Introduction
Patients with non-small cell lung cancer (NSCLC)
harboring epidermal growth factor receptor (EGFR) activating
mutations usually undergo treatment with EGFR-tyrosine kinase
inhibitors (EGFR-TKIs) (1). However,
despite the initially positive impact of such inhibitors, nearly
all patients develop resistance following 8–16 months of treatment
(2). Several acquired-resistance
mechanisms have been identified; the most common is the EGFR
T790M secondary mutation within exon 20, observed in 50–65% of
resistant disease (3). Other
resistant mechanisms are based on the bypassing of transmembrane
kinase receptors and include amplification of the c-MET
oncogene (4), overexpression and
mutation of ErbB2 (5) and
upregulation of Axl, which may activate Akt, mitogen activated
protein kinase or nuclear factor-κB signaling (6). Less common mechanisms of TKI resistance
mechanisms may include small cell histological transformation
(7) and transition to a mesenchymal
phenotype (8,9).
The frequency and possible overlay of these
mechanisms has not yet been elucidated. Currently, the use of
mutant-selective inhibitors of EGFR and the combination of
EGFR-TKIs with drugs inhibiting a specific pathway of resistance
represent a possible clinical approach to overcome EGFR-TKI
resistance (10). Therefore,
rebiopsies of growing tumors during clinical progression are
considered critical to characterize the mechanisms of acquired
resistance to EGFR-TKIs for therapeutic and prognostic reasons
(3,11).
However, a single tumor rebiopsy may not be
representative of the dominant characteristics of the tumor due to
the well-known intratumor heterogeneity of resistant mechanisms in
lung cancer (7). Recently, the
analysis of blood samples has been suggested to reflect the
dominant properties of tumors, and the detection of T790M in plasma
may qualify patients as candidates for treatment with a third
generation EGFR-TKI (12).
The present study describes a case of tumor
heterogeneity of acquired resistance following EGFR-TKI treatment
failure.
Case report
In March 2013, a 45-year-old man with no history of
smoking was subjected to medical examination at the Unit of
Pneumology, University Hospital of Pisa (Pisa, Italy) due to a
