Introduction
Gastric plexiform fibromyxoma (PF), also known as
plexiform angiomyxoid myofibroblastic tumor (PAMT), is a
mesenchymal tumor that typically develops in the pyloric antrum or
duodenal bulb with a multinodular pattern infiltrating the
muscularis propria (1,2). The nodules are composed of bland spindle
cells embedded in a myxoid matrix with myofibroblastic
differentiation and small thin blood vessels (2). Epidemiologically, PF usually affects
young or middle-aged males and females (1,2). PF is
frequently misdiagnosed as gastrointestinal stromal tumor (GIST) in
routine clinical practice, but the prognosis is good with little
recurrence reported (1–8). GIST is a more prevalent mesenchymal
tumor involving the gastrointestinal tract, including the stomach
(60%) and small intestinal tract (30%), and other common locations
include the pancreas, gallbladder and appendix (9). Activation of KIT (80%) and
platelet-derived growth factor receptor α (PDGFRA, 7%) are
the typical molecular characteristics of this tumor (10). The biological behaviors of GIST are
varied and features including the anatomic location, tumor size and
mitotic activity are able to aid the evaluation of patient
prognosis (11). The morphology of
GIST frequently exhibits a sheet pattern with spindle or
epithelioid cells, but plexiform architecture and the presence of
myxoid matrix are uncommon (12).
The current study presents a case of PF and a case
of GIST masquerading as PF by exhibiting a distinctive multinodular
figuration due to a PDGFRA mutation. Therefore, the present
study demonstrates the necessity for considering and eliminating
the possibility of GIST first, when determining the differential
diagnosis of gastrointestinal mesenchymal tumors.
Case report
Case 1
A 51-year-old male was admitted to the First
Affiliated Hospital of Zhengzhou University (Zhengzhou, China) in
January 2013 due to epigastric discomfort and an intermittent
burning sensation in the chest, without nausea, acid reflux or
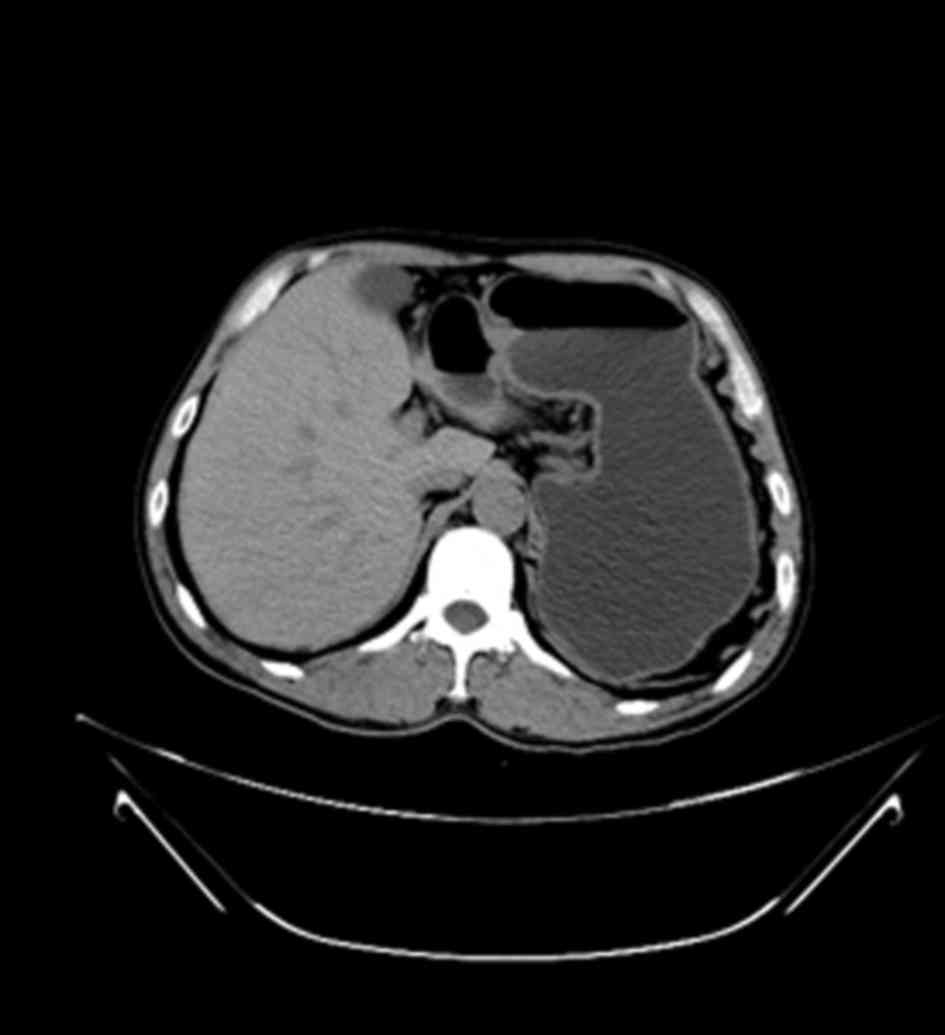
stomach-ache. A gastroscopy and a computed tomography (CT) scan
revealed a mass located in the atrium of the greater curvature of
the stomach and protruding into the gastric cavity, creating a
narrowed outlet (Fig. 1). No
significant biomarkers were identified, including α-fetoprotein,
carcinoembryonic antigen and carbohydrate antigen 19–9, and there
was no relevant family medical history. Total resection of the
tumor was performed, and the patient had no evidence of relapse or
metastasis for 15 months following surgery.
