Introduction
Escape from tumor necrosis factor (TNF)-α-induced
cell apoptosis and necrosis is of importance in tumor development
(1–3).
This process is regulated by a number of intracellular signaling
pathways, including c-jun N-terminal kinase (JNK) and IκB kinase
(IKK), as well as reactive oxygen species (ROS) (4,5). Extensive
studies have indicated that reduced levels of oxidant stress and
ROS promote malignant transformation and oncogenic growth in
hepatocellular carcinoma (HCC) cells (6–9). However,
the key molecules regulating ROS in HCC remain to be elucidated. It
has been reported that scaffolding protein receptor for activated C
kinase 1 (RACK1) enhances JNK activation in HCC, leading to
promotion of the malignant growth of HCC (10). Therefore, it may be assumed that RACK1
affects other aspects of HCC. RACK1 was originally identified to
bind and activate protein kinase C and is now recognized as a
multi-functional scaffold protein (11,12).
Evidence has indicated that RACK1 protects from oxidative
stress-induced cell death in various types of cells, including
fission yeasts (13), shrimp cells
(14), neurons (15), HeLa cells (16) and HL60 cells (17). However, such a role for RACK1 has not
been reported in HCC cells to the best of our knowledge. In the
present study, it was demonstrated that RACK1 knockdown leads to
increased cell death in TNF-α-treated HCC cells in the presence of
cycloheximide (CHX), a protein synthesis inhibitor. Subsequently,
it was observed that RACK1 knockdown promotes intracellular ROS
accumulation upon TNF-α or H2O2 stimulation.
A combination of co-immunoprecipitation (co-IP) and mass
spectrometry analysis indicated that carbonyl reductase 1 (CBR1), a
ubiquitous nicotinamide adenine dinucleotide phosphate-dependent
enzyme, acts as a RACK1-interacting partner in HCC cells. CBR1 has
been reported to provide protection from ROS-induced cellular
damage in HCC and leukemia (4,18), which
suggests that CBR1 serves a role in cellular anti-oxidation. In the
present study, it was reported that overexpression of exogenous
CBR1 in HCC cells reverses enhanced cell death upon silencing of
endogenous RACK1, which indicated that RACK1 may have a pivotal
role in sustaining the protein stability of CBR1.
Materials and methods
Cell culture and treatment
Human hepatic carcinoma cell lines HepG2 and
SMMC7721, and mouse embryonic liver cell line BNL CL.2 were
purchased from American Type Culture Collection (Manassas, VA,
USA). Cells were cultured in Dulbecco's modified Eagle's medium
(catalog no. 12800-058; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% fetal calf serum (Standard Grade;
Lanzhou Bailing Biotechnology Co., Ltd., Lanzhou, China.) at 37°C
and 5% CO2.
Cell transfection
Small interfering RNA (siRNA) targeting human RACK1
(5′-GGATGAGACCAACTATGGAAT-3′) and human CBR1
(5′-ATACGTTCACCACTCTCCCTT-3′) and small hairpin RNA targeting mouse
RACK1 (5′-GTCCCGAGACAAGACCATAAA-3′) were designed and chemically
synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). The
siRNAs were delivered into the SMMC7721 cells at 50% confluence
using Lipofectamine® RNAiMAX transfection reagent
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Cells were transfected for 48 h and then analyzed for
various parameters. SMMC7721 stable clones, which were a gifts from
Dr Wendie Wang (Institute of Medicinal Biotechnology, Chinese
Academy of Medical Sciences, Beijing, China), were selected in 600
µg/ml G418 sulfate (catalog. no. 11811-023; Thermo Fisher
Scientific, Inc.) for approximately 2 months.
Sample preparation, co-IP and western
blot analysis
Cells were harvested and lysed in lysis buffer (20
mM Tris, pH 7.6, 250 mM NaCl, 1% Nonidet P-40, 3 mM EDTA, 1.5 mM
ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid,
10 µg/ml aprotinin, 10 mM p-nitrophenylphosphate, 1 mM
Na3VO4, 1 mM dithiothreitol). Following
clarification by centrifugation at 5,000 × g for 15 min at
4°C, cell lysates were incubated with the indicated antibodies in
the presence of 30 µl [50% (v/v)] of protein A-Sepharose beads
(Sigma-Aldrich; EMD Millipore, Billerica, MA, USA) at 4°C for 4 h.
Precipitates were washed with washing buffer [20 mM Tris (pH 7.6),
250 mM NaCl, 1% Nonidet P-40, 3 mM EDTA, 1.5 mM ethylene
glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid and 1 mM
phenylmethane sulfonyl fluoride) at least three times. For western
blot analysis, cell lysates or co-IP samples underwent 12% SDS-PAGE
for 2 h, followed by transferal to polyvinylidene difluoride
membranes for 3 h and blocking with 5% nonfat milk in TBS
containing 0.1% Tween-20 (TBST) for 1 h at room temperature.