persistent cough. Computed tomography (CT) revealed a 3.8-cm right
middle lobe mass and bilateral nodules, with the largest measuring
8 mm. In addition, enlarged right hilar and subcarinal lymph nodes
were observed. A positron emission tomography (PET) scan exhibited
increased fluorodeoxyglucose uptake in the lung lesions and the
lymphadenopathy. The patient was reported to have a single 13-mm
hepatic metastatic lesion and several bone lesions (in the
thighbone, the scapula, and C4 and D10 lamina).
Broncoscopic biopsy was performed. The obtained
tissue was formalin-fixed, paraffin-embedded and cut into 5 µm
sections, which were subsequently stained with hematoxylin and
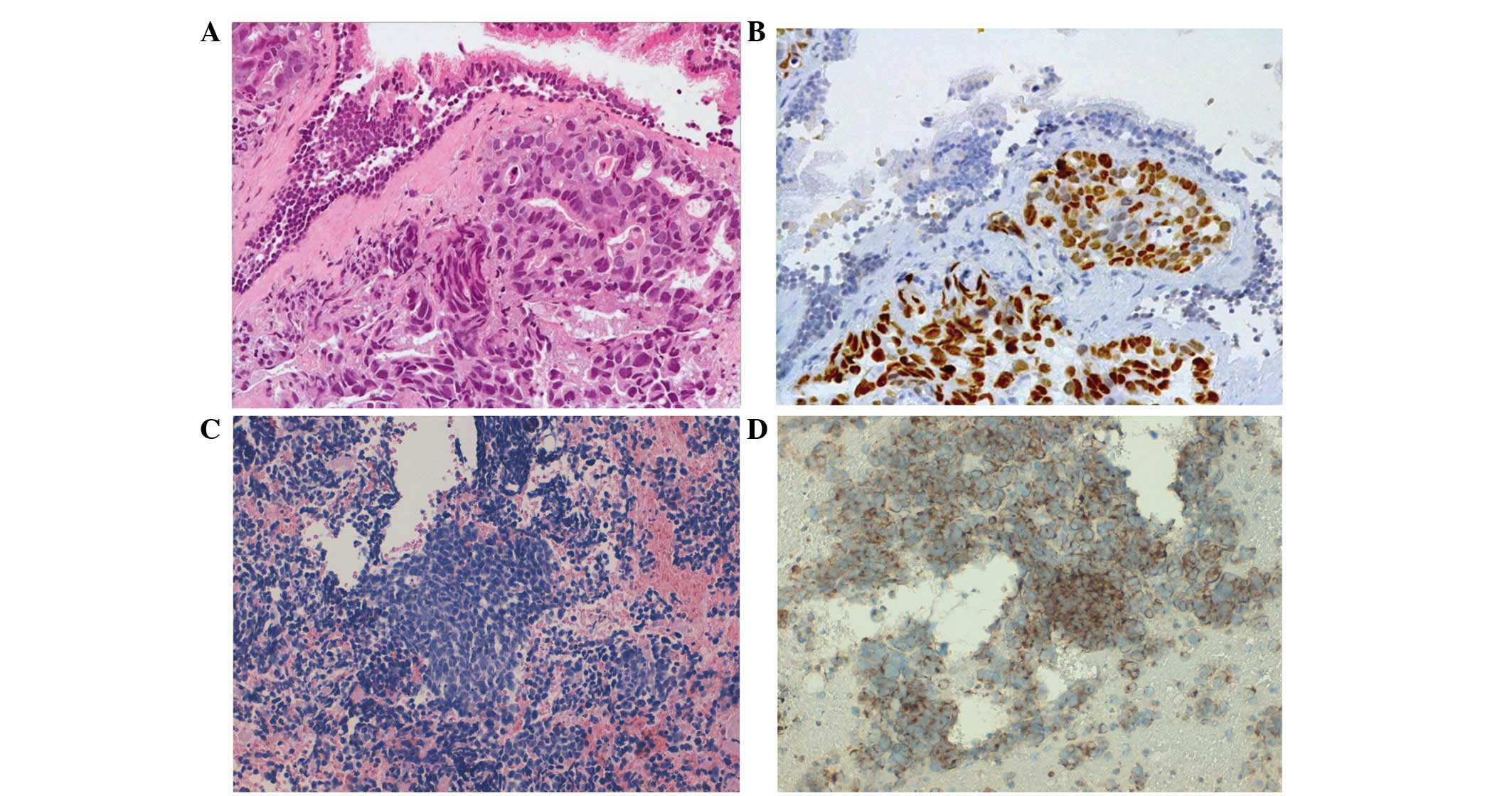
eosin (Fig. 1A). Histological
examination of the biopsy identified the presence of
adenocarcinoma, which was confirmed by further immunohistochemical
examination demonstrating strong nuclear expression of thyroid
transcription factor 1 (TTF1) and negative expression of p63
(Fig. 1A and B). Analysis of
EGFR mutational status, determined by Sanger sequencing,
indicated an EGFR exon 19 deletion.
In April 2013, the patient began treatment with the
TKI inhibitor gefitinib (250 mg/day orally for 11 months) and
zoledronic acid (4 mg every 28 days intravenously for 11 months)
for the T4N2M1b adenocarcinoma. In May 2013, the patient also
received radiation (27 Gy administered in a single fraction) to the
osteoblastic lesion of the thighbone. After 1 month, a CT/PET scan
was performed and indicated a significant decrease in the size of
the right middle lobe mass, the bilateral nodules, and the hilar
and subcarinal lymphadenopathy. CT/PET scans performed in September
and December 2013 were indicative of stable disease. However, by
March 2014, disease progression was observed. Therefore, the
following treatment regimen was initiated in April 2014: 6 cycles
of 200 mg/m2 paclitaxel, 6 AUC carboplatin and 15
mg/m2 bevacizumab administered intravenously every 21
days, followed by 5 cycles of maintenance therapy with 15
mg/m2 bevacizumab administered intravenously every 21
days until December 2014. By that time, the disease had progressed
with slight growth of the right middle lobe lesion and a right
inferior paraesophageal lymphadenopathy (11×13 mm) had appeared.