The dimensions of the submucosal tumor were 4×3×1.8
cm; the cut surface appeared pale tan and gelatinous, and subtle
intramural nodules were visible. The surgical specimen was
routinely fixed in 4% buffered formalin, embedded in paraffin and
then hematoxylin and eosin (H&E) staining was performed using
the Leica ST 5010 Autostainer XL (Leica Microsystems, Inc., Buffalo
Grove, IL, USA). Immunohistochemical studies were performed with
commercial antibodies using the Ventana BenchMark XT instrument
(Ventana Medical Systems, Inc., Tucson, AZ, USA) according to the
manufacturer's protocol. Briefly, the tissue sections were
deparaffinized using EZ prep solution (Ventana Medical Systems,
Inc.), followed by heat-induced epitope retrieval using CC1
solution (20 min at 95°C; Ventana Medical Systems, Inc.).
Subsequently, the slides were incubated with primary antibodies for
1 h at 37°C and Ventana anti-rabbit secondary antibody (ultraView
universal HRP multimer, prediluted; included in the ultraView
Universal DAB Detection kit, no. 760-500; Ventana Medical Systems,
Inc.) for 8 min at 37°C. The immunoreaction was detected under a
light microscope (BX43; Olympus Corporation, Tokyo, Japan)
following use of the ultraView Universal DAB Detection kit and
counterstaining with hematoxylin and bluing reagent (Ventana
Medical Systems, Inc.). The primary antibodies included KIT (no.
A4502; polyclonal; dilution, 1:200; Dako, Glostrup, Denmark), Dog-1
(no. MONX11114; clone K9; dilution, 1:200; Novocastra; Leica
Microsystems Inc.), S-100 (no. Z0311; polyclonal; dilution, 1:400;
Dako), SMA (no. M0851; clone 1A4; dilution, 1:100; Dako) desmin
(no. M0760; clone D33; dilution, 1:50; Dako), Ki-67 (no. M7248;
clone MIB; dilution 1:100; Dako), cluster of differentiation (CD)
34 (no. 550760; clone MY10, dilution, 1:100; BD Biosciences, San
Jose, CA, USA) and cytokeratin AE1/AE3 (no. M3515; clone AE1+AE3;