Membranes were incubated with primary antibodies against RACK1
(catalog no. 610177; BD Biosciences, San Jose, CA, USA), CBR1
(catalog no. ab4148; Abcam, Cambridge, MA, USA), β-actin (catalog
no. 47778; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
GAPDH (catalog no. sc-81545; Cell Signaling Technology, Inc.,
Danvers, MA, USA) at a dilution of 1:1,000 overnight at 4°C.
Following three times washing with TBST (10 min each wash), the
membranes were incubated with horseradish peroxidase-conjugated
secondary antibodies at a dilution of 1:5,000 for 1 h at room
temperature (polyclonal goat anti-rabbit or goat anti-mouse
secondary antibodies; catalog no. ZB2301 and ZB2305, respectively;
OriGene Technologies, Inc., Beijing, China), followed by additional
washing. The membranes were subjected to exposure in the dark and
the immunoreactive bands were visualized with an enhanced
chemiluminescence kit (GE Healthcare Life Sciences, Chalfont, UK).
Quantification of western blot were performed by using Gel-Pro
Analyzer 4.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Cell death assay by flow
cytometry
Cells treated with 10 ng/ml TNF-α (R&D Systems,
Inc., Minneapolis, MN, USA) with or without 10 µg/ml CHX
(Sigma-Aldrich; EMD Millipore), or 1 mM H2O2
for 24 h were digested by 0.25% trypsin for approximately 2 min
with gentle shaking, and subsequently harvested. Following washing
twice with PBS, the cell pellet was resuspended in 200 ml PBS
containing Annexin-V and propidium iodide (PI)/7-aminoactinomycin D
(BD Biosciences) and incubated at 4°C for 30 min, followed by flow
cytometry assay.
ROS assay
Cells were resuspended and incubated in pre-warmed
Hank's balanced salt solution (HBSS) containing 10 mM
carboxy-.2′,7′-dichlorodihydrofluorescein diacetate (Thermo Fisher
Scientific, Inc.) for 30 min at 37°C, followed by incubation with
10 ng/ml TNF-α or 400 µmol/l H2O2 for 30 min
at 37°C. Cells were washed with HBSS twice and subjected to flow
cytometry.
Statistical analysis
Cell death assay experiments were performed
independently at least three times. Statistical differences between
groups were assessed by Students t-test. Descriptive statistics
were computed by using Excel 2007 (Microsoft Corporation, Redmond,
WA, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Knockdown of RACK1 leads to increased
cell death and ROS generation following TNF-α stimulation
The present study initially investigated the
correlation between RACK1 and TNF-α-induced cell death in SMMC7721
cells. SMMC7721 cells were transiently transfected with RACK1 siRNA
or NC siRNA by using the iMAX delivery system. A total of 48 h
later, the cells were treated with 10 ng/ml TNF-α with or without
10 µg/ml CHX for 24 h, followed by cell death assay with Annexin-V
and PI double staining. Transfection with RACK1 siRNA dramatically
decreased RACK1 protein levels (Fig.
1A), leading to an increased cell death rate compared with NC
siRNA following co-treatment with TNF-α and CHX
(P=2.67×10−7; Fig. 1B).
Notably, treatment with TNF-α alone only caused slight cell death,
and cell death was markedly induced in the presence of CHX, a
pan-protein synthesis inhibitor (Fig.
1B), indicating that CHX sensitized SMMC7721 cells to
TNF-α-induced cell death. It was subsequently investigated whether
RACK1 affected ROS responses to stimuli, including TNF-α or
hydrogen peroxide. As demonstrated in Fig. 1C, knockdown of RACK1 resulted in
increased ROS generation in SMMC7721 cells upon TNF-α and hydrogen
peroxide treatment. Taken together, the results of the present
study suggested that RACK1 acted as a ROS suppressor to antagonize
TNF-α-induced cell death in SMMC7721 cells.