Furthermore, a magnetic resonance imaging scan of the brain
identified a frontal lobe lesion.
The patient subsequently underwent radiation
(whole-brain radiation of 30 Gy administered in 10 fractions of 3
Gy for 2 weeks) to the whole brain and received oral third-line
therapy with erlotinib (150 mg/day) for 4 months and intravenous
bevacizumab (15 mg/m2) every 21 days for 6 cycles.
In April 2015, a full-body CT scan was performed and
identified a new right middle lobe mass, a slight increase in the
right lower lobe nodule and a marked dimensional growth of the
right inferior paraesophageal lymphadenopathy (22×22 vs. 11×13 mm
previously).
In June 2015, the patient was subjected to a chest
CT scan that documented a further increase in the size of the
primitive right lower lobe mass and the right inferior
paraesophageal lymphadenopathy (35×28 vs. 22×22 mm previously).
Mutational analysis of the EGFR gene was performed on
circulating free tumor (cft) DNA purified from 4 ml plasma using
the Easy® EGFR Real-time PCR kit (Diatech
Pharmacogenetics SRL, Jesi, Italy) and the Myriapod®
Lung Status kit with Sequenom MassARRAY® technology
(Diatech Pharmacogenetics SRL) according to the manufacturer's
protocol. In addition to the original EGFR exon 19 deletion,
cftDNA analysis identified a secondary resistant mutation, T790M,
occurring in EGFR exon 20.
In July 2015, a CT-guided biopsy and fine-needle
aspiration of the right inferior paraesophageal lymphadenopathy
indicated an emergent small cell morphology (Fig. 1C). Further immunohistochemical
examination for TTF1 (mouse monoclonal primary antibody; clone
8G7G3/1; ready-to-use; #790-438), cluster of differentiation 56
(mouse monoclonal primary antibody; clone 123C3; ready-to-use;
#790–4465) chromogranin (mouse monoclonal primary antibody; clone
LK2H10; ready-to-use; #760–2519) and cytokeratin-pan (mouse
monoclonal primary antibody; clone AE1; ready-to-use; #760–2521)
(Ventana Medical Systems, Inc., Tucson, AZ, USA) expression was
positive (Fig. 1D).
Molecular characterization of the EGFR
mutational status of the rebiopsy, performed using the same methods
employed for cftDNA analysis, detected the presence of an exon 19
deletion alone.