dilution 1:100; Dako). Sanger sequencing was performed to detect
the status of exons 9, 11, 13 and 17 of KIT and exons 12 and 18 of
PDGFRA. The coding regions of these exons were amplified by
polymerase chain reaction (PCR) using HotStart Taq DNA polymerase.
The reaction conditions and primers were used according to
previously published protocol (13–15). The
PCR products were directly sequenced using BigDye Terminator v3.1
Cycle Sequencing kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.) according the manufacturer's protocol on the ABI 4500Dx, and
analyzed using ABI Prism 3500Dx DNA Sequence Analysis Software
version 4.0. The products were sequenced with forward and reverse
primers, as previously reported (16,17).
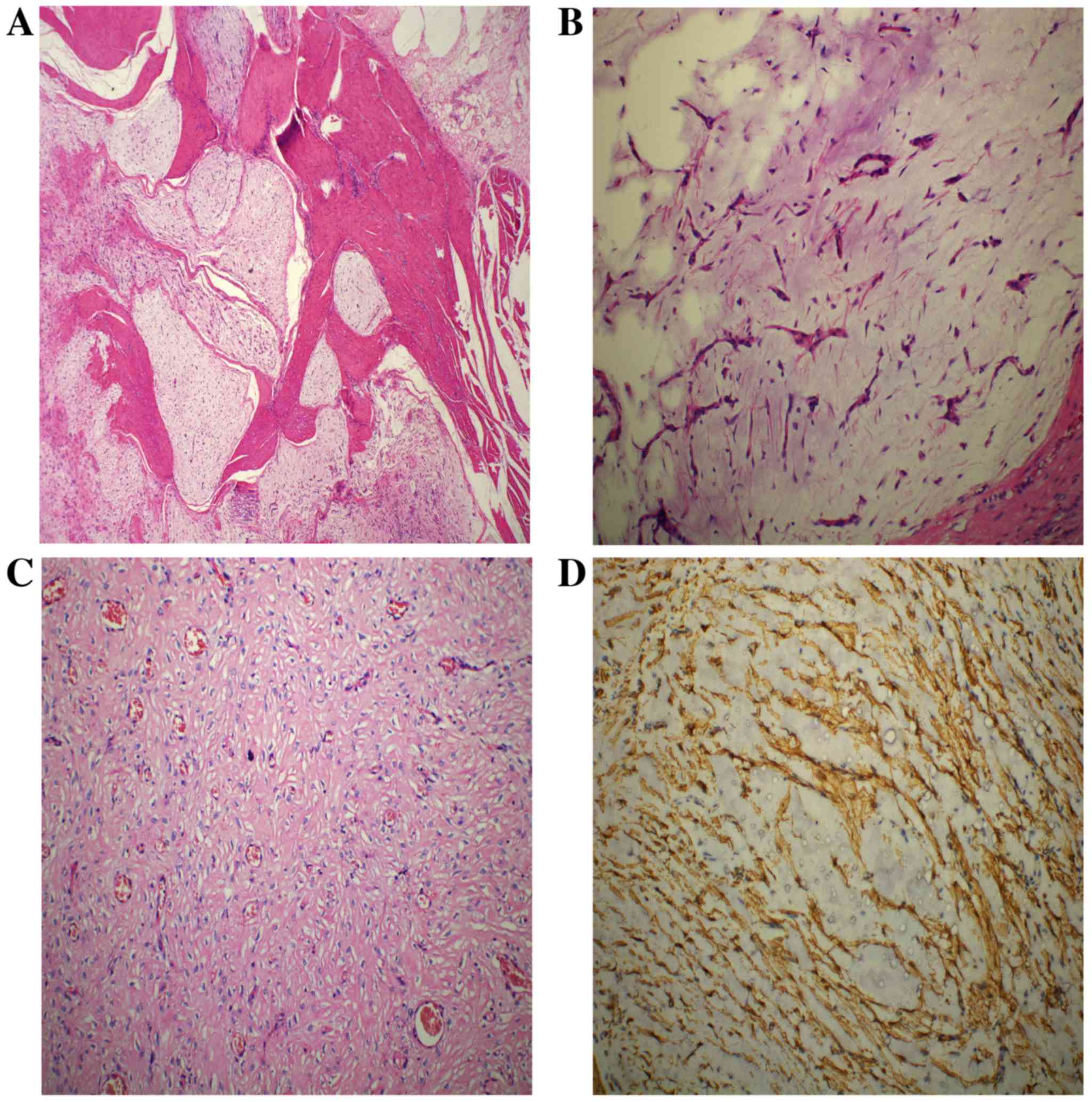
Histological analysis revealed that the tumor had a plexiform and
multinodular involvement in the muscularis propria (Fig. 2A). The nodules were variable in size
with a fine demarcation or infiltrative margin from the remaining
normal tissues, a number of which had coalesced to form sheets.
Within the nodules, the bland spindle cells (with tapering ends,
round or oval nuclei, fine chromatin, indistinct nucleoli, an
eosinophilic or amphophilic cytoplasm, and discrete cell borders)
were dispersed in the matrix (Fig.
2B). Necrosis and mitotic bodies were not detectable using a
light microscope (magnification, 200x; BX43; Olympus Corporation).
An arborizing vascular network of small capillaries was observed in
the tissue (Fig. 2B). In addition,
some tumor cells with indistinct borders were embedded in the dense
collagenous matrix deficient in mucus, and exhibited an epithelioid
appearance reminiscent of epithelioid GIST (Fig. 2C). The tumor cells demonstrated
immunoreactivity for smooth muscle actin (SMA; Fig. 2D), but were negative for mast/stem