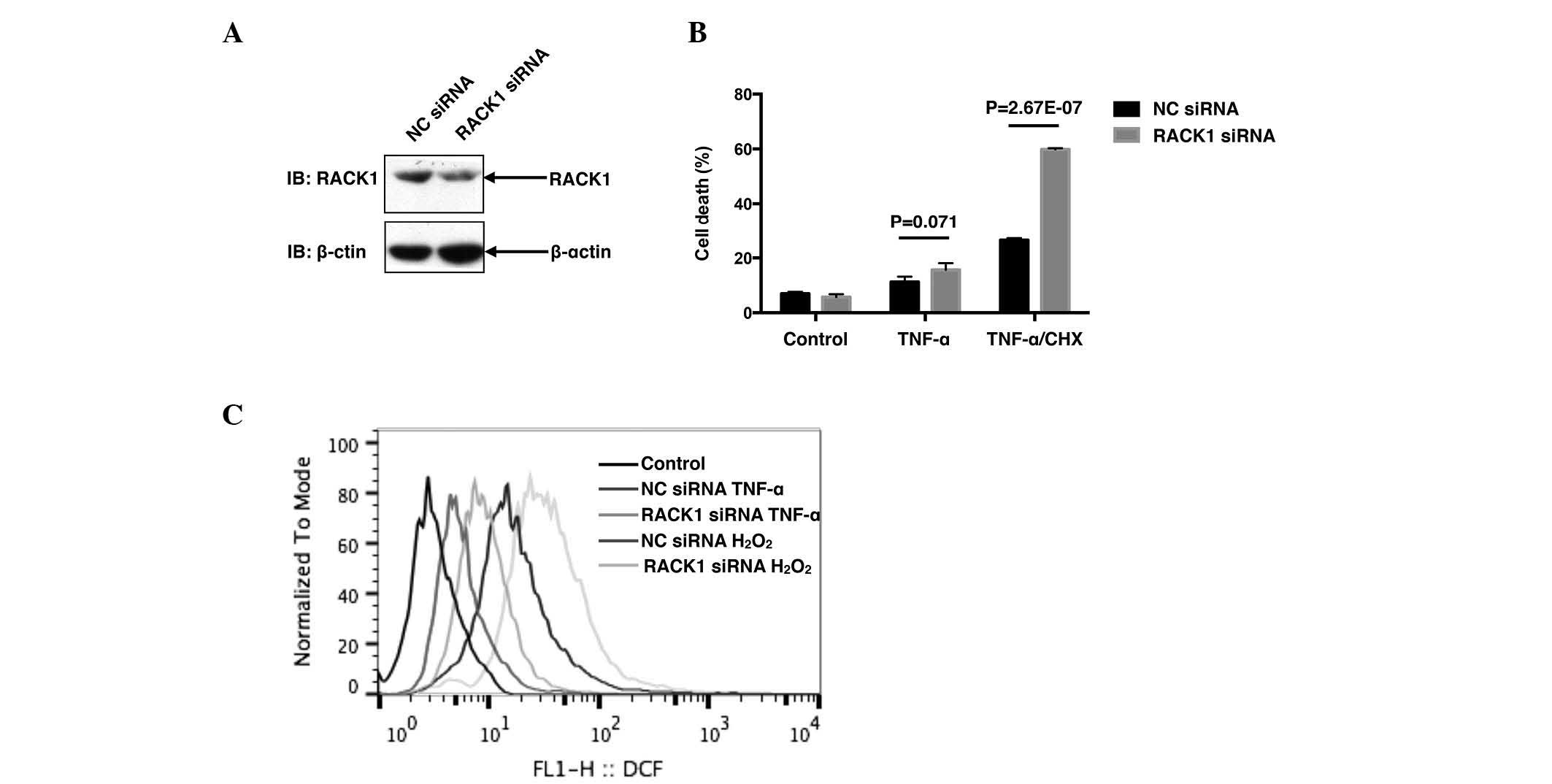 | Figure 1.Knockdown of RACK1 promotes cell death
and ROS generation. (A) SMMC7721 cells were transiently transfected
with NC siRNA or RACK1 siRNA. Cell lysates were subjected to
western blot analysis. (B) SMMC7721 cells were transiently
transfected with NC siRNA or RACK1 siRNA. A total of 48 h later,
cells were stimulated with 10 ng/ml TNF-α with or without 10 µg/ml
CHX, followed by cell death staining and flow cytometry assays.
Cell death was determined by fluorescein-Annexin-V and propidium
iodide double staining, and the results of three independent
experiments are presented as the mean ± standard deviation; n=3.
