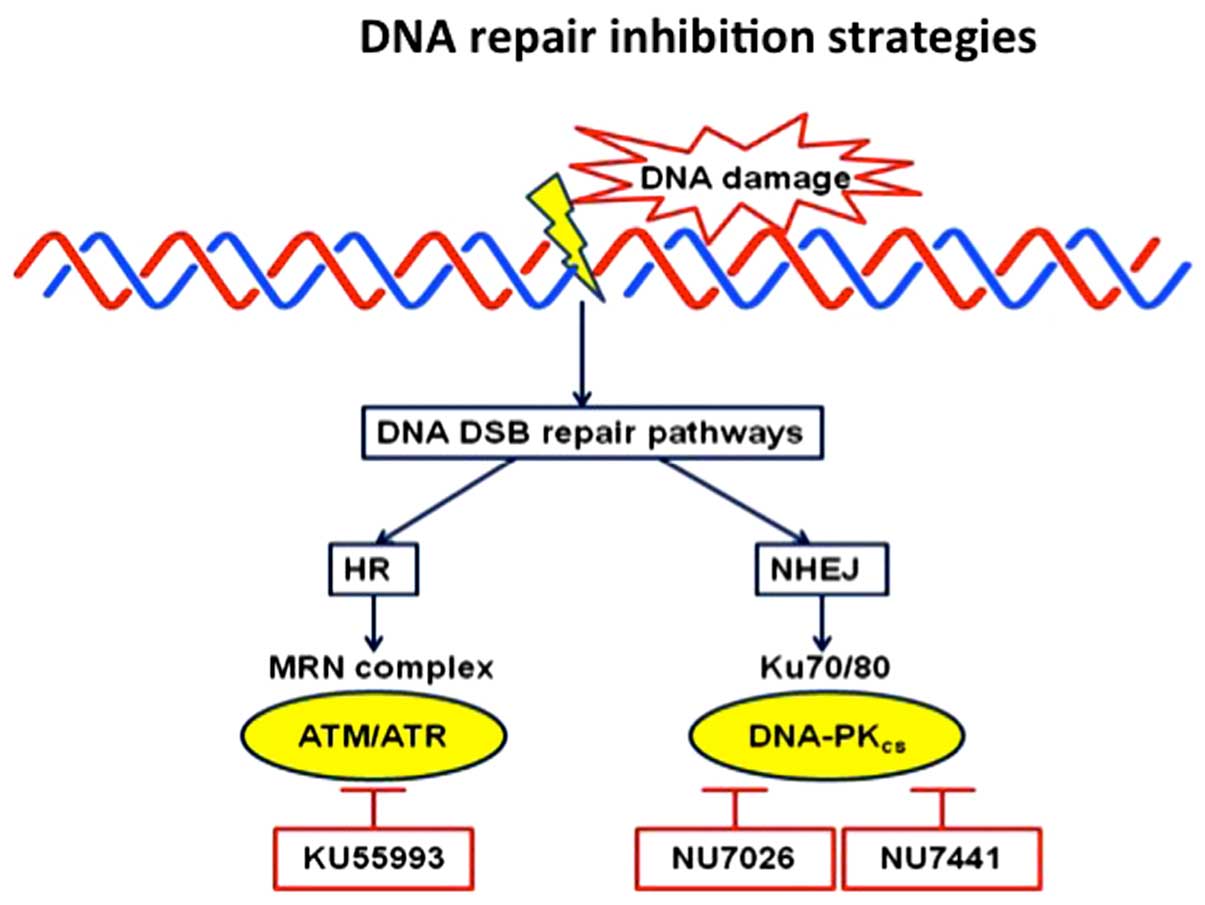
ATM and DNA-PK inhibitors. Inhibition of one of the
main kinases of the phosphatidylinositol 3-kinase-related protein
kinase family, ATM, which play a crucial role in the repair of DNA
DSBs, have also been tested. The rationale is that inhibition of
ATM may result in lack of proper detection of the DNA DSB inflicted
by the chemotherapy and, hence, they may accumulate to a level
leading the tumor cells towards cell death. Attempts have thus
generated small molecules, which in preclinical settings have been
shown to inhibit ATM kinase activity, e.g., KU55933 (AstraZeneca,
Cambridge, UK) (Fig. 1). DNA-PK also
plays a critical role in NHEJ-mediated repair and has been the
focus for small molecule inhibitor development (53). A number of candidates have been
generated, among them NU7441 and NU7026 (Fig. 1). These agents have shown some effect
as monotherapy (54,55). However, they have also been
demonstrated to sensitize tumor cells to DNA DSB-inducing
treatments, i.e., ionizing radiation and etoposide, a topoisomerase
II inhibitor, proving the concept of DNA-PK inhibition in tumor
treatment (56,57). Notably, induced hyper-activation of
DNA-PK causes a chemosensitizing effect in tumor cells (58). Thus, perturbations of DNA-PK kinase
activity, i.e., hypo- or hyper-activation/phosphorylation, may also
increase sensitivity of tumor to standard DNA damaging
treatment.