cell growth factor receptor (KIT), GIST-1 (DOG1), CD34, S-100,
desmin and cytokeratin AE1/AE3; staining for CD34 delineated the
capillary network. The Ki-67 proliferation index was ~1%, and
KIT (exon 9, 11, 13 and 17) and PDGFRA (exon 12 and
18) genetic mutations were not identified. The final diagnosis was
determined to be PF.
Case 2
A 47-year-old female was admitted to The First
Affiliated Hospital of Zhengzhou University in April 2013 due to a
2-month history of intermittent abdominal distension, acid reflux
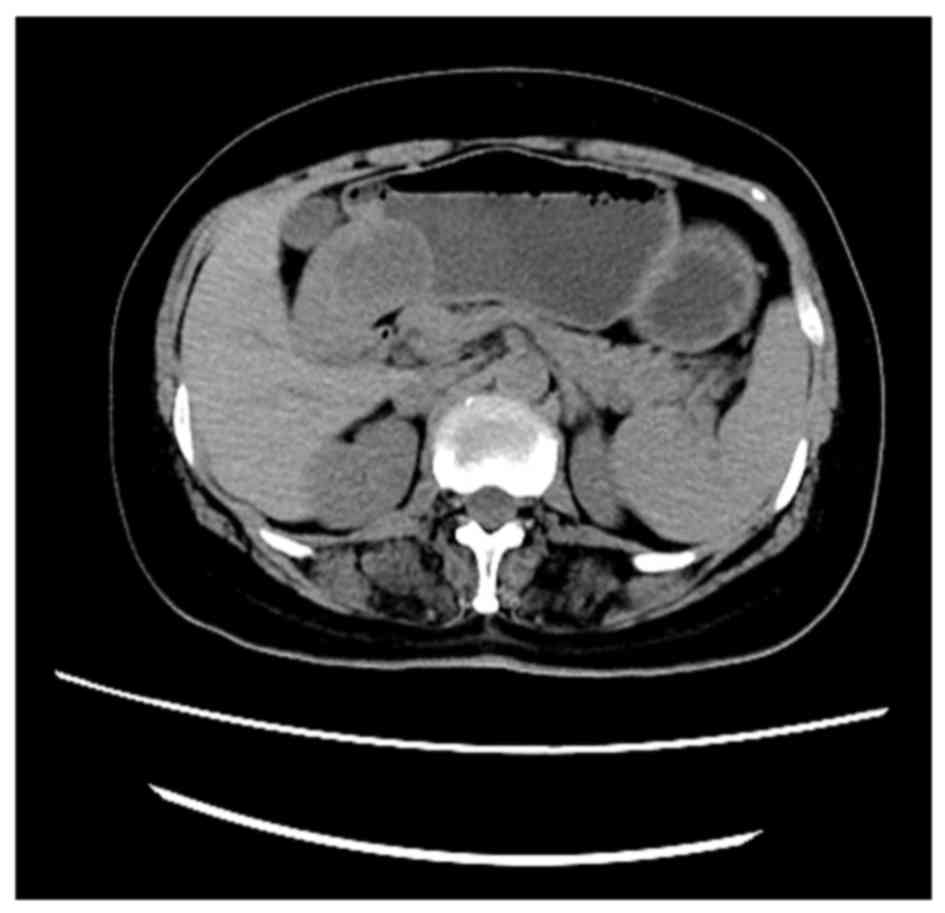
and heartburn. A CT scan revealed a mass (48×36 mm in the maximum
cross-section) located in the gastric antrum of the lesser
curvature (Fig. 3). No significant
biomarkers or relevant family medical history were identified. The
neoplasm was completely resected by a distal gastrectomy without
adjuvant therapies, and the patient had a favorable prognosis
without relapse or metastasis for 12 months.
The submucosal tumor measured 4×3×3 cm, and the cut
surface appeared gray and subtly mucoid. The resected specimen was
examined using H&E and immunohistochemical staining, and the
molecular testing of KIT and PDGFR was performed,
according to the aforementioned protocol in case 1. Under a light
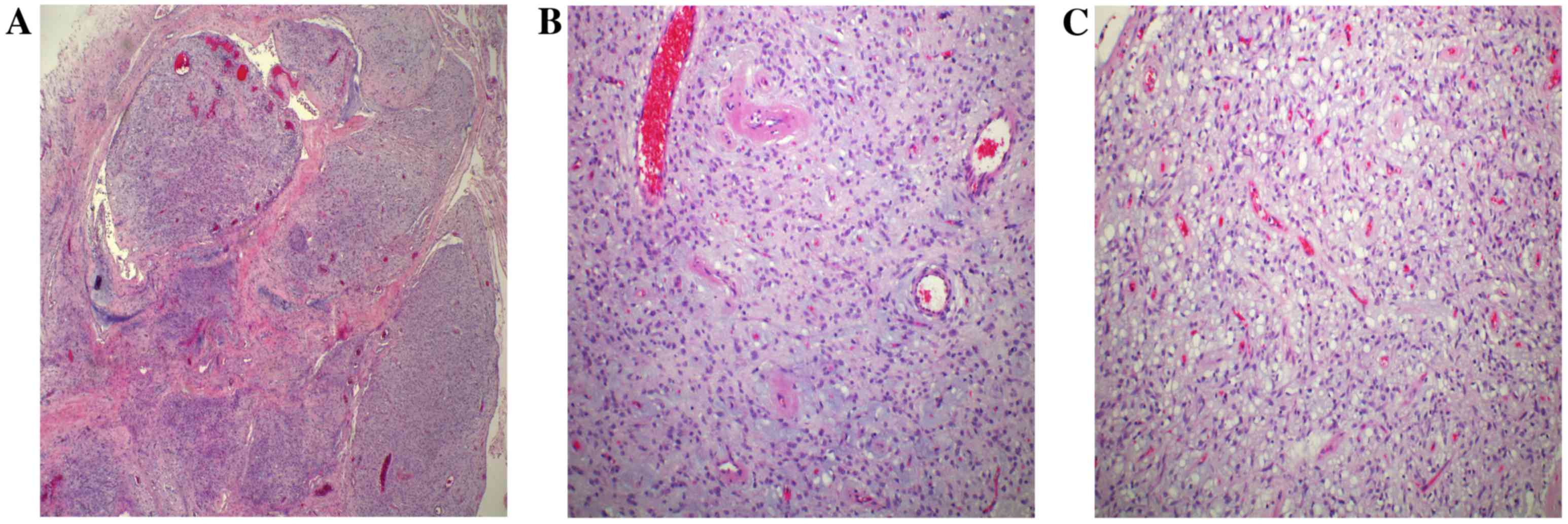
microscope (BX43; Olympus Corporation), the tumor exhibited a
nodular growth pattern involving the muscularis propria (Fig. 4A). The nodules varied in size and
merged together in some areas; they were principally composed of
numerous randomly arranged spindle cells, short capillaries and
dilated small veins in a myxoid matrix (Fig. 4B). Significant hemorrhage was observed
in some of the nodules, but evidence of necrosis and mitotic bodies
was absent. In addition, the tumor cells in some nodules exhibited
prominent cytoplasmic vacuoles, similar to the vacuoles typically
observed in epithelioid GIST (Fig.