(C) SMMC7721 cells were transiently transfected with NC siRNA or
RACK1 siRNA. Prior to flow cytometry assays, cells were treated
with TNF-α or hydrogen peroxide and incubated with 10 mM 5(6)-carboxy-.2′,7′-dichlorodihydrofluorescein
diacetate green probes. Intracellular ROS levels were measured by
flow cytometry. RACK1, receptor for activated C kinase 1; ROS,
reactive oxygen species; NC, negative control; siRNA, small
interfering RNA; TNF, tumor necrosis factor; CHX, cycloheximide;
IB, immunoblotting. |
RACK1 suppresses TNF-α-induced cell
death via CBR1
Although it was observed that knockdown of
endogenous RACK1 promoted ROS generation upon TNF-α and hydrogen
peroxide stimulation, the molecular mechanism(s) by which RACK1
affected ROS remain to be elucidated. Subsequently, a co-IP was
performed, followed by mass spectrometry analysis (performed by
National Center of Biomedical Analysis, Beijing, China) to identify
RACK1-interacting partners (data not shown). Among the candidates
of RACK1-interacting proteins obtained, CBR1 was notable due to its
close association with ROS. CBR1 was considered to be a ROS
suppressor, as it had been reported to protect cells from cytotoxic
drug-triggered cell death in doxorubicin-treated HCC cells and
As2O3-treated leukemia cells (4,18).
Therefore, the functions of CBR1 were investigated in TNF-α-treated
SMMC7721 cells. As expected, transfection with CBR1 siRNA markedly
decreased the protein level of CBR1 (Fig.
2A), leading to increased percentages of cell death induced by
TNF-α (P=0.046) and TNF-α/CHX (P=0.00067; Fig. 2B). Subsequently, the present study
employed RACK1 stably silenced single clones screened from SMMC7721
cells to investigate whether CBR1 affected the enhanced cell death
caused by RACK1 knockdown. As expected, overexpression of green
fluorescent protein (GFP)-CBR1 reversed the enhanced cell death in
the RACK1 stably silenced clone, at least partially, compared with
the clone overexpressing GFP (P=0.00071; Fig. 2C). Cell death upon hydrogen peroxide
stimulation was also investigated in HCC cells. Rather than
apoptotic cell death, hydrogen peroxide stimulation caused a more
necrotic cell death than TNF-α/CHX stimulation (data not shown).
However, hydrogen peroxide caused ~80% cell death, and knockdown of
CBR1 elevated cell death up to >90% (P=6.42×10−5;
Fig. 2D). Thus, the results of the
present study suggested RACK1 may exert its protecting function via
CBR1.
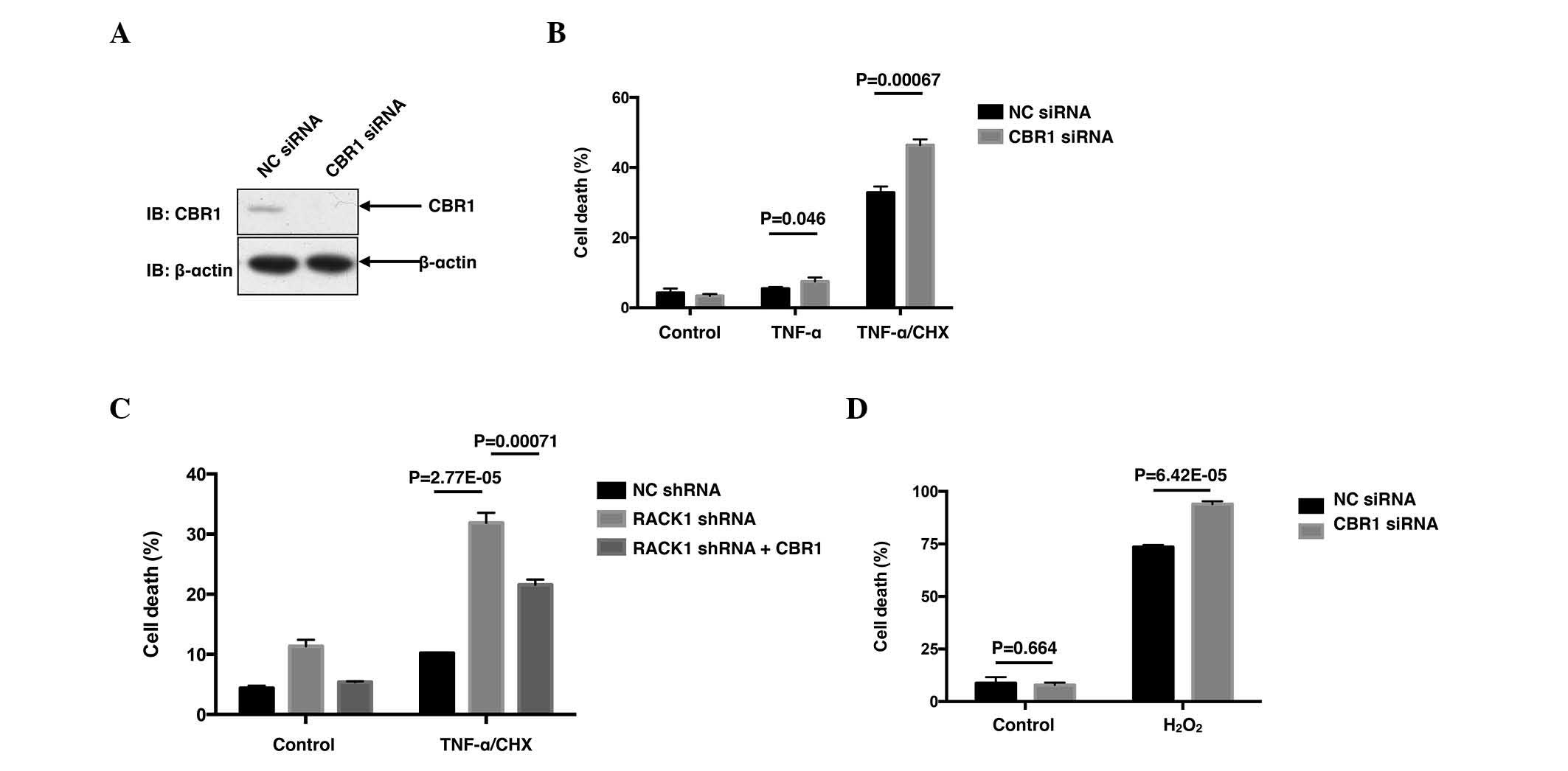 | Figure 2.CBR1 promotes cell survival and
reverses the enhanced cell death caused by RACK1 knockdown. (A)
SMMC7721 cells were transiently transfected with NC siRNA or CBR1
siRNA. The amounts of CBR1 protein were examined by western blot
analysis with the indicated antibodies. (B) SMMC7721 cells were