Epigenetics as a new tool to target DDR
signaling
Recently, a novel, promising approach was introduced
to cancer therapy and there are successful examples that targeting
of alterations in epigenetic signaling in tumor cells may be used
as therapy, as shown by the introduction of HDAC inhibitors (HDACi)
in hematological malignancies (59).
Epigenetic alterations have been shown to be involved in DDR
signaling, e.g., the (NAD+)-dependent histone deacetylase, SIRT1,
was reported to impair repair via the NHEJ pathway (60), SIRT6 was found to stabilize DNA-PK
associated with chromatin and in this way influence DNA DSB repair
(61). Additionally, HDAC1 and HDAC2
were reported to promote DSB repair (62). Previous studies also demonstrated that
HDACi applied in tandem with DNA damaging agents caused increased
cytotoxicity as a consequence of increased DNA damage and/or
impaired DNA repair capacity (63).
One such example is decitabine (2′-deoxy-5-azacytidine), a DNA
demethylating agent, which was combined in tests with
platinum-based drugs (i.e., cisplatin or carboplatin) to reverse
drug resistance in ovarian cancer patients in clinical trials
(64).
Conclusion
DDR signaling targeting therefore holds good
potential in enhancing sensitization in different therapeutic
avenues against cancer.
References
|
1
|
Wang VE, Grandis JR and Ko AH: New
strategies in esophageal carcinoma: Translational insights from
signaling pathways and immune checkpoints. Clin Cancer Res. Jul
01–2016.(Epub ahead of print). View Article : Google Scholar
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
National Cancer Institute, . Cancer
Statistics: Statistics at a Glance: The Burden of Cancer in the
United States. http://www.cancer.gov/statisticsMarch
14–2016
|
|
4
|
Bombardelli L and Berns A: The steady
progress of targeted therapies, promising advances for lung cancer.
Ecancermedicalscience. 10:6382016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Viktorsson K, Lewensohn R and Zhivotovsky
B: Systems biology approaches to develop innovative strategies for
lung cancer therapy. Cell Death Dis. 5:e12602014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morgensztern D, Campo MJ, Dahlberg SE,
Doebele RC, Garon E, Gerber DE, Goldberg SB, Hammerman PS, Heist
RS, Hensing T, et al: Molecularly targeted therapies in
non-small-cell lung cancer annual update 2014. J Thorac Oncol.
10(Suppl 1): S1–S63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weinberg RA: The Biology of Cancer. Sigrid
Masson and Alan Grose: Garland Science; New York, NY: 2007
|
|
10
|
De Palma M and Hanahan D: The biology of
personalized cancer medicine: Facing individual complexities
underlying hallmark capabilities. Mol Oncol. 6:111–127. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hanahan D: Rethinking the war on cancer.
Lancet. 383:558–563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rajagopalan H and Lengauer C: Aneuploidy
and cancer. Nature. 432:338–341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hoeijmakers JH: Genome maintenance
mechanisms for preventing cancer. Nature. 411:366–374. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Friedberg EC, McDaniel LD and Schultz RA:
The role of endogenous and exogenous DNA damage and mutagenesis.
Curr Opin Genet Dev. 14:5–10. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kastan MB: DNA damage responses:
Mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial
Award Lecture. Mol Cancer Res. 6:517–524. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bensimon A, Aebersold R and Shiloh Y:
Beyond ATM: The protein kinase landscape of the DNA damage
response. FEBS Lett. 585:1625–1639. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Khanna KK and Jackson SP: DNA
double-strand breaks: Signaling, repair and the cancer connection.