4C). All these features are similar to those observed in PF, as
depicted in case 1. However, the diagnosis of GIST must be
eliminated for soft tissue neoplasms in the gastrointestinal tract,
due to the changeable morphological patterns observed in this
disease. Accordingly, immunohistochemistry was performed,
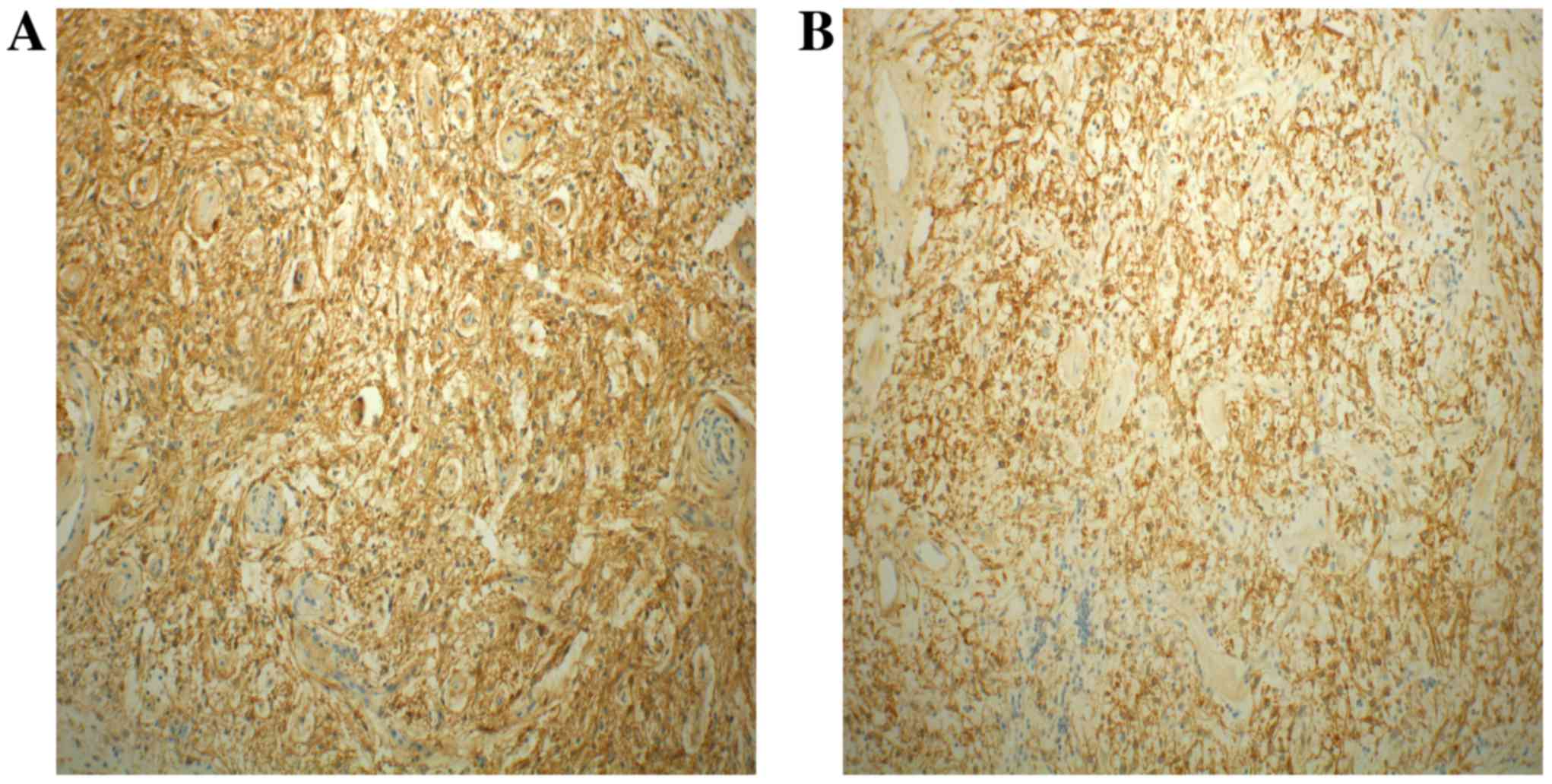
demonstrating that the tumor cells were positive for KIT (Fig. 5A) and DOG1 (Fig. 5B), but were negative for SMA, CD34,
S-100, desmin and AE1/AE3. The Ki-67 proliferation index was ~3%.