transiently transfected with NC siRNA or CBR1 siRNA. Cells were
stimulated with 10 ng/ml TNF-α with or without 10 µg/ml CHX,
followed by cell death staining and flow cytometry assays. Cell
death was determined by FITC-Annexin-V and PI double staining. (C)
Control clone cells and RACK1 stably silenced clone cells were
transfected with GFP or GFP-CBR1 vectors. Cells were stimulated
with 10 ng/ml TNF-α and 10 µg/ml CHX for 24 h, followed by flow
cytometry assays. GFP positive cells were gated and cell death was
analyzed with phycoerythrin-Annexin-V and 7-aminoactinomycin D
double staining. (D) SMMC7721 cells transfected with NC siRNA or
CBR1 siRNA were stimulated with 1 mM H2O2 for
24 h, followed by cell death staining and flow cytometry assays.
Cell death was determined by FITC-Annexin-V and PI double staining.
Results are representative of three independent experiments and are
presented as the mean ± standard deviation; n=3. CBR1, carbonyl
reductase 1; RACK1, receptor for activated C kinase 1; NC, negative
control; siRNA, small interfering RNA; shRNA, short hairpin RNA;
TNF, tumor necrosis factor; CHX, cycloheximide; IB, immunoblotting;
FITC, fluorescein isothiocyanate; PI, propidium iodide; GFP, green
fluorescent protein. |
RACK1 promotes the protein stability
of CBR1
To address whether RACK1 affected the functioning of
CBR1, it was necessary to address whether and how RACK1 regulates
CBR1. Initially, the present study briefly addressed the
interaction between RACK1 and CBR1 in vivo. As shown in
Fig. 3A, when endogenous CBR1 was
immunoprecipitated from SMMC7721 cells with CBR1 antibody, RACK1
was detected in the precipitant, suggesting that RACK1 was able to
interact with CBR1 in SMMC7721 cells (Fig. 3A). Furthermore, in SMMC7721 RACK1
stably silenced single clones, CBR1 protein levels exhibited a
marked decrease compared to control clones (Fig. 3B), suggesting that RACK1 potentially
regulated the protein stability of CBR1. For further investigation,
the present study examined the degradation of CBR1 in HCC cells in
RACK1 stably silenced clones and control clones upon CHX treatment
for various time courses. CBR1 protein exhibited a decrease at 16
and 20 h subsequent to CHX treatment in HCC clones with RACK stable
knockdown but exhibited no change in control HCC clones (Fig. 3C). It has previously been reported
that RACK1 exhibited higher expression in HCC cells compared to
‘normal’ hepatocytes (10). As
expected, an increased level of RACK1 protein was detected in
SMMC7721 cells and HepG2 cells, as compared to BNL CL.2 mouse
embryonic liver cells (Fig. 3D).
Consistent with the idea that RACK1 promotes the stability of CBR1
protein, HepG2 cells exhibited an increased level of CBR1 protein
compared with BNL CL.2 cells (Fig.