Nat Genet. 27:247–254. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ciccia A and Elledge SJ: The DNA damage
response: Making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giglia-Mari G, Zotter A and Vermeulen W:
DNA damage response. Cold Spring Harb Perspect Biol. 3:a0007452011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harper JW and Elledge SJ: The DNA damage
response: Ten years after. Mol Cell. 28:739–745. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Campisi J and d'Adda di Fagagna F:
Cellular senescence: When bad things happen to good cells. Nat Rev
Mol Cell Biol. 8:729–740. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Halazonetis TD, Gorgoulis VG and Bartek J:
An oncogene-induced DNA damage model for cancer development.
Science. 319:1352–1355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aparicio T, Baer R and Gautier J: DNA
double-strand break repair pathway choice and cancer. DNA Repair
(Amst). 19:169–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Negrini S, Gorgoulis VG and Halazonetis
TD: Genomic instability-an evolving hallmark of cancer. Nat Rev Mol
Cell Biol. 11:220–228. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aguilera A and Gómez-González B: Genome
instability: A mechanistic view of its causes and consequences. Nat
Rev Genet. 9:204–217. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bartek J, Bartkova J and Lukas J: DNA
damage signalling guards against activated oncogenes and tumour
progression. Oncogene. 26:7773–7779. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lehmann AR: DNA repair-deficient diseases,
xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy.
Biochimie. 85:1101–1111. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rotman G and Shiloh Y: ATM: A mediator of
multiple responses to genotoxic stress. Oncogene. 18:6135–6144.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tauchi H, Matsuura S, Kobayashi J,
Sakamoto S and Komatsu K: Nijmegen breakage syndrome gene, NBS1,
and molecular links to factors for genome stability. Oncogene.
21:8967–8980. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lavin MF: Ataxia-telangiectasia: From a
rare disorder to a paradigm for cell signalling and cancer. Nat Rev
Mol Cell Biol. 9:759–769. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cerbinskaite A, Mukhopadhyay A, Plummer
ER, Curtin NJ and Edmondson RJ: Defective homologous recombination
in human cancers. Cancer Treat Rev. 38:89–100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen J and Loeb LA: Unwinding the
molecular basis of the Werner syndrome. Mech Ageing Dev.
122:921–944. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Al-Ejeh F, Kumar R, Wiegmans A, Lakhani
SR, Brown MP and Khanna KK: Harnessing the complexity of DNA-damage
response pathways to improve cancer treatment outcomes. Oncogene.
29:6085–6098. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Curtin NJ: DNA repair dysregulation from
cancer driver to therapeutic target. Nat Rev Cancer. 12:801–817.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Darzynkiewicz Z, Traganos F and Wlodkowic
D: Impaired DNA damage response-an Achilles' heel sensitizing
cancer to chemotherapy and radiotherapy. Eur J Pharmacol.
625:143–150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Damia G and D'Incalci M: Targeting DNA
repair as a promising approach in cancer therapy. Eur J Cancer.
43:1791–1801. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
O'Connor MJ, Martin NM and Smith GC:
Targeted cancer therapies based on the inhibition of DNA strand
break repair. Oncogene. 26:7816–7824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Helleday T, Petermann E, Lundin C, Hodgson
B and Sharma RA: DNA repair pathways as targets for cancer therapy.
Nat Rev Cancer. 8:193–204. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Soussi T: p53 alterations in human cancer:
More questions than answers. Oncogene. 26:2145–2156. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bartek J and Lukas J: Pathways governing
G1/S transition and their response to DNA damage. FEBS Lett.