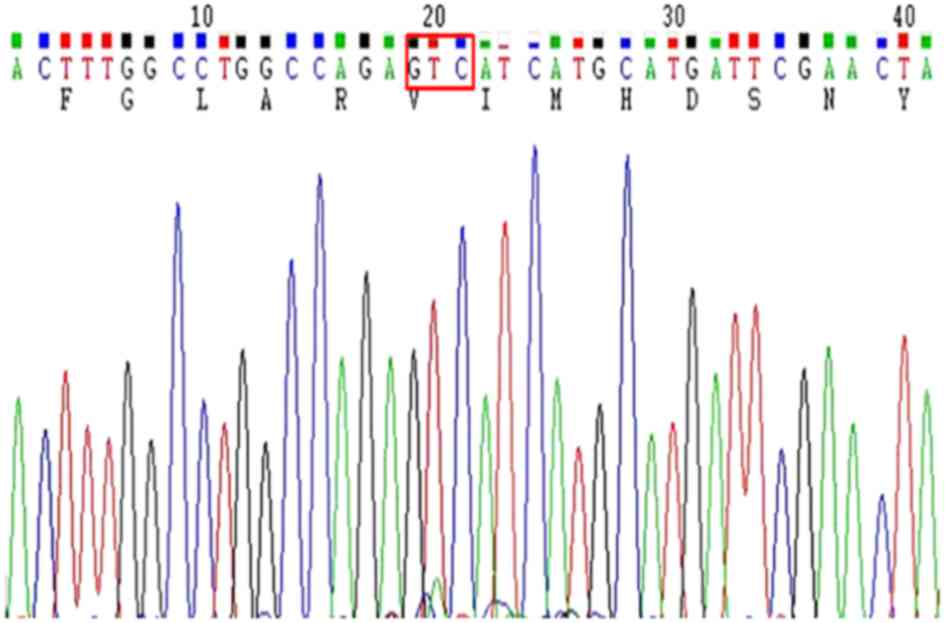
Sequencing revealed a D842V mutation in exon 18 of the
PDGFRA gene; however, mutations in exon 12 of the
PDGFRA and KIT genes were not detected (Fig. 6). Therefore, the final diagnosis was
determined to be GIST.
Discussion
PF is a rare and benign cancer that exclusively
develops in the stomach, particularly in the gastric antrum, and
has previously been designated as myxoma, giant myxoma, fibromyxoma
and plexiform angiomyxoid myofibroblastic tumor (1,3,18,19). PF
has no significant gender distribution and primarily presents in
young and middle-aged adults (7–75 years, mean 41 years) (2,20).
Patients typically present with gastrointestinal bleeding, anemia,
ulcer, hematemesis, an occasional gastric outlet obstruction or
other nonspecific upper digestive tract symptoms (5,8). The
tumors usually range in size from 2 to 15 cm (median, 5.5 cm), and
have a multinodular, pale appearance and a mucoid cut surface
(2).
PF exhibits a histologically distinctive
multinodular plexiform architecture involving the muscularis
propria and the serosa with an infiltrative margin (2,21). The
nodules are usually composed of an abundant myxoid matrix,
prominent arborizing capillaries, and scattered spindle cells with
small nuclei, inconspicuous nucleoli and an eosinophilic cytoplasm
(1,2).
Mitotic activity and necrosis are scarcely apparent (1,22).
Immunohistochemistry typically demonstrates that the PF tumor cells
are consistently positive for SMA, but negative for KIT, DOG1,
CD34, S-100, desmin and caldesmon, indicating myofibroblastic
differentiation and a low Ki-67 proliferation index (20). Previous studies and follow-up data
indicate that PF typically has a favorable prognosis and that a
conservative treatment involving tumor resection is sufficient
(1–8).
As the most common mesenchymal neoplasm in the
gastrointestinal tract, GISTs are considered to originate from the
interstitial cells of Cajal (1). They
are primarily driven by the mutations in KIT or
PDGFRA, which account for 80% and 5–8% of cases,
respectively (23). GISTs may develop
at any age, but typically present in middle-aged or elderly
patients, with a median age of onset of ~60 years without specific
gender distribution (24).
By contrast to PFs, GISTs may involve any tissues
along the gastrointestinal tract, including the gastric antrum, and
have varied biological behaviors that may be assessed using a
specialized system of risk stratification based on anatomic site,
tumor size and mitotic activity (11,25). A
variable histological morphology of GIST tissues may be observed,
including completely spindle shaped, entirely epithelioid shaped or
pleomorphic cell cytomorphologies (25). The spindle cell GISTs of the stomach
are often arranged in fascicles with round or elongated nuclei,
fine chromatin, inconspicuous nucleoli, a moderate to abundant
quantity of eosinophilic cytoplasm, relatively infrequent mitotic
bodies and no cytological pleomorphisms (25,26).
Occasionally, gastric GISTs may exhibit prominent paranuclear
vacuoles and nuclear palisading that mimics schwannoma (26). Epithelioid GIST variants exhibit a
sheet-like or nested growth pattern with round or polygonal cells,
vesicular chromatin and an eosinophilic, clear or vacuolated
cytoplasm, which may occasionally result in a conspicuous signet
ring form (27). Extremely rare
subsets of gastric GISTs have a plexiform growth pattern,
particularly in the case of ‘pediatric-type’ GIST or patients with
Carney triad or Carney-Stratakis syndrome. These cases exhibit
distinctive features including female predilection, young age,
epithelioid tumor cells or a multifocal growth pattern, frequent
lymph node metastasis and serial tumor occurrence, but relatively
indolent behavior even with metastasis, with a low number of cases
being fatal (2,28,29). The
presence of these characteristics indicates that a diagnosis of
GIST may be considered. Other lesions, including extra-adreanal
paragonglioma, pulmonary chondroma and the mutation of succinate
dehyrogenase (SDH) complex B, SDH complex C, or SDH complex D (the
genetic basis of Carney-Stratakis syndrome) may also be detected
(30). Similar to the aforementioned
literature (1,28,29), the
patient discussed in case 2 was diagnosed with GIST and had a
favorable prognosis; however, no evidence of a Carney complex or of
other hereditary diseases was identified.