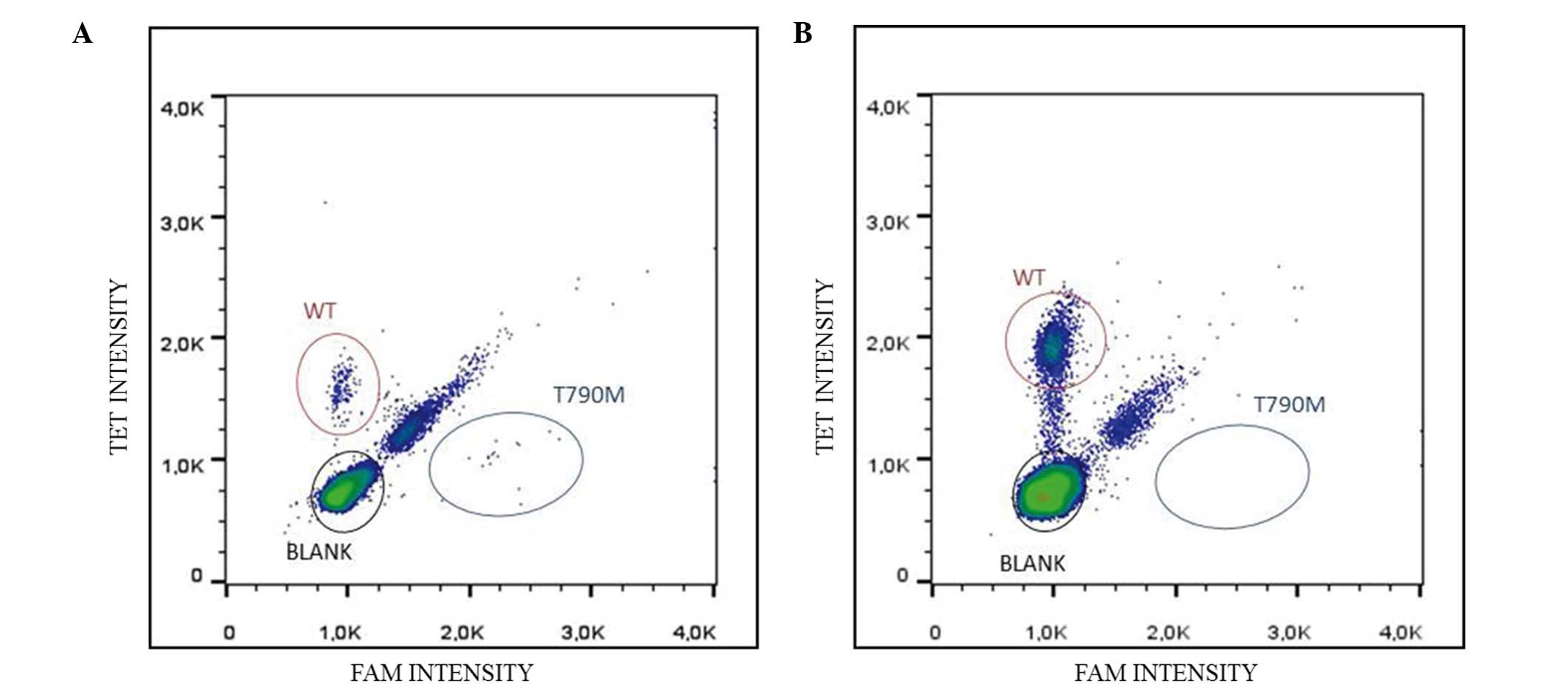
To more effectively assess the presence of the T790M
mutation, further molecular analyses were performed on the cftDNA
and rebiopsy using the RainDrop® Digital PCR system
(Diatech Pharmacogenetics SRL) specifically for T790M, which is
more sensitive than quantitative polymerase chain reaction and
Sequenom MassARRAY technology (13).
The PCR thermal cycling conditions were as follows: Polymerase
activation step at 95°C for 10 min; denaturation step at 95°C for
15 sec for 50 cycles; annealing-extension step at 59°C for 60 sec
for 50 cycles; incubation step at 98°C for 10 min; further
incubation step at 12°C for 10 min; and a final hold at 4°C for no
more than 1 h until digital analysis. The T790M mutation was
detected following liquid biopsy (Fig.
2) with a mutant allele prevalence of 7.15%, which was
consistent with the previous results.
Due to the SCLC transformation, the patient began
fourth-line chemotherapy, which consisted of epirubicin (80
mg/m2) and paclitaxel (160 mg/m2)
administered intravenously every 21 days. Re-evaluation with a
full-body CT scan following 2 cycles of epirubicin plus paclitaxel
identified a significant dimensional decrease in the right inferior
paraesophageal lymphadenopathy (15×20 vs. 35×28 mm previously) and
of the hepatic metastatic lesion (8 vs. 10 mm). Stability of the
lung right lobe lesion, the bilateral satellite nodules, the hilar
and subcarinal lymphadenopathy, and the osteoblastic bone lesions
were all observed.