3E). This finding is consistent with a previous report in which
CBR1 demonstrated overexpression in 56 (72%) out of 78 human HCC
tissues (6). Notably, upon TNF-α/CHX
stimulation, BNL CL.2 cells exhibited a marked increase in cell
death compared to HepG2 cells (P=0.00017; Fig. 3F). These results indicate that
upregulation of RACK1 together with CBR1 may have a significant
role in the transformation of normal liver cells to malignant
cells.
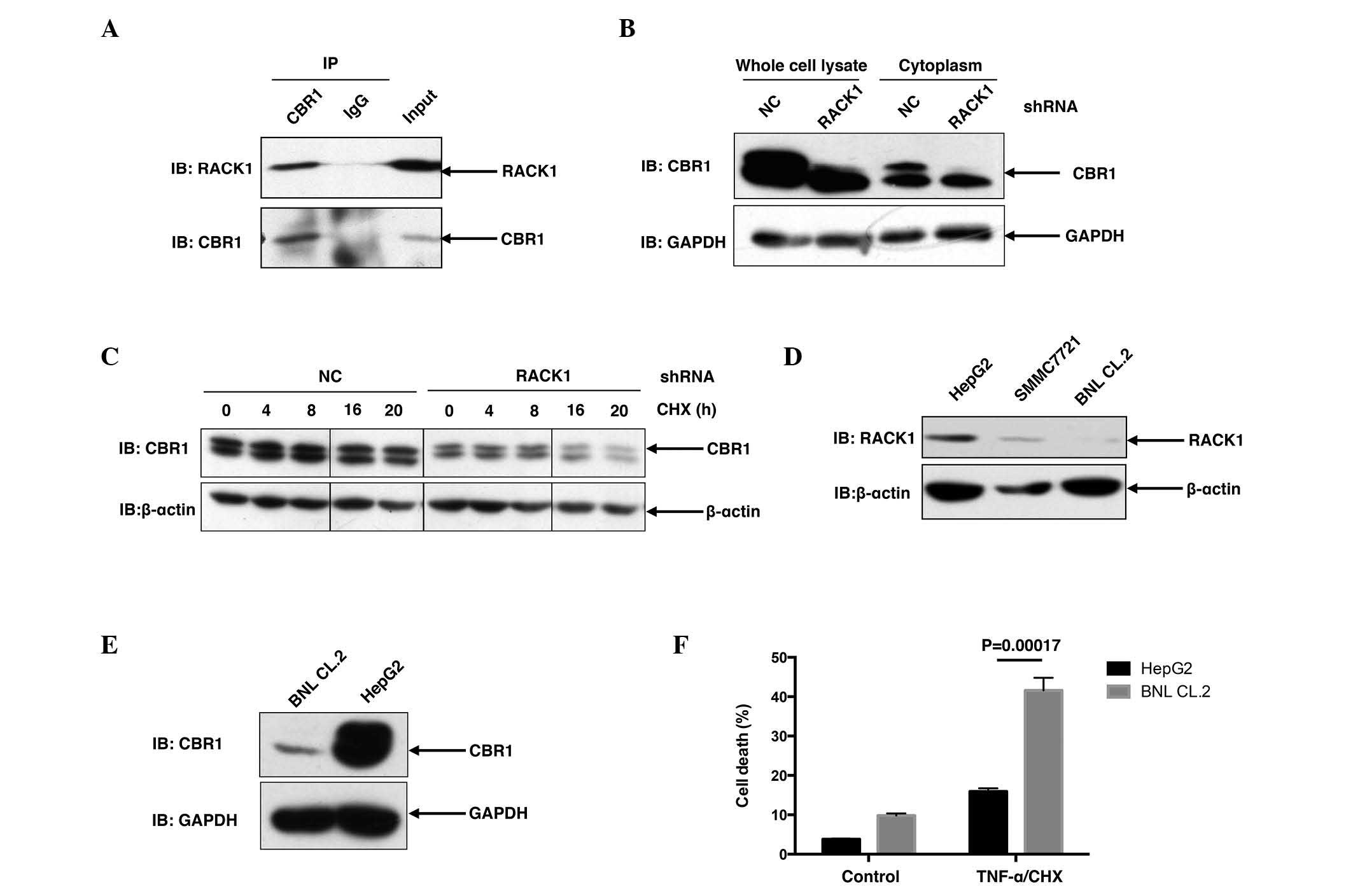 | Figure 3.RACK1 binds to CBR1 and sustains the
protein stability of CBR1. (A) SMMC7721 cells were lysed and the
lysates were subjected to immunoprecipitation with an anti-CBR1
antibody or a control antibody. Following immunoprecipitation,
western blot analysis was performed. (B) Control clone cells and
RACK1 stably silenced clone cells were lysed and subjected to
western blot analysis. (C) Control clone cells and RACK1 stably
silenced clone cells were treated with CHX for various durations,
followed by western blot analysis. (D and E) HepG2 cells, SMMC7721
cells and BNL CL.2 cells were lysed and subjected to western blot
analysis. (F) Following TNF-α/CHX stimulation, HepG2 cells and BNL
CL.2 cells were subjected to flow cytometry assays. Cell death was
determined by fluorescein isothiocyanate-Annexin-V and propidium
iodide double staining, and the results of three independent
experiments are presented as the mean ± standard deviation; n=3.