GIST tissues frequently exhibit variably sized
nodules with irregular margins or broad bands involving the
muscularis propria; the nodules are often composed of variable
areas of epithelioid or mixed spindle and epithelioid cell
morphology, with or without myxoid or collagen matrix (29).
The majority of GISTs have constitutively activating
mutations in the KIT gene with four mutation hotspots,
including on exons 9, 11, 13 and 17, accounting for 65, 10, 1 and
1% of GISTs, respectively (31). The
small molecular inhibitor imatinib has been used as the first-line
drug for the treatment of GIST with KIT mutations (32). However, a minority of GISTs (~5%) have
a PDGFRA mutation, with three mutation hotspots, of which
exon 18 is the most frequently affected (almost always the D842V
mutation), contributing to the clinical resistance to imatinib and
a more favorable disease course (23,31,33).
Similarly, the case of GIST presented in the current study had a
good prognosis following tumor resection without targeted therapy.
GISTs with an epithelioid phenotype or a plexiform growth pattern
usually exhibit a PDGFRA mutation, and are preferentially
located in the stomach (33).
Immunohistochemistry has demonstrated that ~80% of GISTs express
KIT (CD117) and CD34, but SMA S-100 and desmin are expressed in 25,
5 and <1% of GISTs, respectively (34).
The case of gastric GIST discussed in the present
study exhibited a prominent multinodular or plexiform pattern with
myxoid stroma and proliferated capillaries, reminiscent of PF.
However, the case of PF exhibited a more abundant mucous matrix and
a distinctively elongated vascular network that was not present in
the GIST tissues. The nodules of the GIST consisted of sheets of
round cells with prominent cytoplasmic vacuoles, suggesting a
possible diagnosis of a GIST with a distinctive growth pattern,
rather than PF; however, in PF tissues, the presence of some tumor
cells embedded in the dense collagenous stroma with epithelioid
features may superficially resemble GIST. Immunostaining for KIT
and DOG1 and the detection of a D842V mutation in exon 18 of the
PDGFRA gene distinguishes these two types of gastric
cancer.
In conclusion, PF is a rare benign mesenchymal
neoplasm that frequently develops in the gastric antrum, and
exhibits a prominent plexiform growth pattern with an abundant
myxoid matrix and a distinctive vascular network. However, a small
subset of gastric GISTs that present with myxoid and multinodular
histological features may masquerade as PF. Therefore, GIST must be
considered and rejected first when determining a differential
diagnosis for gastrointestinal mesenchymal neoplasms.
Acknowledgements
This study was supported by The First Affiliated
Hospital of Zhengzhou University.
References
|
1
|
Takahashi Y, Shimizu S, Ishida T, Aita K,
Toida S, Fukusato T and Mori S: Plexiform angiomyxoid
myofibroblastic tumor of the stomach. Am J Surg Pathol. 31:724–728.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miettinen M, Makhlouf HR, Sobin LH and
Lasota J: Plexiform fibromyxoma: A distinctive benign gastric
antral neoplasm not to be confused with a myxoid GIST. Am J Surg
Pathol. 33:1624–1632. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fukunaga M: Gastric fibromyxoma, a
distinct entity of pure fibroblastic tumor-an ultrastructural
study. APMIS. 112:304–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ikemura M, Maeda E, Hatao F, Aikou S, Seto
Y and Fukayama M: Plexiform angiomyxoid myofibroblastic tumor
(PAMT) of the stomach. A case report focusing on its characteristic
growth pattern. Int J Clin Exp Pathol. 7:685–689. 2014.PubMed/NCBI
|
|
5
|
Lee PW, Yau DT, Lau PP and Chan JK:
Plexiform fibromyxoma (plexiform angiomyxoid myofibroblastic tumor)
of stomach: An unusual presentation as a fistulating abscess. Int J
Surg Pathol. 22:286–290. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim A, Bae YK, Shin HC and Choi JH:
Plexiform angiomyxoid myofibroblastic tumor of the stomach: A case
report. J Korean Med Sci. 26:1508–1511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kang Y, Jung W, Do IG, Lee EJ, Lee MH, Kim
KM and Choi J: Plexiform angiomyxoid myofibroblastic tumor of the
stomach: Report of two cases and review of the literature. Korean J
Pathol. 46:292–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takahashi Y, Suzuki M and Fukusato T:
Plexiform angiomyxoid myofibroblastic tumor of the stomach. World J
Gastroenterol. 16:2835–2840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de la Roza G, Naqvi A and Clark K:
Gastrointestinal stromal tumors presenting as a prostatic mass. Can
J Urol. 16:4502–4506. 2009.PubMed/NCBI
|
|
10
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: Pathology and prognosis at different sites. Semin
Diagn Pathol. 23:70–83. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: Review on morphology, molecular pathology,
prognosis, and differential diagnosis. Arch Pathol Lab Med.