490:117–122. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Brooks K, Oakes V, Edwards B, Ranall M,
Leo P, Pavey S, Pinder A, Beamish H, Mukhopadhyay P, Lambie D, et
al: A potent Chk1 inhibitor is selectively cytotoxic in melanomas
with high levels of replicative stress. Oncogene. 32:788–796. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sarkaria JN, Busby EC, Tibbetts RS, Roos
P, Taya Y, Karnitz LM and Abraham RT: Inhibition of ATM and ATR
kinase activities by the radiosensitizing agent, caffeine. Cancer
Res. 59:4375–4382. 1999.PubMed/NCBI
|
|
45
|
Garrett MD and Collins I: Anticancer
therapy with checkpoint inhibitors: What, where and when? Trends
Pharmacol Sci. 32:308–316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chabner BA and Roberts TG Jr: Timeline:
Chemotherapy and the war on cancer. Nat Rev Cancer. 5:65–72. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Haince JF, Rouleau M, Hendzel MJ, Masson
JY and Poirier GG: Targeting poly(ADP-ribosyl)ation: A promising
approach in cancer therapy. Trends Mol Med. 11:456–463. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jagtap P and Szabó C: Poly(ADP-ribose)
polymerase and the therapeutic effects of its inhibitors. Nat Rev
Drug Discov. 4:421–440. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sandhu SK, Yap TA and de Bono JS:
Poly(ADP-ribose) polymerase inhibitors in cancer treatment: A
clinical perspective. Eur J Cancer. 46:9–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Helleday T, Bryant HE and Schultz N:
Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination
and as a target for cancer therapy. Cell Cycle. 4:1176–1178. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Turner NC, Lord CJ, Iorns E, Brough R,
Swift S, Elliott R, Rayter S, Tutt AN and Ashworth A: A synthetic
lethal siRNA screen identifying genes mediating sensitivity to a
PARP inhibitor. EMBO J. 27:1368–1377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Boulton S, Kyle S and Durkacz BW:
Interactive effects of inhibitors of poly(ADP-ribose) polymerase
and DNA-dependent protein kinase on cellular responses to DNA
damage. Carcinogenesis. 20:199–203. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Davidson D, Amrein L, Panasci L and Aloyz
R: Small molecules, inhibitors of DNA-PK, targeting DNA repair, and
beyond. Front Pharmacol. 4:52013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Salles B, Calsou P, Frit P and Muller C:
The DNA repair complex DNA-PK, a pharmacological target in cancer
chemotherapy and radiotherapy. Pathol Biol (Paris). 54:185–193.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kashishian A, Douangpanya H, Clark D,
Schlachter ST, Eary CT, Schiro JG, Huang H, Burgess LE, Kesicki EA
and Halbrook J: DNA-dependent protein kinase inhibitors as drug
candidates for the treatment of cancer. Mol Cancer Ther.
2:1257–1264. 2003.PubMed/NCBI
|
|
56
|
Leahy JJ, Golding BT, Griffin RJ,
Hardcastle IR, Richardson C, Rigoreau L and Smith GC:
Identification of a highly potent and selective DNA-dependent
protein kinase (DNA-PK) inhibitor (NU7441) by screening of
chromenone libraries. Bioorg Med Chem Lett. 14:6083–6087. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Willmore E, de Caux S, Sunter NJ, Tilby
MJ, Jackson GH, Austin CA and Durkacz BW: A novel DNA-dependent
protein kinase inhibitor, NU7026, potentiates the cytotoxicity of
topoisomerase II poisons used in the treatment of leukemia. Blood.
103:4659–4665. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Quanz M, Chassoux D, Berthault N, Agrario
C, Sun JS and Dutreix M: Hyperactivation of DNA-PK by double-strand
break mimicking molecules disorganizes DNA damage response. PLoS
One. 4:e62982009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rodríguez-Paredes M and Esteller M: Cancer
epigenetics reaches mainstream oncology. Nat Med. 17:330–339. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dobbin MM, Madabhushi R, Pan L, Chen Y,
Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y, et al: SIRT1
collaborates with ATM and HDAC1 to maintain genomic stability in
neurons. Nat Neurosci. 16:1008–1015. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
McCord RA, Michishita E, Hong T, Berber E,
Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, et
al: SIRT6 stabilizes DNA-dependent protein kinase at chromatin for
DNA double-strand break repair. Aging (Albany NY). 1:109–121. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Miller KM, Tjeertes JV, Coates J, Legube
G, Polo SE, Britton S and Jackson SP: Human HDAC1 and HDAC2
function in the DNA-damage response to promote DNA nonhomologous
end-joining. Nat Struct Mol Biol. 17:1144–1151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Thurn KT, Thomas S, Moore A and Munster
PN: Rational therapeutic combinations with histone deacetylase
inhibitors for the treatment of cancer. Future Oncol. 7:263–283.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zeller C and Brown R: Therapeutic
modulation of epigenetic drivers of drug resistance in ovarian
cancer. Ther Adv Med Oncol. 2:319–329. 2010. View Article : Google Scholar : PubMed/NCBI
|