RACK1, receptor for activated C kinase 1; CBR1, carbonyl reductase
1; CHX, cycloheximide; TNF, tumor necrosis factor; IP,
immunoprecipitation; NC, negative control; IB, immunoblotting;
shRNA, short hairpin RNA. |
Discussion
In the present study, it was initially confirmed
that RACK1 protected HCC cells from TNF-α-induced cell death.
However, it was observed that TNF-α alone only caused slightly
enhanced cell death. It has been reported that metabolic
inhibitors, including actinomycin D or CHX, dramatically sensitized
HCC cells to TNF-α-induced cell death due to their ability to
inhibit the de novo synthesis of anti-apoptotic proteins
(19). Following co-treatment with
TNF-α and CHX, knockdown of RACK1 led to markedly enhanced cell
death in SMMC7721 cells. This may be attributed to increased ROS
generation upon TNF-α stimulation. Hydrogen peroxide stimulation
also increased ROS generation and cell death. Treatment of HCC
cells with TNF-α/CHX is frequently used to mimic acute fulminant
hepatitis (20). Therefore, further
investigation of the mechanisms by which RACK1 affects the cell
death of HCC may support novel clinic treatments for acute
fulminant hepatitis.
Among the signaling pathways mediating TNF-α signal
transduction, three are considered to regulate the process of
TNF-α-induced cell death: Mitogen-activated protein kinase, nuclear
factor (NF)-κB and ROS. In a previous study, JNK activity, which is
frequently involved in promotion of cell death, was downregulated
by the knockdown of RACK1 (10). The
NF-κB signaling pathway is considered to be crucial to
anti-TNF-α-induced cell death, as TNF-α-induced NF-κB activation
induces the expression of numerous anti-apoptotic genes (21,22).
However, it has been reported IKKβ depletion in hepatocytes only
causes slightly increased sensitivity of cells to
lipopolysaccharide challenge (23),
which indicates that the effects of the NF-κB signaling pathway are
compensated by other factors in this process. In the present study,
CBR1 was identified as regulating TNF-α-induced HCC cell death.
Knockdown of CBR1 gave rise to enhanced cell death following
TNF-α/CHX stimulation, and overexpression of CBR1 in RACK1 stably
silenced single clone reversed enhanced cell death due to the
downregulation of RACK1. These data suggested RACK1 exerted its
protecting function via CBR1. Further investigation revealed that
RACK1 and CBR1 were involved in the same complex, indicating that
RACK1 is bound to CBR1. It should be noted that the amount of CBR1
protein in RACK1 stably silenced clone markedly decreased compared
to the control, suggesting that RACK1 sustained the protein level
of CBR1. Further experiments confirmed this observation. As RACK1
and CBR1 have been confirmed to be highly expressed in human HCC by
previous studies (6,10), the results of the present study
suggested that the levels of RACK1 may be positively correlated
with those of CBR1 in human HCC. Additionally, compared to
non-malignant BNL CL.2 cells, malignant HepG2 cells exhibited
increased protein levels of RACK1 and CBR1, and increased
resistance to TNF-α-induced cell death. Therefore, the positive
association between RACK1 and CBR1 may act in the malignant
transformation of normal liver cells to malignant cells. However,
these theories require further investigation.
In conclusion, the results of the present study have
confirmed that the increased expression of RACK1 in HCC cell lines
causes increased protein levels of CBR1 and a reduced ROS response,
thereby leading to increased resistance to TNF-α-induced cell
death.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no., 81472736
to JZ) and Innovation Foundation (grant no., 2015cxjj-011).
References
|
1
|
Wajant H: The role of TNF in cancer.
Results Probl Cell Differ. 49:1–15. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ivanov VN, Bhoumik A and Ronai Z: Death
receptors and melanoma resistance to apoptosis. Oncogene.
22:3152–3161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang S and El-Deiry WS: TRAIL and
apoptosis induction by TNF-family death receptors. Oncogene.