130:1466–1478. 2006.PubMed/NCBI
|
|
13
|
Tryggvason G, Hilmarsdottir B, Gunnarsson
GH, Jónsson JJ, Jónasson JG and Magnusson MK: Tyrosine kinase
mutations in gastrointestinal stromal tumors in a nation-wide study
in Iceland. APMIS. 118:648–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Went PT, Dirnhofer S, Bundi M, Mirlacher
M, Schraml P, Mangialaio S, Dimitrijevic S, Kononen J, Lugli A,
Simon R and Sauter G: Prevalence of KIT expression in human tumors.
J Clin Oncol. 22:4514–4522. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Buleje J, Acosta Ó, Guevara-Fujita M,
Enriquez Y, Taxa L, Machicado E, Lizaraso-Caparó F and Fujita R:
Mutational profile of KIT and PDGFRA genes in gastrointestinal
stromal tumors in Peruvian samples. Rev Esp Enferm Dig. 107:72–78.
2015.PubMed/NCBI
|
|
16
|
Antonescu CR, Sommer G, Sarran L,
Tschernyavsky SJ, Riedel E, Woodruff JM, Robson M, Maki R, Brennan
MF, Ladanyi M, et al: Association of KIT exon 9 mutations with
nongastric primary site and aggressive behavior: KIT mutation
analysis and clinical correlates of 120 gastrointestinal stromal
tumors. Clin Cancer Res. 9:3329–3337. 2003.PubMed/NCBI
|
|
17
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A,
Town A, et al: PDGFRA activating mutations in gastrointestinal
stromal tumors. Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Traverso JP and Vidal HJ: Stomach myxoma.
Prensa Med Argent. 43:881–883. 1956.(In Spanish). PubMed/NCBI
|
|
19
|
Faraoni H, Urruti ER Recagno and Escalante
DA: Giant myxoma of the stomach. Sem Med. 106:135–136. 1955.(In
Spanish). PubMed/NCBI
|
|
20
|
Duckworth LV, Gonzalez RS, Martelli M, Liu
C, Coffin CM and Reith JD: Plexiform fibromyxoma: Report of two
pediatric cases and review of the literature. Pediatr Dev Pathol.
17:21–27. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Galant C, Rousseau E, Ho Minh Duc DK and
Pauwels P: Re: Plexiform angiomyxoid myofibroblastic tumor of the
stomach. Am J Surg Pathol. 32:1910author reply 1912–1913. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sing Y, Subrayan S, Mqadi B, Ramdial PK,
Reddy J, Moodley MS and Bux S: Gastric plexiform angiomyxoid
myofibroblastic tumor. Pathol Int. 60:621–625. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eisenberg BL and Pipas JM:
Gastrointestinal stromal tumor-background, pathology, treatment.
Hematol Oncol Clin North Am. 26:1239–1259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Quek R and George S: Gastrointestinal
stromal tumor: A clinical overview. Hematol Oncol Clin North Am.
2369–78. (viii)2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kitamura Y: Gastrointestinal stromal
tumors: Past, present, and future. J Gastroenterol. 43:499–508.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miettinen M and Lasota J: Histopathology
of gastrointestinal stromal tumor. J Surg Oncol. 104:865–873. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suster S and Fletcher CD: Gastrointestinal
stromal tumors with prominent signet-ring cell features. Mod
Pathol. 9:609–613. 1996.PubMed/NCBI
|
|
28
|
Zhang L, Smyrk TC, Young WF Jr, Stratakis
CA and Carney JA: Gastric stromal tumors in Carney triad are
different clinically, pathologically, and behaviorally from
sporadic gastric gastrointestinal stromal tumors: Findings in 104
cases. Am J Surg Pathol. 34:53–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rege TA, Wagner AJ, Corless CL, Heinrich
MC and Hornick JL: ‘Pediatric-type’ gastrointestinal stromal tumors
in adults: Distinctive histology predicts genotype and clinical
behavior. Am J Surg Pathol. 35:495–504. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pasini B, McWhinney SR, Bei T, Matyakhina
L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie
G, et al: Clinical and molecular genetics of patients with the
Carney-Stratakis syndrome and germline mutations of the genes
coding for the succinate dehydrogenase subunits SDHB, SDHC, and
SDHD. Eur J Hum Genet. 16:79–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Joensuu H: Gastrointestinal stromal tumor
(GIST). Ann Oncol. 17:(Suppl 10). x280–x286. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Siehl J and Thiel E: C-kit, GIST, and
imatinib. Recent Results Cancer Res. 176:145–151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang J, Du X, Lazar AJ, Pollock R, Hunt K,
Chen K, Hao X, Trent J and Zhang W: Genetic aberrations of
gastrointestinal stromal tumors. Cancer. 113:1532–1543. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miettinen M, Sobin LH and Lasota J:
Gastrointestinal stromal tumors of the stomach: A
clinicopathologic, immunohistochemical, and molecular genetic study
of 1765 cases with long-term follow-up. Am J Surg Pathol. 29:52–68.
2005. View Article : Google Scholar : PubMed/NCBI
|