At present, the patient is completing 6 cycles of
chemotherapy with epirubicin plus paclitaxel and is waiting to
begin radiotherapy on the hepatic metastatic lesion. Furthermore,
due to the presence of non-responsive lesions and according to the
T790M mutation detected in cftDNA, the patient is currently being
considered for treatment with a third generation TKI. Written
informed consent for the publication of the current study was
obtained from the patient.
Discussion
Acquired resistance to TKI therapy may be due to
multiple biological mechanisms (2,8,13). The use of mutant-selective inhibitors
of EGFR, in addition to a combination of treatments and
multi-targeting drugs, constitutes current clinical approaches for
overcoming EGFR-TKI resistance in NSCLC (14,15).
Therefore, the characterization of all the molecular resistant
mechanisms occurring in the same patient is crucial to define a
more effective therapeutic regimen.
The present study described the case of a patient
diagnosed with metastatic lung adenocarcinoma harboring a deletion
within exon 19 of EGFR, who developed two resistant
mechanisms against TKI: A small cell histological transformation
and the EGFR T790M mutation. Small cell transformation and
the T790M mutation are clinically relevant mechanisms of drug
resistance (7), however, their
interaction and overlapping is not yet fully understood.
The coexistence of SCLC transformation and the
EGFR T790M mutation in response to EGFR-TKI therapy has been
described in various studies (3,16–19); however, the majority of these refer to
autopsy cases or to cases where a direct analysis of repeat tumor
biopsies was possible.
To the best of our knowledge, in all previously
reported cases, SCLC metastatic lesions harbored only the
EGFR activating mutation, while in the current case the
adenocarcinoma metastases harbored the T790M mutation together with
the original EGFR mutation. Only one study, by Yu et
al (3), describes SCLC
transformation and the EGFR T790M mutation occurring in the same
tumor rebiopsies in response to EGFR-TKIs.
The current study and published literature suggest
that SCLC and adenocarcinoma components arise from the same origin
due to them both possessing the identical activating mutation in
EGFR, reflecting the importance of tumor heterogeneity in
acquired resistance to TKIs (16–18,20). In
the present study, molecular and histological analyses of the tumor
rebiopsy and molecular analysis of the corresponding blood sample
were performed, enabling a clearer image of TKI resistance. If only
the rebiopsy or the cftDNA had been analyzed, coexistence of the
two resistant mechanisms would not have been detected.
In comparison with previous cases, the patient
described in the present study is currently alive and only one
rebiopsy was performed. The plasma sample highlighted tumor
properties that were unable to be identified in tissue, therefore
making it possible to improve the efficacy of the therapeutic
regimen.
In conclusion, on the basis of the histological
analysis of the paraesophageal lymph nodes, the current patient was
treated for neuroendocrine carcinoma and experienced a clinical
response for lymphadenopathy and hepatic metastasis. All other
neoplastic lesions, including the primary tumor, did not respond to
this treatment. This lack of responsiveness may be due to the
heterogeneous EGFR molecular status of the tumor, confirmed
by the presence of the T790M mutation in cftDNA, which most likely
reflects unresponsive lesions. As such, the patient may benefit
from a third generation T790M-specific EGFR-TKI. Therefore, the
present study underlines the importance of performing, whenever
possible, tumor biopsies following the development of TKI
resistance, together with monitoring drug resistance by
plasma-based assessments of cftDNA.
References
|
1
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et
al: Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): An open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cappuzzo F, Jänne PA, Skokan M,
Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi
L, Toschi L, et al: MET increased gene copy number and primary
resistance to gefitinib therapy in non-small-cell lung cancer
patients. Ann Oncol. 20:298–304. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takezawa K, Parazzoli V, Arcila ME, Nebhan
CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ,
Melnick MA, et al: HER2 amplification: A potential mechanism of
acquired resistance to EGFR inhibition in EGFR-mutant lung cancers
that lack the second-site EGFR T790M mutation. Cancer Discov.
2:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang L and Fu L: Mechanisms of resistance
to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 5:390–401.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lim J and Thiery JP:
Epithelial-mesenchymal transitions: Insights from development.
Development. 139:3471–3486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Janjigian YY, Smit EF, Groen HJ, Horn L,
Gettinger S, Camidge DR, Riely GJ, Wang B, Fu Y, Chand VK, et al:
Dual inhibition of EGFR with afatinib and cetuximab in kinase
inhibitor-resistant EGFR-mutant lung cancer with and without T790M
mutations. Cancer Discov. 4:1036–1045. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cross DA, Ashton SE, Ghiorghiu S, Eberlein
C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ,
et al: AZD9291, an irreversible EGFR, TKI, overcomes T790M-mediated
resistance to EGFR inhibitors in lung cancer. Cancer Discov.
4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ishii H, Azuma K, Sakai K, Kawahara A,
Yamada K, Tokito T, Okamoto I, Nishio K and Hoshino T: Digital PCR
analysis of plasma cell-free DNA for non-invasive detection of drug
resistance mechanisms in EGFR mutant NSCLC: Correlation with paired
tumor samples. Oncotarget. 6:30850–30858. 2015.PubMed/NCBI
|
|
13
|
Bordi P, Del Re M, Danesi R and Tiseo M:
Circulating DNA in diagnosis and monitoring EGFR gene mutations in
advanced non-small cell lung cancer. Transl Lung Cancer Res.
4:584–597. 2015.PubMed/NCBI
|
|
14
|
Jorge SE, Kobayashi SS and Costa DB:
Epidermal growth factor receptor (EGFR) mutations in lung cancer:
Preclinical and clinical data. Braz J Med Biol Res. 47:929–939.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Piotrowska Z, Niederst MJ, Karlovich CA,
Wakelee HA, Neal JW, Mino-Kenudson M, Fulton L, Hata AN, Lockerman
EL, Kalsy A, et al: Heterogeneity underlies the emergence of
EGFRT790 wild-type clones following treatment of T790M-Positive
cancers with a third-generation EGFR inhibitor. Cancer Discov.
5:713–722. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morinaga R, Okamoto I, Furuta K, Kawano Y,
Sekijima M, Dote K, Satou T, Nishio K, Fukuoka M and Nakagawa K:
Sequential occurrence of non-small cell and small cell lung cancer
with the same EGFR mutation. Lung Cancer. 58:411–413. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Furugen M, Uechi K, Hirai J, Aoyama H,
Saio M, Yoshimi N, Kinjo T, Miyagi K, Haranaga S, Higa F, et al: An
autopsy case of two distinct, acquired drug resistance mechanisms
in epidermal growth factor receptor-mutant lung adenocarcinoma:
Small cell carcinoma transformation and epidermal growth factor
receptor T790M mutation. Intern Med. 54:2491–2496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Popat S, Wotherspoon A, Nutting CM,
Gonzalez D, Nicholson AG and O'Brien M: Transformation to ‘high
grade’ neuroendocrine carcinoma as an acquired drug resistance
mechanism in EGFR-mutant lung adenocarcinoma. Lung Cancer. 80:1–4.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fallet V, Ruppert AM, Poulot V, Lacave R,
Belmont L, Antoine M, Cadranel J, Wislez M and Lavolé A: Secondary
resistance to erlotinib: Acquired T790M mutation and small-cell
lung cancer transformation in the same patient. J Thorac Oncol.
7:1061–1063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Costa DB and Kobayashi SS: Whacking a
mole-cule: Clinical activity and mechanisms of resistance to third
generation EGFR inhibitors in EGFR mutated lung cancers with
EGFR-T790M. Transl Lung Cancer Res. 4:809–815. 2015.PubMed/NCBI
|