22:8628–8633. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: Signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schwabe RF and Brenner DA: Mechanisms of
liver injury. I. TNF-alpha-induced liver injury: Role of IKK, JNK,
and ROS pathways. Am J Physiol Gastrointest Liver Physiol.
290:G583–G589. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tak E, Lee S, Lee J, Rashid MA, Kim YW,
Park JH, Park WS, Shokat KM, Ha J and Kim SS: Human carbonyl
reductase 1 upregulated by hypoxia renders resistance to apoptosis
in hepatocellular carcinoma cells. J Hepatol. 54:328–339. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim YS, Seo HW and Jung G: Reactive oxygen
species promote heat shock protein 90-mediated HBV capsid assembly.
Biochem Biophys Res Commun. 457:328–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin B, Tan X, Liang J, Wu S, Liu J, Zhang
Q and Zhu R: A reduction in reactive oxygen species contributes to
dihydromyricetin-induced apoptosis in human hepatocellular
carcinoma cells. Sci Rep. 4:70412014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xia L, Mo P, Huang W, Zhang L, Wang Y, Zhu
H, Tian D, Liu J, Chen Z, Zhang Y, et al: The
TNF-α/ROS/HIF-1-induced upregulation of FoxMI expression promotes
HCC proliferation and resistance to apoptosis. Carcinogenesis.
33:2250–2259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo Y, Wang W, Wang J, Feng J, Wang Q, Jin
J, Lv M, Li X, Li Y, Ma Y, et al: Receptor for activated C kinase 1
promotes hepatocellular carcinoma growth by enhancing
mitogen-activated protein kinase kinase 7 activity. Hepatology.
57:140–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ron D and Mochly-Rosen D: An
autoregulatory region in protein kinase C: The pseudoanchoring
site. Proc Natl Acad Sci USA. 92:492–496. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gibson TJ: RACK1 research - ships passing
in the night? FEBS Lett. 586:2787–2789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mos M, Esparza-Franco MA, Godfrey EL,
Richardson K, Davey J and Ladds G: The role of the RACK1 ortholog
Cpc2p in modulating pheromone-induced cell cycle arrest in fission
yeast. PLoS One. 8:e659272013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saelee N, Tonganunt-Srithaworn M, Wanna W
and Phongdara A: Receptor for activated C kinase-1 protein from
Penaeus monodon (Pm-RACK1) participates in the shrimp antioxidant
response. Int J Biol Macromol. 49:32–36. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma J, Wu R, Zhang Q, Wu JB, Lou J, Zheng
Z, Ding JQ and Yuan Z: DJ-1 interacts with RACK1 and protects
neurons from oxidative-stress-induced apoptosis. Biochem J.
462:489–497. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Polosukhina D, Singaravelu K and Padanilam
BJ: Activation of protein kinase C isozymes protects LLCPK1 cells
from H2O2 induced necrotic cell death. Am J Nephrol. 23:380–389.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Korchak HM and Kilpatrick LE: Roles for
beta II-protein kinase C and RACK1 in positive and negative
signaling for superoxide anion generation in differentiated HL60
cells. J Biol Chem. 276:8910–8917. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jang M, Kim Y, Won H, Lim S, Kr J,
Dashdorj A, Min YH, Kim SY, Shokat KM, Ha J and Kim SS: Carbonyl
reductase 1 offers a novel therapeutic target to enhance leukemia
treatment by arsenic trioxide. Cancer Res. 72:4214–4224. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Okano H, Shiraki K, Inoue H, Kawakita T,
Yamanaka T, Deguchi M, Sugimoto K, Sakai T, Ohmori S, Fujikawa K,
et al: Cellular FLICE/caspase-8-inhibitory protein as a principal
regulator of cell death and survival in human hepatocellular
carcinoma. Lab Invest. 83:1033–1043. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pathil A, Warth A, Chamulitrat W and
Stremmel W: Comparison of different bile acid-phospholipid
conjugates in acute hepatitis. Eur J Clin Invest. 42:130–138. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Van Antwerp DJ, Martin SJ, Kafri T, Green
DR and Verma IM: Suppression of TNF-alpha-induced apoptosis by
NF-kappaB. Science. 274:787–789. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang G, Minemoto Y, Dibling B, Purcell NH,
Li Z, Karin M and Lin A: Inhibition of JNK activation through
NF-kappaB target genes. Nature. 414:313–317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maeda S, Chang L, Li ZW, Luo JL, Leffert H
and Karin M: IKKbeta is required for prevention of apoptosis
mediated by cell-bound but not by circulating TNFalpha. Immunity.
19:725–737. 2003. View Article : Google Scholar : PubMed/NCBI
|














